
Abstract
Current nomenclature for the taxonomic classification of melon cultivars (Cucumis melo L.) at the horticultural group level relies on morphological variation in certain key characters. However, the reliability of current infraspecific classification scheme in considering horticultural groups as botanical taxa was not fully understood. In the present study, the information of horticultural group classification in melon was assessed at the molecular level using genome-wide single nucleotide polymorphisms (SNPs). A total of 143 melon accessions of 15 horticultural groups in two subspecies, subsp. melo and subsp. agresti were collected and genotyped by using Genotyping-By-Sequencing (GBS). From the filtering of resultant sequence variants, 10,949 SNPs were selected and used for downstream genetic analysis including population structure, principle component analysis, and hierarchical clustering of 143 melon accessions. Our genetic analyses indicated that the distribution of accessions at the molecular level generally matched the subspecies classification and no substantial contradictions existed between the division of accessions based on horticultural group information and genetic relatedness revealed by the GBS. However, the distinction between horticultural groups was not clear-cut, implying the limitation of considering horticultural groups as botanical taxa. To improve the resolution of horticultural group classification in melon, our SNP data may be useful as supporting information in conjunction with morphological characters.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Melon (Cucumis melo L. 2n = 2 × = 24) belongs to the genus Cucumis and has two geographical centers of origin, Africa and Asia (Sebastian et al. 2010). Cultivated melons are an economically important fruit vegetable crop, which differ from wild or feral-type melons in some domesticated traits such as large fruits, non-bitter taste, and thicker flesh. Cultivated melons include sweet ‘dessert’ melons, as well as non-sweet forms that are consumed raw, pickled, or cooked. Due to extensive phenotypic diversity, botanists have proposed an infraspecific classification scheme for melon (Robinson and Decker-Walters 1997). Generally, C. melon is classified into two sub-species, subsp. melo and subsp. agrestis, depending on the length of the pubescence (hair) of the ovary (Jeffrey 1980). These two subspecies are further divided into diverse infraspecific or horticultural (varietal or cultivar) groups based on key flower and fruit characteristics, such as sex expression, fruit size and shape, sweetness, flavor, color, and climacteric attributes (Munger and Robinson 1991; Robinson and Decker-Walters 1997; Pitrat et al. 2000).
Botanists have suggested and revised different schemes and nomenclature for the taxonomic classification of melon at the horticultural group level. A European taxonomy suggested by Pitrat (2008) is widely accepted for ‘infraspecific taxa’, which classifies cultivated melons, including Asian types, into 16 different horticultural groups; 11 groups for subsp. melo (adana, ameri, cantalupensis, chandalak, chate, chito, dudaim, flexuosus, inodorus, reticulatus, and tibish) and five groups for subsp. agrestis (acidulous, chinensis, conomon, makuwa, and momordica). However, distinctions between horticultural groups are not clear and the assignment of a cultivar to a specific horticultural group is not easy. Thus, more efficient methods to improve the current taxonomy scheme are needed (Esteras et al. 2013). Molecular polymorphisms are widely used to assess genetic diversity due to their abundance among individuals and the robustness of the data (Bae et al. 2015; Sim et al. 2015). In melon, molecular markers such as RAPD, SSR, and single nucleotide polymorphisms (SNPs) have been used as reference data to validate and refine the scheme of current taxonomic classification (Stepansky et al. 1999). However, case studies based on genome-wide genotyping in a broad range of melon accessions remain limited.
In melon, 83.3% (375 Mb) of the genome of a double-haploid line DHL92 has been sequenced via NGS, and the information is publicly available as a reference genome (Cucumis melo L. pseudo-molecules v3.5.1, https://melonomics.cragenomica.es) (Garcia-Mas et al. 2012). This work paved the way for whole-genome resequencing (WGRS) of diverse melon germplasm. While WGRS is costly and time-consuming, genotyping-by-sequencing (GBS) is a high-throughput genotyping method that can generate sufficient sequence variants by analyzing a small part of the genome with a relatively simple protocol at lower cost (Elshire et al. 2011; Poland and Rife 2012). GBS has been widely used to mine large quantities of SNPs and for genome analysis, including the construction of high-density genetic maps, genome-wide association studies (GWAS), and for analysis of genetic diversity in germplasm collections.
In the present study, GBS was performed using 143 melon accessions assigned to 15 horticultural groups. Genetic relationships between melon accessions were assessed based on genome-wide SNP data produced from the GBS and evaluated for consistency with information of infraspecific taxa. We discuss the current scheme of infraspecific taxa in melon in comparison to relationships determined by DNA sequence analysis.
2 Materials and methods
2.1 Plants materials
A melon germplasm collection comprising 143 melon accessions of 15 horticultural groups in two C. melo subspecies (subsp. melo and subsp. agrestis) was used as the plant material (Table 1). Among these, 93 accessions were selected from the melon germplasm inventory of the Gyeongnam Agricultural Research and Extension Service (GARES) (Jinju, Gyeongnam, Korea). An additional set of 50 accessions comprising 13 horticultural groups was selected based on the curation reported by Pitrat (2008). These 50 accessions were used as a reference array to validate the consistency between DNA sequence variations and infraspecific classification. Seeds of 93 accessions in the GARES inventory were provided by GARES, while reference array accessions were obtained from the Germplasm Resources Information Network (GRIN), USA, and the Leibniz Institute of Plant Genetics and Crop Plant Research (IPK), Germany.
2.2 Construction of GBS library and NGS
Five seedlings for each accession were grown on a plastic cell tray until the third true leaves were fully expanded. Leaf samples were collected and ground in liquid nitrogen. Genomic DNA was extracted using cetyltrimethylammonium bromide (CTAB) following the method described by Saghai-Maroof et al. (1984), with modifications. Purified DNA samples were quantified using a NANODROP 1000 (Thermo Scientific, Waltham, MA, USA) and diluted to 100 ng〮μL−1 for GBS. To construct the GBS library, each genomic DNA sample was digested with 3.6 U of ApeKI restriction enzyme in 20 μL reaction volume for 4 h at 75 °C, and 20U T4 DNA ligase was added to ligate the adapter-containing barcode to a common adapter. Then, ligated DNA samples were pooled and cleaned using the QIAquick PCR purification kit, and PCR was carried out as follows: 2 min at 95 °C; 16 cycles of 30 s at 95 °C, 30 s at 62 °C; 30 s at 68 °C; and 5 min at 68 °C. The GBS library was sequenced by Theragen Etex Bio Institue (Daejeon, Korea) using Illumina Hiseq 2000.
2.3 SNP detection
Barcodes and adapter sequences were removed from demultiplexed reads using Cutadapt (v.1.8.3) (Martin 2011), while sequence quality trimming was conducted with DynamicTrim and LengthSort of the SolexaQA (v.1.13) package (Cox et al. 2010). Cleaned reads were mapped using the BWA (0.6.1-r104) program based on a melon reference genome (C. melo L. pseudo-molecules v3.5.1) (Li and Durbin 2009), and then SAM files were prepared for the detection of raw SNPs between the reference genome and sequenced samples. An integrated SNP matrix of all DNA samples was constructed after SNP filtering using SAMtools (v.0.1.16) under the following conditions; biallelic SNP loci, min. depth ≥ 10, minor allele frequency (MAF) ≥ 5%, missing data < 20%.
2.4 Population structure and genetic relationship
Population structure was analyzed based on the admixture-based clustering model (Bayesian model-based Markov Chain Monte Carlo model) of STRUCTURE 2.3.4. The K value ranged from 1 to 10, and K value analyses were each repeated 10 times by setting a burn-in period of 10,000. The optimal minimum number of subpopulations was calculated based on second-order rate of change of likelihood (delta K). PCA was conducted using the SNPRelate R package (Zheng et al. 2012). A hierarchical clustering tree was calculated based on Nei’s genetic distance using the Poppr R package (Kamvar et al. 2014) and bootstrap values were determined based on 1000 replications.
3 Results
3.1 GBS and SNP detection
NGS of the GBS library for 143 accessions resulted in ~ 58 M raw reads, and the number of trimmed reads was ~ 52 M (88.2%). For each accession, the average number of trimmed reads was ~ 3.63 M, and ~ 2.65 M (73.5%) reads were mapped to the reference genome, indicating 1.4% genome coverage. The average length of mapped reads was 81.35 bp and their average depth was 27.91 X. After SNP filtering of mapped reads, 10,949 SNPs were selected and used for subsequent analyses. Among those SNPs, 15% were located in intergenic regions, 32% in introns, 44% in exons, and 9% in untranslated regions (UTRs). The transition/transversion ratio of those genome-wide SNPs was 2.03. Relatively higher SNP distribution was observed from the marginal regions of each chromosome.
3.2 Population structure
Genetic analyses of population structure, PCA, and hierarchical clustering were conducted for 143 melon accessions comprising 16 horticultural groups by engaging 10,949 SNPs. In each genetic analysis, 50 accessions of the reference array were analyzed to assess the sustainability of the results between the two population settings (50 reference array accession and a total of 143 accessions).
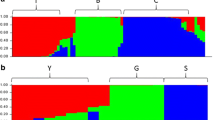
Population structure analysis of 50 accessions of a reference array found that estimation of the deltaK parameter inferred the most suitable model with four subpopulations (K = 4) (Fig. 1a, b). Distinct subpopulation structures were observed for the accessions adana, cantalupensis, chandalak, and inodorus of subsp. melo (yellow color-coded), for the accessions conomon and makuwa of subsp. agrestis (red color-coded), for the accessions flexuosus (blue color-coded), and for one accession of dudaim (green color-coded). Many other accessions showed an admixture of two subspecies. The best-fit model for the total 143 accessions was found for two subpopulations (K = 2) (Fig. 1a, c). The distribution of the accessions in the two subpopulations generally matched the subspecies classification, subsp. melo (red color-coded) and subsp. agrestis (yellow color-coded). However, many accessions presented membership fraction values (q) close to 0.5, indicating that their genetic structures are an admixture of both subspecies. The second-best fit was found for subgrouping the accessions into eight subpopulations (K = 8), from which the division of each accession into its horticultural group taxonomic classification was not clear (Fig. 1d).

Population structure of a melon (Cucumis melo L.) germplasm collection was analyzed using the admixture-based clustering model of STRUCTURE. Each accession is represented by a vertical line. Each color indicates a different subpopulation inferred by estimation of the deltaK parameter. a Estimation of the number of population by delta K value for 50 accessions of the reference array (Left) and 143 accessions (Right), b Population structure for 50 accessions of the reference array showing the number of subpopulations (K) = 4, which was most suitable for the data. c Population structure for 143 accessions showing K = 2, which was most suitable for the data. d Population structure for 143 accessions showing K = 8, which was the second-most suitable for the data
3.3 Principle component analysis
A two-dimensional PCA plot using PCA component 1 (PC1, explained genetic variation 22%) and PCA component 2 (PC2, explained genetic variation 7.7%) is presented in Fig. 2, in which the 15 horticultural groups are color-coded. Based on the results of the PCA for the reference array accessions (Fig. 2a), individuals could be divided into four distinct groups: Group 1 for most accessions of subsp. melo, Group 2 for most accessions of subsp. agrestis, Group 3 for momordica and dudaim accessions, and Group 4 for flexuosus accessions. PCA of all the 143 accessions (Fig. 2b) formed three major groups: Group 1 for reticulatus and cantalupensis accessions of subsp. melo, Group 2 for the accessions of remaining horticultural groups in subsp. melo, Group 3 for most of horticultural groups in subsp. agrestis, and dudaim accessions in subsp. melo.

Principe component analysis (PCA) of a melon (Cucumis melo L.) germplasm collection using the SNPRelate R package. a PCA plot for 50 accessions of the reference array. b PCA plot for 143 accessions
The PCA plots retained the results obtained with STRUCTURE. Generally, good consensus was observed for a separation between the melo and agrestis subspecies. As shown in the population structure analysis, many admixed accessions were also observed in the PCA, which scattered at the center of the plots. In addition, distinct population structures of flexuosus and dudaim were sustained in the PCA.
3.4 Hierarchical clustering
The hierarchical clustering analysis dissected 50 reference array accessions into two major clusters (Cluster I and Cluster II) at a genetic distance coefficient of 0.2 (Fig. 3a). Cluster I was further divided into subcluster 1, which comprised mainly accessions for subsp. melo including flexuous, and subcluster 2, which comprised a mixture of accessions for subsp. melo (chate and dudaim) and subsp. agrestis (makuwa and momordica). Cluster II mainly comprised accessions for subsp. agrestis. Generally, for reference array accessions, there was separation between the subsp. melo and subsp. agrestis; however, accessions of different horticultural groups were admixed and accessions of the same horticultural group dispersed in clusters, as shown in the population structure analysis and PCA.

Hierarchical clustering analysis of a melon (Cucumis melo L.) germplasm collection calculated based on Nei’s genetic distance using the Poppr R package. a Dendrogram for 50 accessions of the reference array. b Dendrogram for 143 accessions
A detailed genetic relationship could be assessed from the hierarchical clustering analysis of 143 accessions, which dissected those accessions into two major clusters (Cluster I and Cluster II) at a genetic distance coefficient of 0.2 (Fig. 3b). Cluster I comprised most accessions of subsp. melo. including three major cultivated melon groups, cantalupensis, inodorus, and reticulatus. Two subclusters I-1 and I-2 were identified from Cluster I. In Cluster I-l, all reticulatus accessions from KOR and JPN were tightly clustered (I-la) under the same node, whereas reticulatus accessions from around the world (USA, Uzbekistan, Taiwan, and East Asia) dispersed into different clusters (Cluster I-1b and I-2a). Six cantalupensis accessions from France and USA also formed a tight subcluster (Cluster I-1b) under Cluster I-1, while other cantalupensis accessions from diverse countries including Afghanistan, Iran, United Kingdom, Japan, and Ukraine) were separated within Cluster I-2a. Accessions of inodorus, which are dominant in Spain and Central Asia, dispersed within Cluster I-2a. Five inodorus accessions (25, 33, 74, 75, 124) from Turkey were closely related, whereas the remaining inodorus accessions (7,12, 34, 124) from Uzbekistan and India were dispersed. Accessions of chandalak, which are mainly cultivated from Central Asia to India, tended to be grouped under the same node in Cluster I-2a regardless of their country of origin, while two chandalak accessions (97, 107) were spread in Cluster I-2a. One tibish accession that is endemic in Sudan and eaten raw in salads was grouped with CAN accessions (127, 132, 133) from JPN and FRC. For the Group flexousus, which is cultivated in a large area from Morocco to India, all accessions except for two (122, 113) were grouped in Cluster I-2b, but were distantly related to other subsp. melo types located in Cluster I-2.
Cluster II mainly comprised melon accessions for conomon, makuwa, and momordica, which belong to subsp. agristes. Group conomon and makuwa are both cultivated in the Far East (China, Japan) and show andromonoecious sex expression, no netting, and no winkled fruit. An F1 hybrid cultivar ‘Gumsaragi’ (68), which is a Korean melon called ‘Chamwae’ is known as makuwa and appeared in Cluster II. Interestingly, all chito and dudaim accessions, which were classified in subsp. melo by Pitrat (2008), were closely related to the accessions of subsp. agristes and located in Cluster II. Four dudaim accessions were grouped together and formed a subcluster (Cluster II-2).
4 Discussion
Since Pangalo (1929) proposed the subdivision of C. melo into two homologous subspecies, melo and agrestis, this infraspecific classification scheme remains the most recent taxonomy in melons. However, schemes for the further division of subspecies into horticultural groups have been controversial and continue to be revised (Munger and Robinson 1991; Pitrat et al. 2000; Pitrat 2008; Pitrat 2017), mainly because of limitations in the nomenclature of botanical taxa based on morphological variation in certain key-characters. Genotype data derived from sophisticated DNA fingerprint techniques such as GBS can help assess how the current infraspecific classification scheme reflects biologically significant demarcation between accessions.
Here, genetic analyses of 143 melon accessions based on GBS revealed discrete separation at the level of two subspecies. However, the distinction between horticultural groups as infraspecific taxa was not strong or clear: a few accessions of the same group dispersed into different clusters and subclusters, and an admixture of ancestors was observed from accessions of different groups. Although there were no substantial contradictions among the results, there was inconsistency between the division of accessions based on horticultural group and genetic relatedness assessed by genome-wide DNA sequence variation, making the current classification system for cultivar grouping somewhat arbitrary. This pattern of variation can be explained by the fact that no reproductive barriers evolved between melon varieties, which can be easily outcrossed. A breeding process that combines genetic materials from different groups may also facilitate horizontal transfer between clusters, causing an apparent poor resolution of molecular phylogeny (Stepansky et al. 1999).
A low level of variation among reticulatus accessions from Korea and Japan in Cluster I-1 suggests an erosion of genetic variability caused by drift and/or inbreeding, implying that reticulatus breeding lines from the GARES in this study were derived from common ancestors that are close to reticulatus melons from Japan. In addition, some reticulatus accessions were closely related to cantalupensis accessions, as shown in Cluster I-1. These two groups represent the dessert melon group termed ‘net melon’, which are mainly cultivated in the USA, Japan, and Europe. Netting is the main difference between reticulatus and cantalupensis; however, Munger and Robinson (1991), and Pitrat (2017) suggested that these two groups could be merged as there is a continuum for netting. However, in Cluster I-2, several other reticulatus (2, 29, 76, 53, 81, 77) and cantalupensis accessions (127,132,133, 107, 125, 118, 142, 28) from different countries showed a wide dispersion and inter-mix with accessions of other horticultural groups. Therefore, it is difficult to merge these groups when genome-wide sequence variation is considered for infraspecific classification.
The large Group inodorus is highly polymorphic in fruit traits and is cultivated in different regions such as Asia, Spain, and Mediterranean countries (Pitrat 2017). Highly dispersed inodorus accessions in Cluster I-2a may explain genetic variation in this group. Pitrat (2017) proposed splitting inodorus into three groups (casaba, ibericus, and inodorus), which included the ‘Honeydew’ and ‘Earl’s’ previously defined as reticulatus: no strong association between these two groups was observed in our clustering analysis. In addition, our results indicated that the Group flexousus is distinctive from other groups in subsp. melo in terms of morphology as well as genome sequence-based polymorphisms. A long snake-type flexousus was first described as an independent species (C. flexuosus) by Linnaeus.
In the present study, dudaim accessions of subsp. melo were positioned in Cluster II for subsp. agrestis. Group dudaim possesses small, round, and highly fragrant flesh, and is cultivated from Turkey to Afghanistan. This group was first described as an independent species (C. dudaim) by Linnaeus, but is now considered a synonym of C. melo together, the same as flexousus (Pitrat 2017). In the years since then, dudaim was classified into subsp. ageistis (Stepansky 1999), but was later reclassified into subsp. melo by Pitrat (2008). A feral-type chito accession of subsp. melo present in Central America and the Caribbean Islands was also positioned in Cluster II. Group chito has short hairs on the ovary, which is a characteristic of subsp. agrestis; however, current taxonomy classifies it as subsp. melo. In a genetic diversity analysis using SSR, Monforte et al. (2003) suggested that dudaim and chito groups should be included in subsp. agrestis according to the observed SSR variability. This is also supported by our GBS-based clustering. Kirkbride (1993) noted that the two sub-species defined by pubescence of the ovary (short and appressed hair versus long spreading hairs) are not always relevant, as both types are encountered in several groups such as agrestis, kachri, and flexousus. It may not be necessary to maintain the subspecies rank in the infraspecific classification of melon; one may need to use only the Group level and, in some cases, the subgroup level. Korean melon type ‘Chamwae’, such as the F1 cultivar ‘Gumsaragi’, is currently known as makuwa; however, it has been suggested to separate it to conomon or a new variety group chinensis. In our dendrogram, it was not possible to assign ‘Gumsaragi’ into either conomon or makuwa, because no clear division between these two groups was observed in Cluster II.
In conclusion, our study supported an admixed genetic background based on DNA variation among different horticultural groups. The distinction between horticultural groups as ‘infraspecific taxa’ was not clear, indicating that a ‘horticultural’ rather than a ‘botanical’ approach should be applied to meet the current infraspecific classification scheme. Nevertheless, our molecular data may be useful as supporting information to improve the resolution of horticultural group classification in melon.
References
Bae KM, Sim SC, Hong JH, Choi KJ, Kim DH, Kwon YS (2015) Development of genomic SSR markers and genetic diversity analysis in cultivated radish (Raphanus sativus L.). Hort Environ Biotechnol 56:216–224. https://doi.org/10.1007/s13580-015-0089-y
Cox MP, Peterson DA, Biggs PJ (2010) SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinform 11:485. https://doi.org/10.1186/1471-2105-11-485
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. https://doi.org/10.1371/journal.pone.0019379
Esteras C, Formisano G, Roig C, Díaz A, Blanca J, Garcia-Mas J, Picó B (2013) SNP genotyping in melons: genetic variation, population structure, and linkage disequilibrium. Theor Appl Genet 126:1285–1303
Garcia-Mas J, Benjak A, Sanseverino W, Bourgeois M, Mir G, González VM, Alioto T (2012) The genome of melon (Cucumis melo L.). Proc Natl Acad Sci U S A 109:11872–11877
Jeffrey C (1980) A review of the Cucurbitaceae. Bot J Linn Soc 81:233–247. https://doi.org/10.1111/j.1095-8339.1980.yb01676.x
Kamvar Z, Tabima JF, Grunwald NJ (2014) Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2:e281. https://doi.org/10.7717/peerj.281
Kirkbride JH (1993) Biosystematic monograph of the genus Cucumis (Cucurbitaceae). Parkway Publishers, North Carolina
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. https://doi.org/10.1093/bioinformatics/btp324
Martin M (2011) Cutadapter removes adapter sequences from high-throughput sequencing reads. ENBnet.j 17:10. https://doi.org/10.14806/ej.17.1.200
Monforte AJ, Garcia-Mas J, Arus P (2003) Genetic variability in melon based on microsatellite variation. Plant Breed 122:7. https://doi.org/10.1046/j.1439-0523.2003.00848.x
Munger HM, Robinson RW (1991) Nomenclature of Cucumis melo L. Cucurbit Genet Coop Rep 14:43–45
Pangalo KJ (1929) Critical review of the main literature on the taxonomy, geography and origin of cultivated and partially wild melons. Trudy Prikl Bot 23:397–442
Pitrat M (2008) Melon(Cucumis melo L.). In: Prohens J, Nuez F (eds) Handbook of plant breeding-vegetables, vol. 1. Springer: New York, pp 283–315. doi:10.1007/978-0-387-30443-4_9
Pitrat M (2017) Melon genetic resources: phenotypic diversity and horticultural taxonomy. In: Grumet R, Katzir N, Garcia-Mas J (eds) Genetics and genomics of Cucurbitaceae. Plant genetics and genomics: crops and models, vol 20. Springer, Cham
Pitrat M, Hanelt P, Hammer K (2000) Some comments on infraspecific classification of cultivars of melon. Acta Hort 510:29–36. https://doi.org/10.17660/ActaHortic.2000.510.4
Poland JA, Rife TW (2012) Genotyping-by-sequencing for plant breeding and genetics: the plant. Genome 5:92–102. https://doi.org/10.3835/plantgenome2012.05.0005
Robinson RW, Decker-Walters DS (1997) Cucurbit. CAB International, Wallingford
Saghai-Maroof MA, Soliman KM, Jerenson RA, Allard RW (1984) Ribosomal DNA spacer length polymorphism in barley: Mendelian inheritance, chromosomal location and population dynamics. Proc Natl Acad Sci USA 81:8014–8018
Sebastian P, Schaefer H, Telford IRH, Renner S (2010) Cucumber (Cucumis sativus) and melon (C. melo) have numerous wild relatives in Asia and Australia, and the sister species of melon is from Australia. Proc Natl Acad Sci USA 107:14269–14273. https://doi.org/10.1073/pnas.1005338107
Sim SC, Hong JH, Kwon YS (2015) DNA profiling of commercial pumpkin cultivars using simple sequence repeat polymorphism. Hort Environ Biotechnol 56:811–820. https://doi.org/10.1007/s13580-015-0123-0
Stepansky A, Kovalski I, Perl-Treves R (1999) Infraspecific classificaiotn of melon (Cucumis melo L.) in view of their phenotypic and molecular variation. Plant Syst Evol 217:313–333
Zheng X, Levine D, Shen J, Gogarten S, Laurie C, Weir B (2012) A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformtics 26:3326–3328. https://doi.org/10.1093/bioinformatics/bts606
Acknowledgements
This work was supported by the Korea Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry (IPET) through the Agri-Bio Industry Technology Development Program funded by the Ministry of Agriculture, Food, and Rural Affairs (MAFRA) (317011–04-2-HD040) to Y. Park.
Author information
Authors and Affiliations
Contributions
J. Jung, G. Park, and J. Oh prepared seeds and plant materials and carried out DNA extraction; J. Jung and J. Oh J. Jung, E. Shim, S. Chung, and G. Lee performed the GBS data analysis and population genetics analysis; J. J ung and Y. Park wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
All authors confirm that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Communicated by Sung-Chur Sim
Rights and permissions
About this article

Cite this article
Jung, J., Park, G., Oh, J. et al. Assessment of the current infraspecific classification scheme in melon (Cucumis melo L.) based on genome-wide single nucleotide polymorphisms. Hortic. Environ. Biotechnol. 61, 537–547 (2020). https://doi.org/10.1007/s13580-020-00230-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13580-020-00230-0