
Abstract
Nucleoporin 93 (NUP93) is an important component of the nuclear pore complex, exhibiting pro-tumorigenic properties in some cancers. However, its function in esophageal squamous cell carcinoma (ESCC) has not been elucidated. This study aimed to investigate the effects of NUP93 in ESCC and the underlying mechanisms involved. Through analysis of public human cancer datasets, we observed higher expression of NUP93 in esophageal cancer tissues than in normal tissues. Stable ESCC cell lines with NUP93 overexpression or knockdown were established by lentiviral vector transduction and puromycin selection. NUP93 knockdown suppressed the proliferation, colony formation, cell cycle transition, migration, and invasion of ESCC cells, while the overexpression of NUP93 displayed opposite effects. NUP93 positively regulated epithelial–mesenchymal transition and AKT signaling transduction in ESCC cells. In addition, NUP93 increased the expression of programmed death ligand 1 (PD-L1) in ESCC cells and attenuated NK cell-mediated lysis of ESCC cells. In vivo experiments demonstrated that NUP93 promotes the growth of ESCC in nude mice, enhances Ki67 and PD-L1 expression, and promotes AKT signaling transduction in xenografts. Mechanistically, we demonstrated that the HECT domain E3 ubiquitin protein ligase 1 (HECTD1) contributes to the ubiquitination and degradation of NUP93 and acts as a tumor suppressor in ESCC. To conclude, this study has shown that NUP93 has pro-tumor properties in ESCC and that HECTD1 functions as an upstream regulator of NUP93 in ESCC. These findings may contribute to the investigation of potential therapeutic targets in ESCC.
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Esophageal cancer (ESCA) is a common type of malignant neoplasm of the upper aerodigestive tract. According to global cancer statistics, it is estimated that there will be 604,100 new cases and 544,100 deaths from ESCA in 2020. Esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC) are two major histologic subtypes of ESCA, of which ESCC accounts for approximately 85% [1]. Despite advances in diagnosis and treatment over the past decades, survival rates for ESCA remain low, ranging from 10% to 30% 5 years after diagnosis [2]. Investigation of the molecular mechanism will help improve our understanding of the initiation and progression of ESCA and provide clues for the development of therapeutic targets.
The nuclear pore complex (NPC) controls the transport of nucleocytoplasmic cargo, such as RNAs and proteins [3]. Recent research has shown that inhibiting NPC formation induces cancer cell death and leads to tumor regression, suggesting the important role of NPC in tumor progression [4]. Nucleoporin 93 (NUP93) is an important component of the NPC. It has been reported that NUP93 is highly expressed and exhibits pro-tumor functions in breast cancer [5, 6], cervical cancer [7], and hepatocellular carcinoma [8] by promoting the proliferation, migration, and invasion of cancer cells. However, it is not clear whether it plays a pro-tumor role in ESCA. We also note that Monwan et al. have identified the function of NUP93 in mediating antiviral immune responses [9], whereas little is known about the effects of NUP93 on anti-tumor immunity, which merits further investigation.
The ubiquitin–proteasome pathway represents one of the most predominant pathways for protein degradation in eukaryotic cells. HECT domain E3 ubiquitin protein ligase 1 (HECTD1) contributes to the ubiquitination and degradation of substrates [10]. It has previously been determined that lower expression of HECTD1 is associated with a worse prognosis in patients with breast cancer [11]. Functionally, the silencing of HECTD1 promotes migration, invasion, and epithelial–mesenchymal transition (EMT) of breast and cervical cancer cells while increasing their resistance to cisplatin [12, 13]. These reports suggest that HECTD1 may have a tumor suppressor role. After analyzing public datasets, we observed that HECTD1 expression in ESCA tissues was significantly lower than in normal control tissues. In addition, an interaction between HECTD1 and NUP93 was identified. These findings suggest that HECTD1 may promote NUP93 degradation, and therefore act as a tumor suppressor in ESCA.
This study primarily investigates the roles of NUP93 in ESCC, specifically concerning cancer cell proliferation, migration, invasion, epithelial–mesenchymal transition (EMT), and oncogenic signaling transduction. We further examined the effects of NUP93 on programmed death ligand 1 (PD-L1) expression and natural killer (NK) cell-mediated lysis of cancer cells. Mechanistically, we identified HECTD1 as an upstream regulator of NUP93 via the regulation of NUP93 ubiquitination and degradation in ESCC cells.
Methods
Cells
All cell lines used in this study were obtained from Shanghai iCell Bioscience. KYSE-410 cells were grown in RPMI-1640 medium supplemented with 10% FBS. KYSE-30 cells were grown in RPMI-1640/ F-12 medium supplemented with 2% FBS. KYSE-410 and KYSE-30 were established in the laboratory of Dr. Shimada in 1992 [14] and have been used in equivalent original research papers [15,16,17,18]. Primary NK cells were grown in a primary NK cell culture system (Shanghai iCell Bioscience, China).
RNA extraction, reverse-transcription PCR (RT-PCR), and real-time PCR (qPCR)
Total RNA was extracted from cultured cells using TRIpure (BioTeke Co. Ltd., Beijing, China) and then used for cDNA synthesis. qPCR was performed using a PCR master mix supplemented with SYBR Green (Solarbio Ltd, China). Relative mRNA levels were calculated using the standard 2-ΔΔCt method. The primers used were NUP93 F: 5’-GTCAGTTCTCCTCGGGTCT-3’ and R: 5’-TTCGCTGTTTCACTTGCTC-3’.
Western blot and immunoprecipitation
For Western blot, total protein extracts were prepared using lysis buffer (Solarbio Ltd.). Protein concentrations were determined using a bicinchoninic acid (BCA) kit (Solarbio Ltd.). Loading samples were prepared using 6 × loading buffer (Solarbio Ltd.), denatured for 5 min using a boiling water bath, and then subjected to SDS-PAGE electrophoresis. After that, proteins were transferred onto polyvinylidene fluoride membranes (Millipore Co., USA) and probed with primary antibodies and subsequent HRP-conjugated secondary antibodies. The primary antibodies included NUP93 (sc-374399, Santa Cruz, USA), HECTD1 (20605-1-AP, Proteintech Company, USA), AKT (AF4718, Affbiotech, China), p-AKTSer473 (AF0016, Affbiotech), p-AKTThr308 (AF3262, Affbiotech), cyclin D1 (A19038, ABclonal Biotech, China), p-GSK3βSer9 (AP0039, ABclonal Biotech), PD-L1 (28076-1-AP, Proteintech Company), E-cadherin (AF0131, Affbiotech), N-cadherin (A19083, ABclonal Biotech), and Vimentin (A19607, ABclonal Biotech). Secondary antibodies included goat anti-rabbit IgG (SE134, Solarbio Ltd.) and goat anti-mouse IgG (SE131, Solarbio Ltd.). Finally, protein bands were visualized with an enhanced chemiluminescence substrate (Solarbio Ltd.). For immunoprecipitation, cell lysates were prepared using native lysis buffer (Solarbio) and incubated with specific antibodies immobilized on AminoLink Plus Coupling Resin using the Pierce™ co-immunoprecipitation kit (ThermoFisher co. Ltd., USA). The protein–antibody-AminoLink Plus Resin complexes were then eluted, and the immunoprecipitates were used for Western blot.
Lentiviral transduction and siRNA transfection
Lentiviruses were packaged using lentiviral vectors PLJM1-EGFP-Puro (containing NUP93 coding sequence) or pLKO.1-EGFP-Puro (containing NUP93-targeting shRNA), or their respective negative controls. The lentiviral vectors were obtained from Hunan Fenghui Biotechnology Co., Ltd (Changsha, China). ESCC cells were infected with the indicated lentiviruses. Stable cell lines were generated by puromycin resistance selection. The target sequences (5’ to 3’) of NUP93-sh1 and NUP93-sh2 were GGGAGTCAATGTTGGTTGAGT and AGGAGACGGCAGATGTCAAGG, respectively. The non-specific target sequences of shNC were TTCTCCGAACGTGTCACGT.
siRNA transfection was performed using Lipofectamine 3000 transfection reagent (Invitrogen Co., USA) according to the manufacturer’s protocol. 48 h after transfection, cells were used for further experiments. The sequences (5’ to 3’) of the siRNAs are listed below. si-HECTD1-1 (sense): GGAUGAUGAUUAUGUGCUAtt and si-HECTD1-1 (anti-sense): UAGCACAUAAUCAUCAUCCtt; si-HECTD1-2 (sense): GAAACAAGAUUGUAGUCAAtt and si-HECTD1-2 (anti-sense): UUGACUACAAUCUUGUUUCtt; si-NUP93 (sense): GGAUGAGAAAGAUAGUCAAtt and si-NUP93 (anti-sense): UUGACUAUCUUUCUCAUCCtt; si-NC (sense): UUCUCCGAACGUGUCACGUtt and si-NC (anti-sense): ACGUGACACGUUCGGAGAAtt.
Cell proliferation and colony formation
To evaluate cell proliferation, cells were seeded in 96-well culture plates at 4,000 cells per well, and cell viability was evaluated at 24-h intervals using the CCK-8 reagent (Keygen Co., China). For colony formation assay, cells were seeded at 400 cells per culture dish and cultured for 2 weeks. The cells were then fixed and stained. After washing, the cells were observed under a microscope. The colony formation ratio was calculated as (number of colonies/number of inoculated cells) × 100%.
Analysis of cell cycle distribution
Cell cycle analysis was performed using a cell cycle detection kit (Keygen Co.). ESCC cells were fixed with cold ethanol overnight. The next day, the fixed cells were washed with PBS, stained with PI/RNase A solution, and then analyzed by flow cytometry.
Migration and invasion assays
Cell migration was evaluated using a wound-healing assay. Wound tracks were scratched with a pipette tip after cells were confluent. Wound closure was observed and photographed at 0 and 24 h. Cell invasion was evaluated using the Transwell assay. The 24-well Transwell was pre-coated with Matrigel (Costar, Corning Inc., USA). Cells cultured in serum-free medium were seeded on the upper chamber at 50,000 cells per well. The bottom chamber was filled with culture medium supplemented with 10% FBS. After 24 h, the invaded cells were fixed with 4% paraformaldehyde (PFA), stained with crystal violet (Amresco Ltd., USA), and photographed under a microscope.
Immunofluorescence (IF), immunohistochemistry (IHC), and hematoxylin and eosin (H&E) staining
Cells were seeded onto the coverslips, fixed with 4% PFA, permeabilized with 0.1% triton X-100, and blocked with 1% bovine serum albumin (BSA). The cells were then incubated with the indicated primary antibodies overnight. After that, the cells were washed and incubated with secondary antibodies and stained with DAPI in the dark. Finally, the cells were observed under a fluorescence microscope. Primary antibodies included HECTD1 (20605-1-AP, Proteintech Company), NUP93 (sc-374399, Santa Cruz), N-cadherin (22018-1-AP, Proteintech Company), and E-cadherin (20874-1-AP, Proteintech Company).
Paraffin-embedded tissue sections were deparaffinized and hydrated through decreasing grades of ethanol. After antigen retrieval and blocking of endogenous peroxidase activity, sections were incubated overnight with the indicated primary antibodies (NUP93, sc-374399, Santa Cruz; Ki-67, A20018, ABclonal Biotech). The next day, sections were washed and incubated with HRP-conjugated secondary antibodies. A DAB substrate kit (MXB Biotechnologies, China) was used for visualization. For evaluation of histopathology, sections were subjected to H&E staining.
NK cell-specific lysis
NK cells were activated with recombinant 1000 IU/mL IL-2 and then co-cultured with target ESCC cells at different E:T ratios. After 4 h, cell viability was measured by the CCK-8 method, and the percentage of specific lysis was calculated.
Subcutaneous tumor xenograft
BALB/c nude mice (6 weeks old) were used in this study. Animal experiments were approved by the Ethics Committee of Harbin Medical University (Approval number 2020-50-IIT) and conducted according to the Guide for the Care and Use of Laboratory Animals, 8th edition by National Research Council (US) Committee (Available from: https://www.ncbi.nlm.nih.gov/books/NBK54050/) [19]. Approximately 5 × 106 viable ESCC cells were injected subcutaneously into each mouse. Tumor growth was monitored at 4-d intervals for 4 weeks. Tumor weight was recorded and the dissected xenograft tumors were used for further experiments.
Online databases
Pan-cancer NUP93 expression analysis was performed using the TIMER web portal (http://timer.cistrome.org) [20]. Two Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) datasets were used for gene expression analysis in ESCA tissues and normal tissues. GSE5364 contains 16 tumor samples and 13 normal samples [21]. GSE70409 contains 17 pairs of tumor samples and adjacent normal samples [22]. NUP93 expression analysis in ESCA cell lines was performed based on Cancer Cell Line Encyclopedia (CCLE) data via the DepMap portal (https://depmap.org/portal/ccle/) [23]. The relationship between NUP93 expression and EMT pathway activity was analyzed using the Gene Set Cancer Analysis (GSCA) platform (http://bioinfo.life.hust.edu.cn/GSCA/) [24].
Statistical analysis
Quantitative results in this study are presented as mean ± SD. Data were analyzed using GraphPad Prism version 8 software. The expression of the housekeeping gene GAPDH was used for normalization. Statistical significance was determined by the two-tailed Student’s t test and one-way or two-way ANOVA. Post hoc multiple comparison tests were used when appropriate. P values < 0.05 indicate statistically significant differences (*P < 0.05 and **P < 0.01).
Results
NUP93 is highly expressed in ESCA
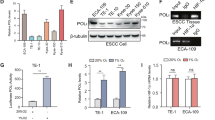
The results of the TCGA pan-cancer analysis showed that aberrant expression of NUP93 was observed in multiple cancer types, and we noticed that NUP93 mRNA levels were significantly higher in ESCA tumors than in normal tissues (Fig. 1A). Similar results were obtained from two GEO datasets (Fig. 1B, C). The CCLE data showed that among the four ESCC cell lines (KYSE-30, KYSE-150, KYSE-410, and TE-1), KYSE-30 had the lowest expression level of NUP93 mRNA (Fig. 1D). Interestingly, based on the data collected from the Cellosaurus database [25], KYSE-30 seemed to have the longest doubling time among the four cell lines, suggesting that NUP93 expression may correlate with the proliferation of ESCC cells.

High expression of NUP93 in ESCA. A Pan-cancer analysis of NUP93 expression based on the TIMER online database. B, C Expression analysis of NUP93 in normal and ESCA tissues based on data from GEO datasets GSE5364 and GSE70409. D Expression profile of NUP93 in ESCA cell lines based on CCLE data via the DepMap portal. Results are presented as mean ± SD. **P < 0.01
NUP93 facilitates proliferation and colony formation of ESCC cells
Next, we established a NUP93-overexpressing KYSE-30 cell line and a NUP93-silenced KYSE-410 cell line using EGFP-tagged lentiviral vectors. Transduction efficiency was determined by observing EGFP under a fluorescence microscope (Fig. 2A, E). Expression of NUP93 mRNA and protein in the cell lines was confirmed by RT-qPCR (Fig. 2B, F) and Western blot (Fig. 2C, G), respectively. The results of cell viability analysis showed that NUP93 overexpression promoted cell proliferation (Fig. 2H), whereas NUP93 knockdown suppressed cell proliferation (Fig. 2D). NUP93 showed similar effects on the colony-forming ability of ESCC cells (Fig. 2I, K). We subsequently examined whether NUP93 affected the cell cycle progression of ESCC cells. As shown in Fig. 2J, NUP93-silenced cells exhibited a higher proportion of G0/G1-phase cells and a lower proportion of S-phase cells compared with control cells (Fig. 2J), whereas opposite results were observed in NUP93-overexpressing cells (Fig. 2L). And NUP93 did not significantly alter the proportion of G2/M-phase cells (Fig. 2J, L).

NUP93 promotes proliferation and colony formation of ESCC cells. ESCC cells were transduced with EGFP-tagged lentiviral vectors overexpressing NUP93 or NUP93-specific shRNA. Stable cell lines were generated by puromycin resistance selection. A, E Representative fluorescence microscopy images of ESCC cells transduced with EGFP lentivirus. Magnification: 100-fold. B, F Relative mRNA expression of NUP93 by real-time PCR analysis. C, G Protein expression analysis of NUP93 by Western blot analysis. D, H Cell viability analysis of ESCC cells by CCK-8 assay after culturing for the indicated times. I, K Colony formation assay of ESCC cells. Left: representative images of colony formation. Right: quantitative results of colony formation ratio. J, L Cell cycle analysis of ESCC cells by flow cytometry. Top: representative images of cell distribution in different phases. Bottom: quantitative results of cell cycle distribution. N = 3 per group. Results are presented as mean ± SD. **P < 0.01
NUP93 promotes migration, invasion, and EMT and activates AKT pathway
Next, we assessed whether NUP93 affects the migration and invasion of ESCC cells. By performing wound-healing and Transwell assays, we confirmed that cell migration and invasion were suppressed in NUP93-silenced cells (Fig. 3A, B) and enhanced in NUP93-overexpressing cells (Fig. 3C, D). Furthermore, correlation analysis based on TCGA-ESCA data showed that higher NUP93 was associated with higher EMT pathway activity scores (Fig. 3E). Turning to the experimental evidence in ESCC cells, we found that overexpression of NUP93 led to a decrease in E-cadherin expression and an increase in N-cadherin expression in ESCC cells, which was supported by the results of Western blot (Fig. 3F) and IF staining (Fig. 3G). Moreover, activation of the oncogenic AKT signaling pathway was significantly impaired in NUP93-silenced cells (Fig. 3H) but was promoted in NUP93-overexpressing cells (Fig. 3I), as evidenced by the changes in the phosphorylation of AKT and its downstream p-GSK3β and cyclin D1 levels.

NUP93 enhances migration, invasion, and EMT of ESCC cells and activates AKT signaling. A, C Wound-healing assay of ESCC cells. Left: representative images of wound closure. Magnification: 200-fold. Right: quantitative results of migration rate. B, D Transwell assay of ESCC cells. Left: representative images of invaded cells. Magnification: 200-fold. Right: quantitative results of the number of invaded cells. E Correlation analysis between NUP93 expression and EMT pathway activity in TCGA-ESCA using the GSCA database. F Western blot analysis of E-cadherin, N-cadherin, and Vimentin in ESCC cells. G Representative images of IF staining of E-cadherin and N-cadherin in ESCC cells. Magnification: 400-fold. H, I Western blot analysis of p-AKT, AKT, and AKT signaling effectors (cyclin D1 and p-GSK3β). N = 3 per group. Results are presented as mean ± SD. **P < 0.01 and *P < 0.05
NUP93 increases PD-L1 expression in ESCC cells and attenuates NK cell lysis of ESCC cells
Next, we evaluated the impacts of NUP93 on PD-L1 expression in ESCC cells and their lysis by NK cells. Decreased PD-L1 was observed in NUP93-silenced cells (Fig. 4A), whereas increased PD-L1 expression was observed in NUP93-overexpressing cells (Fig. 4C). We also noticed that the lysis of ESCC cells was increased in all groups with the increase in the E:T ratio (Fig. 4B, D). Noteworthy, strong lysis by NK cells was observed in NUP93-silenced cells (Fig. 4B), whereas opposite results were observed in NUP93-overexpressing cells (Fig. 4D).

NUP93 increases PD-L1 expression in ESCC cells and attenuates NK cell lysis of ESCC cells. A, C Protein expression of PD-L1 in ESCC cells by Western blot analysis. B, D Quantitative results of NK cell-mediated specific lysis of ESCC cells. N = 3 per group. Results are presented as mean ± SD. **P < 0.01 and *P < 0.05
NUP93 promotes ESCC growth in vivo
To investigate the functions of NUP93 in vivo, we subcutaneously implanted ESCC cells into nude mice. The growth and weight of xenografts were decreased in mice bearing NUP93-silenced cells compared to those in mice bearing control cells (Fig. 5A, B). IHC results showed that the xenograft tumors with silenced NUP93 displayed weaker staining of Ki67 (Fig. 5C). In contrast, an increase in tumor growth and tumor weight was observed in mice bearing NUP93-overexpressing cells (Fig. 5D, E), and the xenograft tumors with NUP93 overexpression also displayed stronger staining of Ki67 (Fig. 5F). In addition, H&E staining showed that knockdown of NUP93 seemed to slightly increase the extent of necrosis, whereas NUP93 overexpression exhibited contrary effects (Supplementary Fig. 1). Furthermore, AKT signaling activation and PD-L1 expression were suppressed in NUP93-silenced tumors (Fig. 5G) but were promoted in NUP93-overexpressing tumors (Fig. 5H).

NUP93 promotes ESCC xenograft tumor growth in nude mice. A, D Tumor growth curve of ESCC xenograft tumors in nude mice. B, E Analysis of ESCC xenograft tumors in nude mice. Left: tumor weight. Right: representative images of ESCC xenograft tumors. C, F Representative images of IHC staining for NUP93 and Ki67 in ESCC xenograft tumors. Magnification: 200-fold. G, H. Western blot analysis of p-AKT, AKT, p-GSK3β, and PD-L1 in ESCC xenograft tumors. N = 5 per group. Results are presented as mean ± SD. **P < 0.01
HECTD1 functions as an upstream modulator of NUP93 in ESCC
Next, we performed a Co-IP assay to verify the interaction between NUP93 and HECTD1. As shown in Fig. 6A, NUP93 was immunoprecipitated from ESCC cell lysates by the HECTD1 antibody. Double IF staining of HECTD1 and NUP93 revealed co-localization of NUP93 and HECTD1 in ESCC cells (Fig. 6B). HECTD1 siRNA caused a reduction in NUP93 expression in KYSE-30 cells (Fig. 6C). In the presence of the proteasome inhibitor MG132, the ubiquitination level of NUP93 was significantly reduced after HECTD1 knockdown (Fig. 6D). Results from the cycloheximide (CHX) chasing assay showed an obvious accumulation of NUP93 in HECTD1-silenced cells (Fig. 6E). These findings suggest that HECTD1 regulates the stability of NUP93 through the ubiquitin–proteasome pathway.

HECTD1 functions as an upstream modulator of NUP93 in ESCC. A Co-IP assay showing the interaction between NUP93 and HECTD1 in ESCC cells. B Double immunofluorescence staining of HECTD1 and NUP93 in ESCC cells. Magnification: 400-fold. ESCC cells were transfected with HECTD1-specific siRNA (si-HECTD1-1/2). C Western blot analysis of HECTD1 and NUP93 in ESCC cells 48 h after transfection. D Co-IP assay showing the ubiquitination of NUP93 in ESCC cells after MG132 treatment for 8 h. E Western blot analysis of NUP93 after CHX treatment for indicated time points. F Expression analysis of HECTD1 in normal and ESCA tissues based on data from GEO dataset GSE70409. ESCC cells were co-transfected with NUP93-specific siRNA (siNUP93) and HECTD1-specific siRNA (si-HECTD1-1/2). G, I Cell viability analysis of ESCC cells 48 h after transfection. H, J Transwell assay of ESCC cells. Left: representative images of invaded cells. Magnification: 200-fold. Right: quantitative results of the number of invaded cells. N = 3 per group. Results are presented as mean ± SD. **P < 0.01 and *P < 0.05
Interestingly, HECTD1 expression in ESCC tissues was significantly lower than that of paired adjacent normal tissues (Fig. 6F), suggesting that HECTD1 may be the upstream regulator of NUP93 in ESCC. Therefore, we investigated the functions of HECTD1 in KYSE-30 cells. Knockdown of HECTD1 increased cell viability (Fig. 6G) and enhanced cell invasion (Fig. 6H). Furthermore, the knockdown of NUP93 partially reversed the effects of HECTD1 siRNA on cell viability (Fig. 6I) and invasion (Fig. 6J).
Discussion
Several studies have demonstrated that NUP93 promotes cancer cell proliferation in vitro and tumor growth in vivo [5,6,7,8]. In contrast, reducing the expression of NUP93 was found to significantly inhibit DNA synthesis in cancer cells [6]. Likewise, the present study showed that NUP93 promotes the proliferation and G1/S transition of ESCC cells, and these effects are likely due to the NUP93-mediated upregulation of cyclin D1, a crucial protein in controlling the G1/S transition [26]. Interestingly, NUP93 silencing was found to reduce the expression of G2-M transition regulator cyclin-dependent kinase 1 (CDK1) [10, 27], indicating that further investigation is necessary to confirm the regulation of G2-M transition by NUP93.
In addition, it has been found that NUP93 influences the metastatic potential of cancer cells. According to the work of Bersini et al., inhibition of cancer cell migration caused by NUP93 silencing is associated with NUP93-mediated modulation of actin cytoskeleton remodeling. Furthermore, in NUP93-silenced cells, several molecules related to the upregulation of cell motility and EMT were significantly decreased. In addition, a high expression level of NUP93 has been associated with facilitated tumor metastasis in mouse models [5]. Consistent with these findings, we demonstrated the pro-metastatic properties of NUP93 in ESCC cells. In addition, we observed a positive correlation between NUP93 expression and EMT pathway activity in ESCA tissues. Experimental results further confirmed this correlation: overexpression of NUP93 reduced the expression of the epithelial E-cadherin and increased the expression of the mesenchymal N-cadherin and Vimentin. Taken together, these findings support the therapeutic potential of targeting NUP93 to prevent tumor metastasis.
AKT signaling is a well-recognized oncogenic pathway that regulates various crucial cellular processes, including proliferation, cell survival, angiogenesis, and metabolism. A previous report has also confirmed its role in promoting the progression of ESCC [28]. Our findings suggest that NUP93 positively regulates AKT signaling transduction since we observed that NUP93 knockdown led to a significant downregulation of the phosphorylation of AKT at Ser473 and Thr308, which is required for the full activation of AKT [29, 30]. To verify this observation, we tested the effects of NUP93 on two commonly known downstream effectors of the AKT pathway, cyclin D1, and phosphorylated GSK3β at Ser9 [31, 32]. Consistent with expectations, the levels of cyclin D1 and p-GSK3β were reduced in NUP93-silenced cells and xenografts. Taken together, these findings demonstrate that NUP93 has a positive regulatory effect on AKT signal transduction in ESCA. It is worth noting that CDK1, a gene previously identified as downregulated in NUP93-silenced cells [10], can interact with AKT and induce its phosphorylation at Ser473 and Thr308 [33]. Thus, NUP93 may modulate the phosphorylation of AKT through a mechanism related to CDK1.
Previous studies have shown an association between the pro-tumor traits of NUP93 and the activation of other signaling pathways. For instance, NUP93 has been found to facilitate the nuclear transport of β-catenin, which activates the expression of the oncogene MYC [6]. Ouyang et al. have reported that NUP93 is implicated in IL-6/STAT3 signaling. The silencing of NUP93 caused a reduction of p-STAT3 in cervical cancer cells [7]. Since STAT3 strongly activates the transcription of the CD274 gene encoding PD-L1 in various types of cancer, including ESCA [34, 35], NUP93-mediated upregulation of PD-L1 is likely correlated with STAT3 activation. However, further investigation is needed to confirm this.
Currently, multiple proteins are recognized as substrates of HECTD1. It has been reported that HECTD1 regulates the stability of EMT transcription factor SNAIL through ubiquitination, thereby acting as a negative regulator of EMT in cervical cancer cells [13]. Similarly, HECTD1 targets ACF7 and facilitates its proteasomal degradation in breast cancer cells. The depletion of HECTD1 not only promotes in vitro EMT in cancer cells but also in vivo tumor metastasis in mice [12]. Furthermore, HECTD1 contributes to the ubiquitination of adenomatous polyposis coli (APC), which negatively regulates the Wnt signaling cascade. Silencing HECTD1 resulted in decreased ubiquitination of APC, interrupting the APC–Axin interaction, which contributed to the stabilization of the Wnt effector β-catenin and its subsequent signaling transduction [36]. This mechanism partially explains how inhibition of Wnt signaling activity in glioma cells is mediated by HECTD1 [37]. In this work, we have identified NUP93 as a new HECTD1 substrate. Our findings indicate that HECTD1 is a tumor suppressor in ESCC, and its functions may be partly due to the negative regulation of NUP93 expression.
In conclusion, our study demonstrates that NUP93 promotes proliferation, migration, invasion, EMT, immune evasion, and AKT signaling activation in ESCC cells. We also uncovered the regulation of NUP93 ubiquitination and degradation mediated by HECTD1. Given these results, NUP93 and HECTD1 may be potential therapeutic targets in ESCC.
Data availability
The data used and analyzed in the current study are available from the corresponding author upon reasonable request.
References
Morgan E, Soerjomataram I, Rumgay H, Coleman HG, Thrift AP, Vignat J, et al. The global landscape of esophageal squamous cell carcinoma and esophageal adenocarcinoma incidence and mortality in 2020 and projections to 2040: new estimates from GLOBOCAN 2020. Gastroenterology. 2022;163(3):649-58.e2. https://doi.org/10.1053/j.gastro.2022.05.054.
Allemani C, Matsuda T, Di Carlo V, Harewood R, Matz M, Nikšić M, et al. Global surveillance of trends in cancer survival 2000–14 (CONCORD-3): analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet (London, England). 2018;391(10125):1023–75. https://doi.org/10.1016/s0140-6736(17)33326-3.
Kosinski J, Mosalaganti S, von Appen A, Teimer R, DiGuilio AL, Wan W, et al. Molecular architecture of the inner ring scaffold of the human nuclear pore complex. Science (New York, NY). 2016;352(6283):363–5. https://doi.org/10.1126/science.aaf0643.
Sakuma S, Raices M, Borlido J, Guglielmi V, Zhu EYS, D’Angelo MA. Inhibition of nuclear pore complex formation selectively induces cancer cell death. Cancer Discov. 2021;11(1):176–93. https://doi.org/10.1158/2159-8290.Cd-20-0581.
Bersini S, Lytle NK, Schulte R, Huang L, Wahl GM, Hetzer MW. Nup93 regulates breast tumor growth by modulating cell proliferation and actin cytoskeleton remodeling. Life Sci Alliance. 2020. https://doi.org/10.26508/lsa.201900623.
Nataraj NB, Noronha A, Lee JS, Ghosh S, Mohan Raju HR, Sekar A, et al. Nucleoporin-93 reveals a common feature of aggressive breast cancers: robust nucleocytoplasmic transport of transcription factors. Cell Rep. 2022;38(8): 110418. https://doi.org/10.1016/j.celrep.2022.110418.
Ouyang X, Hao X, Liu S, Hu J, Hu L. Expression of Nup93 is associated with the proliferation, migration and invasion capacity of cervical cancer cells. Acta Biochim Biophys Sin. 2019;51(12):1276–85. https://doi.org/10.1093/abbs/gmz131.
Lin CS, Liang Y, Su SG, Zheng YL, Yang X, Jiang N, et al. Nucleoporin 93 mediates β-catenin nuclear import to promote hepatocellular carcinoma progression and metastasis. Cancer Lett. 2022;526:236–47. https://doi.org/10.1016/j.canlet.2021.11.001.
Monwan W, Kawasaki T, Hasan MZ, Ori D, Kawai T. Identification of nucleoporin 93 (Nup93) that mediates antiviral innate immune responses. Biochem Biophys Res Commun. 2020;521(4):1077–82. https://doi.org/10.1016/j.bbrc.2019.11.035.
Lv K, Gong C, Antony C, Han X, Ren JG, Donaghy R, et al. HectD1 controls hematopoietic stem cell regeneration by coordinating ribosome assembly and protein synthesis. Cell Stem Cell. 2021;28(7):1275-90.e9. https://doi.org/10.1016/j.stem.2021.02.008.
Uemoto Y, Katsuta E, Kondo N, Wanifuchi-Endo Y, Fujita T, Asano T, et al. Low HECTD1 mRNA expression is associated with poor prognosis and may be correlated with increased mitochondrial respiratory function in breast cancer. Am J Cancer Res. 2022;12(4):1593–605.
Duhamel S, Goyette MA, Thibault MP, Filion D, Gaboury L, Côté JF. The E3 ubiquitin ligase HectD1 suppresses EMT and metastasis by targeting the +TIP ACF7 for degradation. Cell Rep. 2018;22(4):1016–30. https://doi.org/10.1016/j.celrep.2017.12.096.
Wang X, De Geyter C, Jia Z, Peng Y, Zhang H. HECTD1 regulates the expression of SNAIL: Implications for epithelial-mesenchymal transition. Int J Oncol. 2020;56(5):1186–98. https://doi.org/10.3892/ijo.2020.5002.
Shimada Y, Imamura M, Wagata T, Yamaguchi N, Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69(2):277–84. https://doi.org/10.1002/1097-0142(19920115)69:2%3c277::aid-cncr2820690202%3e3.0.co;2-c.
Twarock S, Reichert C, Bach K, Reiners O, Kretschmer I, Gorski DJ, et al. Inhibition of the hyaluronan matrix enhances metabolic anticancer therapy by dichloroacetate in vitro and in vivo. Br J Pharmacol. 2019;176(23):4474–90. https://doi.org/10.1111/bph.14808.
Amin MN, Hussain MS, Sarwar MS, Rahman Moghal MM, Das A, Hossain MZ, et al. How the association between obesity and inflammation may lead to insulin resistance and cancer. Diabetes Metab Syndr. 2019;13(2):1213–24. https://doi.org/10.1016/j.dsx.2019.01.041.
Zargari S, Negahban Khameneh S, Rad A, Forghanifard MM. MEIS1 promotes expression of stem cell markers in esophageal squamous cell carcinoma. BMC Cancer. 2020;20(1):789. https://doi.org/10.1186/s12885-020-07307-0.
Khales SA, Mozaffari-Jovin S, Geerts D, Abbaszadegan MR. TWIST1 activates cancer stem cell marker genes to promote epithelial-mesenchymal transition and tumorigenesis in esophageal squamous cell carcinoma. BMC Cancer. 2022;22(1):1272. https://doi.org/10.1186/s12885-022-10252-9.
National Research Council Committee for the Update of the Guide for the C, Use of Laboratory A. The National Academies Collection: Reports funded by National Institutes of Health. Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US) Copyright © 2011, National Academy of Sciences. 2011.
Li T, Fu J, Zeng Z, Cohen D, Li J, Chen Q, et al. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020;48(W1):W509-w14. https://doi.org/10.1093/nar/gkaa407.
Yu K, Ganesan K, Tan LK, Laban M, Wu J, Zhao XD, et al. A precisely regulated gene expression cassette potently modulates metastasis and survival in multiple solid cancers. PLoS Genet. 2008;4(7): e1000129. https://doi.org/10.1371/journal.pgen.1000129.
Chen YK, Tung CW, Lee JY, Hung YC, Lee CH, Chou SH, et al. Plasma matrix metalloproteinase 1 improves the detection and survival prediction of esophageal squamous cell carcinoma. Sci Rep. 2016;6:30057. https://doi.org/10.1038/srep30057.
Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the cancer cell line encyclopedia. Nature. 2019;569(7757):503–8. https://doi.org/10.1038/s41586-019-1186-3.
Liu C-J, Hu F-F, Xie G-Y, Miao Y-R, Li X-W, Zeng Y, et al. GSCA: an integrated platform for gene set cancer analysis at genomic, pharmacogenomic and immunogenomic levels. Brief Bioinform. 2022. https://doi.org/10.1093/bib/bbac558.
Bairoch A. The cellosaurus, a cell-line knowledge resource. J Biomol Tech JBT. 2018;29(2):25–38. https://doi.org/10.7171/jbt.18-2902-002.
Nakagami H, Kawamura K, Sugisaka K, Sekine M, Shinmyo A. Phosphorylation of retinoblastoma-related protein by the cyclin D/cyclin-dependent kinase complex is activated at the G1/S-phase transition in tobacco. Plant Cell. 2002;14(8):1847–57. https://doi.org/10.1105/tpc.002550%JThePlantCell.
Qiao M, Shapiro P, Fosbrink M, Rus H, Kumar R, Passaniti A. Cell cycle-dependent phosphorylation of the RUNX2 transcription factor by cdc2 regulates endothelial cell proliferation. J Biol Chem. 2006;281(11):7118–28. https://doi.org/10.1074/jbc.M508162200.
Dong D, Zhang W, Xiao W, Wu Q, Cao Y, Gao X, et al. A GRN autocrine-dependent FAM135B/AKT/mTOR feedforward loop promotes esophageal squamous cell carcinoma progression. Can Res. 2021;81(4):910–22. https://doi.org/10.1158/0008-5472.Can-20-0912.
Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR. Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha. Biochem J. 1998;331(1):299–308. https://doi.org/10.1042/bj3310299.
Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci USA. 1998;95(19):11211–6. https://doi.org/10.1073/pnas.95.19.11211.
Fatrai S, Elghazi L, Balcazar N, Cras-Méneur C, Krits I, Kiyokawa H, et al. Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55(2):318–25. https://doi.org/10.2337/diabetes.55.02.06.db05-0757%JDiabetes.
Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 1997;416(3):307–11. https://doi.org/10.1016/s0014-5793(97)01235-0.
Lu X, Pang Y, Cao H, Liu X, Tu L, Shen Y, et al. Integrated screens identify CDK1 as a therapeutic target in advanced gastrointestinal stromal tumors. Can Res. 2021;81(9):2481–94. https://doi.org/10.1158/0008-5472.Can-20-3580.
Marzec M, Zhang Q, Goradia A, Raghunath PN, Liu X, Paessler M, et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7–H1). Proc Natl Acad Sci USA. 2008;105(52):20852–7. https://doi.org/10.1073/pnas.0810958105.
Zhang H, Qin G, Zhang C, Yang H, Liu J, Hu H, et al. TRAIL promotes epithelial-to-mesenchymal transition by inducing PD-L1 expression in esophageal squamous cell carcinomas. J Exp Clin Cancer Res. 2021;40(1):209. https://doi.org/10.1186/s13046-021-01972-0.
Tran H, Bustos D, Yeh R, Rubinfeld B, Lam C, Shriver S, et al. HectD1 E3 ligase modifies adenomatous polyposis coli (APC) with polyubiquitin to promote the APC-axin interaction. J Biol Chem. 2013;288(6):3753–67. https://doi.org/10.1074/jbc.M112.415240.
Oikonomaki M, Bady P, Hegi ME. Ubiquitin specific peptidase 15 (USP15) suppresses glioblastoma cell growth via stabilization of HECTD1 E3 ligase attenuating WNT pathway activity. Oncotarget. 2017;8(66):110490–502. https://doi.org/10.18632/oncotarget.22798.
Acknowledgements
This research was funded by the Beijing Medical Award Foundation (No. YXJL-2022-0080-0184).
Funding
This research was funded by the Beijing Medical Award Foundation (No. YXJL-2022–0080-0184).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors declare no conflicts of interest.
Ethical approval
Animal experiments were approved by the Ethics Committee of Harbin Medical University (Approval number 2020-50-IIT) and conducted according to the Guide for the Care and Use of Laboratory Animals.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhang, J., Xin, Y., Ling, X. et al. Nucleoporin 93, a new substrate of the E3 ubiquitin protein ligase HECTD1, promotes esophageal squamous cell carcinoma progression. Human Cell 37, 245–257 (2024). https://doi.org/10.1007/s13577-023-01005-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-023-01005-2