
Abstract
CIC-DUX4 sarcoma (CDS), an aggressive soft tissue sarcoma, is characterized by a CIC and DUX4 rearrangement. It has a dismal clinical course and high metastatic rate and shows chemotherapy resistance; therefore, a novel therapeutic strategy is required. Patient-derived cell lines are indispensable tools for basic and preclinical research. However, only a few patient-derived CDS cell lines have been currently reported. Therefore, in this study, we aimed to establish and characterize a novel cell line of CDS. We successfully established the NCC-CDS2-C1 cell line by using surgically resected tumor tissue from a patient with CDS. The NCC-CDS2-C1 cells harbored a CIC-DUX4 fusion gene without insertion and exhibited rapid growth, spheroid formation, and invasion. We screened the antiproliferative effects of small anticancer agent compounds, which included FDA-approved anticancer drugs, on NCC-CDS2-C1 cells in comparison with those on the two previously reported patient-derived CDS cell lines, NCC-CDS1-X1-C1 and NCC-CDS1-X3-C1. The response profile of NCC-CDS2-C1 was similar to but distinct from those of the other cell lines for the small anticancer agent compounds. Therefore, we conclude that the NCC-CDS2-C1 cell line will be a useful tool for basic and preclinical studies of CDS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CIC-DUX4 sarcoma (CDS) is an aggressive malignancy characterized by the presence of a unique fusion gene composed of CIC (19q13) and DUX4 (4q35 or 10q26) [1,2,3]. CDS predominantly occurs in the soft tissues of children and young adults. CDS belongs to an aggressive subset of small round cell sarcomas. CDS shares partial morphology overall with Ewing sarcomas, such as undifferentiated and mostly monotonous proliferation of round to ovoid cells [4]. The differential diagnosis of CDS from Ewing sarcomas requires the detection of CIC-related fusions [2, 5]. Despite the histological similarities to Ewing sarcomas, the entities and clinical characteristics of CDS are significantly distinct from those of Ewing sarcomas [2, 4,5,6]. CDS exhibits resistance to chemotherapy, and rapid development to lethal metastatic disease, and has a worse clinical course than Ewing sarcomas; a 5-year survival rate was 43% for the entire group of CDS, which was significantly lower than the 76% 5-year survival in the control Ewing sarcomas [4]. Because no standard treatment has been established yet, novel therapeutic strategies are required for CDS.
Patient-derived cancer cell lines are critical tools for both basic and preclinical studies. Cell lines derived from tumor tissue and maintained for a short period may retain the original genetic and phenotype characteristics. They are adequate in vitro models for examining the etiological factors of disease, the functions of intriguing genes and proteins, and the effects and mode of action of novel drugs. CDS is extremely rare; therefore, only three patient-derived cell lines of CDS had been reported [7, 8] until our current study. Considering the general diversity of malignant tumors, a greater number of cell lines from distinct patients with CDS are required.
Previously, we reported two novel cell lines of CDS [7]. They were the first cell lines established from the tumor tissues of CIC-DUX4 sarcoma. We characterized their phenotypes and molecular backgrounds and evaluated the antiproliferative effects of small compounds on the cell lines established [7]. Studying the molecular background of CDS using those cell lines helped establish a novel indication for dinaciclib, an inhibitor of cyclin-dependent kinase [9].
Here, we report the establishment of NCC-CDS2-C1, a novel CDS cell line established from surgically resected tumor tissue from a patient with CDS. We characterized the NCC-CDS2-C1 cells in terms of proliferation, spheroid formation, and invasion. We also examined the antiproliferative effects of small compounds on NCC-CDS2-C1, comparing them with the effects on the previously reported two CDS cell lines, NCC-CDS1-X1-C1 and NCC-CDS1-X3-C1 [7].
Materials and methods
Patient history
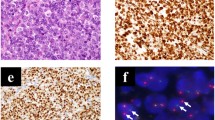
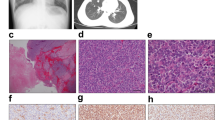
The donor patient was a 50-year-old woman with CIC-rearranged sarcoma. She visited the hospital with the major symptoms of pain and swelling at the back of the left thigh on presentation. Magnetic resonance imaging detected a tumor with heterogeneous components at the back of the left thigh (Fig. 1a–d). Computed tomography and positron emission tomography detected lung metastasis (Fig. 1e, f). CIC-rearranged sarcoma was confirmed by histopathological observation and FISH analysis performed using a needle biopsy sample (Fig. 1g, h). The tumor consisted of diffuse proliferation of relatively monotonous small round cells, and most tumor cells harbored split green and orange signals, indicating the presence of CIC gene rearrangement. The tumor was immunohistochemically positive for CD99 (focal, I), WT1 (nuclear, J). ETV4 (diffuse strong, data not shown), whereas it was negative for NKX2-2 and myogenin (data not shown).

Clinical imaging findings for patient with CIC-rearranged sarcoma. MRI showed an inhomogeneous mass (20 × 12 × 11 cm) at the back of the left thigh, which presented with low to iso intensity in the T1 axial view (a) and coronal view (b) and iso to high intensity in the T2 axial view (c) and coronal view (d). These findings may reflect partial hemorrhage and necrosis in the mass. Computed tomography showed an enhanced mass in the lung (e). Positron emission tomography demonstrated metastasis in the lung (f). Histological features of tumor tissue showing typical appearance of CIC-rearranged sarcoma (g). FISH analysis demonstrated the presence of the CIC gene rearrangement (h). The tumor was immunohistochemically positive for WT1 (i) and ETV4 (j)
The patient underwent three courses of neoadjuvant chemotherapy (vincristine, doxorubicin, cisplatin, ifosfamide, and etoposide [VDC-IE]). Decrease in tumor size was observed after chemotherapy, and wide resection was performed. The tumor tissues obtained at the time of surgery were used to establish the cell line. The use of these clinical materials for this study was approved by the ethical committee of National Cancer Center (Tokyo, Japan), and written informed consent was obtained from the donor patient with CDS.
Histological analysis
Tumor tissues were fixed in 10% buffered formalin and embedded in paraffin. The 4-μm-thick sections were stained with hematoxylin and eosin. For immunohistochemistry, heat-induced epitope retrieval method was used. The primary antibodies used were: CD99, O13,1:50, Signet, Dedham, MA; WT1, 6F-H2,1:50, Dako, Glostrup, Denmark; ETV4, 1:50, Santa Cruz Biotechnology, Santa Cruz, CA; NKX2-2, 74.5A5, 1:250, Developmental Studies Hybridoma Bank, Iowa City, IA. The reactions were detected with the EnVision system (Dako).
Cell culture procedure
The surgically resected tumor tissue was used for cell line establishment as previously described [7]. In brief, the tumor tissue was minced with scissors and seeded into tissue culture plates (Thermo Fisher Scientific, Waltham, MA, USA). The tumor tissue was treated with collagenase type II (Worthington Biochemical Corporation, Lakewood, NJ, USA). The cells were maintained in DMEM/F12 (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 U penicillin G, and 100 µg/mL streptomycin (Gibco) at 37 °C in a humidified atmosphere and 5% CO2. When the cell monolayers reached subconfluence, the cells were dispersed with 0.1% Trypsin–EDTA (Gibco) and seeded into new culture plates. The cells were maintained for more than 3 months under tissue culture conditions and passaged more than 20 times.
Authentication and quality control of the cell line established
The cell line established was authenticated by examining short tandem repeats (STRs) in 10 loci by using the GenePrint 10 system (Promega, Madison, WI, USA) according to the manufacturer’s instructions and the procedure described in a previous study [9]. In brief, genomic DNA was obtained from original tumor tissue or tissue-cultured cells by using AllPrep DNA/RNA Mini kits (Qiagen, Hilden, Germany). The genomic DNA quantity was determined using a NanoDrop 8000 instrument (Thermo Fisher Scientific). Subsequently, the genomic DNA was stored at − 80 °C until use. The STRs were amplified using this genomic DNA (500 pg), and the PCR products were analyzed using a 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). Data were analyzed using the GeneMapper software (Applied Biosystems), and the STR profiles obtained were compared to those of cell lines deposited in public cell banks by using a function of Cellosaurus, a cell line database [10].
When the cells reached 70–90% confluence, mycoplasma contamination was examined using the DNA in the tissue culture medium of the cell line established, as previously described [9]. This DNA was heated at 95 °C for 10 min, and then the DNA sequence unique to mycoplasma was amplified using the e-Myco Mycoplasma PCR Detection Kit (Intron Biotechnology, Gyeonggi-do, Korea). The amplified DNA was separated by gel electrophoresis performed using a 1.5% agarose gel. Gel images were obtained by staining with Midori Green Advance stain (Nippon Genetics, Tokyo, Japan) and analyzed using an Amersham Imager 600 system (GE Healthcare Biosciences, Little Chalfont, UK).
Genetic analysis
Genomic DNA and total RNA were extracted from the excised tissue or cultured cells with the AllPrep DNA/RNA Mini Kit (Qiagen). Total RNA (1 μg) was used for the reverse transcription (RT) reaction with Superscript III reverse transcriptase (Invitrogen), according to the manufacturer’s instructions. The CIC–DUX4 fusion transcript was amplified with the CIC forward primer CIC2F (5′-ATCATGCAGGCTGCCACT-3′) and DUX4 reverse primer DUX4R2 (5′-ATGCCTTGCATCTGCCC-3′), using KOD-Plus-Neo DNA polymerase (Toyobo, Osaka, Japan). For Sanger sequence analysis, the products were further amplified with the same forward and reverse primers for the junction. The PCR products were purified with ExoSAP-IT (Affymetrix), and direct sequencing was performed using the BigDye v3.1 Cycle Sequencing Kit (Applied Biosystems) on the Applied Biosystems 3130xL by Genewiz (Japan). The sequence data obtained were matched against the CIC (NM_015125.4) and DUX4 (NM_001293798.2) sequences by using BLAST.
FISH analysis was performed on 4-µm-thick sections of a needle biopsy sample. Break-apart probes (Chromosome Science Labo, Sapporo, Japan) were used for CIC genes as previously described [7]. FISH images were obtained using the Metafer Slide Scanning Platform (MetaSystems, Newton, MA, USA). The presence of split 5′ and 3′ signals or isolated 5′ signals in more than 20% of tumor cells was considered positive for CIC rearrangement.
Single nucleotide polymorphism (SNP) array genotyping was performed using the Infinium OmniExpressExome-8 v1.4 BeadChip (Illumina, San Diego, CA). In brief, genomic DNAs were extracted from the original tumor and established cell line, and subjected to amplification and hybridization. Array scan was performed using the iScan system (Illumina), and Log R ratios and B allele frequencies were calculated using Genome Studio 2011.1 + cnvPartition v3.2.0 (Illumina). The results were visualized using KaryoStudio Data Analysis Software version 1.0 (Illumina).
Cell proliferation assay
The tissue-cultured cells were seeded at 4 × 104 cells/well in 24-well culture plates at day 0, and the cell number in each well was counted at the indicated time points in the figure. The doubling time was calculated on the basis of the growth curve. All the experiments were performed in triplicate.
Spheroid formation assay
The spheroid formation assays were performed as previously described [7]. Briefly, 1 × 104 cells were seeded into 96-well plates (96-well Clear Flat Bottom Ultra Low Attachment Microplate; Corning, Inc., Corning, NY, USA) in DMEM/F12 containing 10% FBS. The seeded cells were maintained in a humidified atmosphere of 5% CO2 at 37 °C for 5 days. Then, spheroid formation was confirmed by microscopic analysis (Keyence, Osaka, Japan). All the assays were performed in duplicate.
Transwell cell invasion assay
Cell invasion assays were performed with BD Biocoat Matrigel Invasion Chambers (BD Biosciences, Franklin Lakes, NJ, USA) according to the manufacturer’s instructions; NCC-CDS2-C1 was used. Briefly, 1 × 105 or 2 × 105 cells were plated in the upper chamber in serum-free medium. DMEM/F12 containing 10% FBS was added to the bottom chamber. The cells were incubated for 24 and 48 h. Invading cells on the bottom surface were stained using the Diff-Quik stain (Sysmex, Kobe, Japan); three separate areas were counted under a microscope at a magnification of 100 ×.
Screening for the antiproliferative effects of anticancer reagents
The cells were seeded in a 384-well plate (Thermo Fisher Scientific, Fair Lawn, NJ, USA) at 1 × 104 cells/well in DMEM/F12 supplemented with 10% FBS and incubated at 37 °C in a humidified atmosphere of 5% CO2 with the Bravo automated liquid handling platform (Agilent Technologies, Santa Clara, CA, USA). NCC-CDS2-C1 and the previously reported CDS cell lines, i.e., NCC-CDS1-X1-C1 and NCC-CDS1-X3-C1, were used [7]. On the following day, 195 anticancer agent compounds, including FDA-approved drugs (Selleck Chemicals, Houston, TX), were added at 10 µM concentration with the Bravo automated liquid handler. A list of the anticancer agents has been provided in Supplementary Table 1. After 72 h, survival rates were assessed with the CCK-8 reagent according to the manufacturer’s protocol. The response readout was calculated relative to the DMSO control, in terms of relative growth inhibition %.
Dose–response experiments were performed to validate available hits in the pilot screening. IC50, the sample concentration required to inhibit cell growth by 50% in comparison with the growth of the cell control, was determined from the dose–response curves. The compounds were dispensed in 384-well plates with serial dilution of compounds at 10 different concentrations covering 0.1–100,000 nM by using the Bravo Automated liquid handler. The cells were collected by trypsinization. Twenty-five microliters of single cell suspensions (1 × 104 cells) was dispensed into each well by using the Bravo Automated liquid handler. The plates were incubated for 72 h at 37 °C and 5% CO2 in a humidified environment.
Cell viability was determined using CCK-8. A multimode multiplate reader (Epoch, BioTek, USA) was used for signal quantification. The fluorescence values resulting from normalized cell numbers were plotted against the drug concentrations tested to obtain IC50 values with the GraphPad Prism 8.1.0 software. This analysis was performed in duplicate.
Data analysis
Cluster analysis was performed using the R software version 3.4.0 [11]. The distance matrix was determined using the dist function with method = “euclidean”. Hierarchical clustering was achieved using the hclust function with method = “ward.D.” Heat maps were visualized using the “gplots” package of R by using the script “heatmap.2.”
Results
Authentication of the cell line established
The NCC-CDS2-C1 cell line was authenticated by analyzing the STR status of 10 microsatellites in the NCC-CDS2-C1 cells and the corresponding original tumor tissue. All the STRs examined were identical between the NCC-CDS2-C1 cells and the corresponding original tumor tissue (Table 1). The STR patterns of NCC-CDS2-C1 cells did not match those of any cell lines in the public cell banks examined using a function of the cell line database, Cellosaurus. We did not detect the DNA sequence unique to Mycoplasma in the tissue culture medium of the NCC-CDS2-C1 cells (data not shown).
Characterization of the cell line
The presence of the CIC-DUX4 fusion gene was confirmed in the original tumor tissue as well as in the NCC-CDS2-C1 cells (Fig. 2). The fusion gene in NCC-CDS2-C1 cells was identical to that in the original tumor, and distinct from those in previously reported cell lines [7, 8]. The results of SNP array experiments demonstrated that the overall features of copy number alterations were similar between the original tumor and NCC-CDS2-C1. However, there are aberrations of chromosomal arms or entire chromosomes, which were unique to the tumor or NCC-CDS1-C1: NCC-CDS2-C1: amplified: 9d, 16p-q, and 20 q, deleted: 1p, 9q, and 20p, the tumor tissue: amplified: 8p-q, 16p-q, 20q, deleted: 20p) (Fig. 3).

Fusion gene analysis of NCC-CDS2-C1 cells. Nucleotide sequences of the CIC-DUX4 fusion gene. Sanger sequence analysis of the transcript showing CIC–DUX4 fusion: original tumor tissue (upper panel) and NCC-CDS2-C1 cells (lower panel)

Study of single nucleotide polymorphism (SNP) array. Log R ratios represent the normalized signal intensity, and B allele frequencies represent the normalized ratio of the number of the B allele to the total quantity of both A and B alleles combined. Ideograms of chromosomes are positioned below the plot
NCC-CDS2-C1 cells exhibited the fibroblastic appearance in 2D culture conditions under the phase contrast microscopic observation (Fig. 4a). NCC-CDS2-C1 cell proliferation was monitored using a growth curve based on a cell counting assay. The population doubling time during the logarithmic growth phase was determined as approximately 28 h on the basis of the assay (Fig. 4b). We also evaluated the ability of the NCC-CDS2-C1 cells to form spheroids, which are used in cancer research as in vitro three-dimensional tissue microanalogs [12], and confirmed that the cells formed spheroids when they were seeded on 96-well spheroid microplates (Fig. 4c). We also found that NCC-CDS2-C1 cells exhibited invasion ability and that it varied depending on the number of seeded cells and time after seeding (Fig. 4d).

Characterization of NCC-CDS2-C1 cells. a NCC-CDS2-C1 cells showed fibroblastic appearance under the 2D culture conditions. The images were taken at day 2 after passaging, when the cells were at passage 20. b Growth curve of NCC-CDS2-C1 cells. Each point represents the mean ± SD value (n = 3). The y-axis indicates the cell proliferation plot of NCC-CDS2-C1 cells, and the y-axis represents the day after passage. c Spheroid formation of NCC-CDS2-C1 cells was observed in 96-well spheroid microplates. d The invasion ability of NCC-CDS2-C1 cells was analyzed in comparison to that of the previously reported CDS cell lines, NCC-CDS1-X1-C1 and NCC-CDS1-X3
Sensitivity to anticancer drugs
The sensitivity of the cells to 195 FDA-approved anticancer drugs was monitored (Supplementary Table 1). The cells were treated with the drugs at 10 µM, and the proliferation-suppressive effects were recorded (Supplementary Table 2). Using the data of inhibitory effects, the cluster analysis of anticancer agent compounds was performed (Fig. 5, Supplementary Table 3). Overall, the anticancer agent compounds were grouped into clusters A and B. Cluster A included the anticancer agent compounds which had growth inhibitory effects in at least two of three cell lines examined, and those in cluster B included the anticancer agent compounds which had obvious effects on a single cell line. In addition, there are two groups of anticancer agent compounds which showed inhibitory effects on growth only in NCC-CDS2-C1. Those included (1) romidepsin, paclitaxel, megestrol acetate, and methoxsalen in cluster A, and (2) cobicistat, idarubicin HCl, belinostat, procarbazine HCl, imatinib mesylate, vandetanib, crizotinib, bortezomib, arsenic (III) oxide, and homoharringtonine in cluster B. Dexrazoxane, nintedanib, ponatinib, and carboplatin showed strong inhibitory effects on all three CDS cell lines. The IC50 values of these drugs are listed in Table 2, and the growth curve based on which the IC50 values were calculated are demonstrated in Fig. 6.

Overall features of effects of anticancer agents on cell proliferation. The cell lines were treated with 195 anticancer agents (10 µM) for 72 h. The inhibitory effects of anticancer agents were calculated on the basis of the data from the CCK-8 assay. Data for the NCC-CD2-C1 cells and the previously reported patient-derived CDS cell lines, NCC-CDS1-X1-C1 cells and NCC-CDS1-X3-C1, have been demonstrated in the heat map format. The anticancer agents in clusters A and B have been described in Supplementary Table 3

Growth curves for IC50 value calculation of investigated anticancer agent compounds. a–c Results for NCC-CDS1-X1-C1, NCC-CDS1-X3-C1, and NCC-CDS2-C1, respectively. d–v Eighteen of the compounds inhibited cell growth. The viability of the cells treated with arsenic (III) oxide, belinostat, bortezomib, carboplatin, cobicistat, crizotinib, dexrazoxane, homoharringtonine, idarubicin HCl, imatinib mesylate, megestrol acetate, methoxsalen, nintedanib, paclitaxel, ponatinib, procarbazine HCl, romidepsin, or vandetanib was examined. The IC50 value for each anticancer agent compound is shown in Table 2
Discussion
In this study, we established NCC-CDS2-C1, a novel cell line of CDS derived from another patient with this disease. This paper is the third report about the establishment of patient-derived cell lines of CIC-DUX4 sarcoma [7, 8].
NCC-CDS1-C1 and original tumor had an identical fusion gene. However, NCC-CDS2-C1 and the original tumor had a similar, but slightly different genetic backgrounds in terms of copy number alterations. Because the passage duration of NCC-CDS2-C1 is relatively short, this difference might not be due to the genetic change of tumor cells in the tissue culture condition, but due to the selection of tumor cells in the process of cell line establishment.
We characterized the NCC-CDS2-C1 cells in terms of proliferation, spheroid formation, and invasion. The aggressive characteristics of NCC-CDS2-C1 were equivalent to those of NCC-CDS1-X1-C1 and NCC-CDS1-X3-C1 cells [7]. Their in vitro features could be relevant in relation to the aggressive clinical features of CDS, and the NCC-CDS2-C1 cells can be used for in vitro experiments to assess the effects of genes or proteins whose expression levels are significantly associated with clinical observations.
We previously reported the establishment of two novel patient-derived cell lines of CDS such as NCC-CDS1-X1-C1 and NCC-CDS1-X3-C1 [7]. Those two cell lines were the first patient-derived cancer cell lines of CDS. In the current study, we compared the effects of anticancer agent compounds on proliferation in these three CDS cell lines. We found that the response to treatments were similar but distinct among the three cell lines, with more similarity noted between the two previously reported cell lines that were established from the same patient; thus, our cell lines may reflect the clinical features of CDS. To prove this hypothesis, a greater number of cell lines derived from different patients are required. The anticancer agent compounds whose effects were examined in this study could be divided into three categories: those with inhibitory effects on all three cell lines, those with inhibitory effects on NCC-CDS2-C1 cells, and those without effects on all three cell lines. Comprehensive genetic profiling may be required to understand the molecular basis of the different responses to treatments in these cell lines.
Although the donor patient underwent three courses of neoadjuvant chemotherapy (vincristine, doxorubicin, cisplatin, ifosfamide, and etoposide [VDC-IE]), the NCC-CDS1-C1 cells did not respond to those chemotherapy agents. The NCC-CDS2-C1 cells were established from the tumor tissues after the treatments with those chemotherapy reagents. Tumor cells that were sensitive to chemotherapy had deadly damage after treatment, and the tumor cells which were resistant to the anticancer drugs survived after the neoadjuvant treatment and selectively grew as NCC-CDS2-C1 cells in the tissue culture condition.
We found that a group of drugs that showed antiproliferative effects only in NCC-CDS2-C1 cells included cobicistat, idarubicin HCl, belinostat, procarbazine HCl, imatinib mesylate, vandetanib, crizotinib, bortezomib. arsenic (III) oxide, and homoharringtonine (Fig. 5). These drugs were grouped in cluster B. They belong to different categories of drugs, and we did not find any mechanisms underlying their unique effects on NCC-CDS2-C1 cells. Investigation of the genes and proteins, whose expression and activity are affected by these drugs, may lead a clue to understand this intriguing difference among the cell lines derived from identical sarcoma types.
The clinical features of CIC-DUX4 sarcoma are diverse, and experimental findings obtained using a limited number of cell lines cannot provide conclusive results; therefore, more cell lines from more patients are required for basic and preclinical studies of CDS. Considering the extreme rarity of the disease and renewability of models, information on patient-derived cancer cell lines should be shared in the research community.
We conclude that NCC-CDS2-C1 will be a useful patient-derived cancer cell line for investigating the molecular backgrounds underlying the tumor characteristics and to develop novel therapeutic strategies for CDS.
References
Italiano A, Sung YS, Zhang L, et al. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosom Cancer. 2012;51:207–18.
Yoshida A, Goto K, Kodaira M, et al. CIC-rearranged sarcomas: a study of 20 cases and comparisons with Ewing sarcomas. Am J Surg Pathol. 2016;40:313–23.
Kawamura-Saito M, Yamazaki Y, Kaneko K, et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet. 2006;15:2125–37.
Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC-rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41:941–9.
Specht K, Sung YS, Zhang L, Richter GH, Fletcher CD, Antonescu CR. Distinct transcriptional signature and immunoprofile of CIC-DUX4 fusion-positive round cell tumors compared to EWSR1-rearranged Ewing sarcomas: further evidence toward distinct pathologic entities. Genes Chromosom Cancer. 2014;53:622–33.
Yoshimoto T, Tanaka M, Homme M, et al. CIC-DUX4 induces small round cell sarcomas distinct from Ewing sarcoma. Cancer Res. 2017;77:2927–37.
Oyama R, Takahashi M, Yoshida A, et al. Generation of novel patient-derived CIC- DUX4 sarcoma xenografts and cell lines. Sci Rep. 2017;7:4712.
Nakai S, Yamada S, Outani H, et al. Establishment of a novel human CIC-DUX4 sarcoma cell line, Kitra-SRS, with autocrine IGF-1R activation and metastatic potential to the lungs. Sci Rep. 2019;9:15812.
Okimoto RA, Wu W, Nanjo S, et al. CIC-DUX4 oncoprotein drives sarcoma metastasis and tumorigenesis via distinct regulatory programs. J Clin Investig. 2019;129:3401–6.
Bairoch A. The Cellosaurus, a cell-line knowledge resource. J Biomol Tech. 2018;29:25–38.
R: a language and environment for statistical computing. Vienna. http://www.R-project.org. Accessed 29 Dec 2019.
Nath S, Devi GR. Three-dimensional culture systems in cancer research: focus on tumor spheroid model. Pharmacol Ther. 2016;163:94–108.
Acknowledgements
We thank Drs. Y. Minami, K. Shimizu, T. Mori, T. Uehara M. Sugawara, Y. Araki, and T. Hirose from the Division of Musculoskeletal Oncology, National Cancer Center Hospital, for sampling tumor tissue specimens from surgically resected materials. We appreciate the technical assistance provided by Mr. T. Ono and K. Tanoue. We would like to thank Editage (www.editage.jp) for English-language editing and their constructive comments on the manuscript. This research was financially supported by the National Cancer Center Research and Development Fund (Grant no. 29-A-2).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
Consent for publication was provided by the patient.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yoshimatsu, Y., Noguchi, R., Tsuchiya, R. et al. Establishment and characterization of NCC-CDS2-C1: a novel patient-derived cell line of CIC-DUX4 sarcoma. Human Cell 33, 427–436 (2020). https://doi.org/10.1007/s13577-019-00312-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13577-019-00312-x