
Abstract
Transformation of rice was done through Agrobacterium mediated, utilizing a binary vector pBS, harboring a fungal gene endo α-1, 3-glucanase from Trichoderma harzianum under rice constitutive promoter actin2 and Agrobacterium nopaline synthase (nos) transcriptional terminator in a T-DNA. Likewise, a selectable marker gene hygromycin phosphotransferese (hpt) resistant to Hygromycin B was cloned in the middle of actin2 and nos terminator in a similar T-DNA. The expression of endo α-1, 3-glucanase was affirmed by cloning gfp gene after the fungal gene in the transformation DNA cassette. In the first generation of transgenic rice lines, out of 912 just 209 plants were false positive affirmed through PCR based screening for the transgene. The positive transgenic lines were tried with fungal infection by leaf cut test in vitro and foliar leaf shower technique in vivo. They demonstrated an exceptional protection against sheath blight disease. Further, seeds of all positive transgenic plants of the first generation were developed and screened in next generation. Just 62 plants were false positive out of 873 transgenic lines in this generation. In the comparative way, they were tried against the fungal disease and they demonstrated the exceptional protection once more. In this way, in this investigation, a fungal gene endo α-1, 3-glucanase was transformed into rice (IR 64) effectively which demonstrated protection against fungus Rhizoctonia solani.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice is a cereal grain which produced for the most part in warm place of the world like Asia, Africa, Northern Italy, North America and so forth. It is broadly the most consumable cereal as a staple food by total population. The generation of rice is intensely influenced by impact blast after by sheath blight. Sheath blight is caused by soil-borne pathogenic fungus Rhizoctonia solani, which influences the generation of numerous monetarily vital farming and agricultural products overall (Mayo et al. 2015). The comprehension of contamination component of this disease may control flare-up and movement of it in fields. In this way a change strategy which exchanges the trans-gene from fungus in plant genome with a constitutive DNA cassette may give protection against sheath blight. Rhizoctonia solani is isolated into fourteen hereditarily particular gatherings (AG1 to AG13 and AGBI) based on have specificity and reproducibility (Chen et al. 2016a, b). There is no any gene (s) revealed yet in rice genome which encodes for α-1, 3-glucanase. The parasitic cell wall is made up of chitin, mannans, α and β-glucan (polysaccharides), cell layer sterols and so forth. It was beforehand detailed that Magnaporthe oryzae utilize α-1, 3-glucan in veiling surfaces of their cell wall to attack the host plants (Fujikawa et al. 2012). The organization of the cell wall of plant parasitic pathogen is comprehensively in comparative patterns. In host plant, some pathogen related receptors (PRRs) are available which perceives the pathogen associated molecular patterns (PAMPs) show in pathogens. After acknowledgment, the immune system of host plant got activated and discharge antifungal chemicals, hostile to microbial metabolites, and reactive oxygen species and so forth. However, pathogens built up a few mechanisms which get away from the detection of their PAMPs by the host PRRs. In this manner surface α-1, 3-glucan shields the contagious cell wall from being exposed to compounds delivered by plant cells. In different plant transformation techniques, Agrobacterium tumafaciens mediated genetic change is the effective technique in plant biotechnology. This strategy is broadly utilized as a part of the creation of safe transgenic plants. Officially, some other techniques like biolistic (Tang et al. 2006), Agrobacterium tumefaciens mediated co-transformation (Sengupta et al. 2010), site specific recombination (Scaramelli et al. 2009), many genes vector framework (Svitashev et al. 2015), transposon-intervened (de Vetten et al. 2003), intra-chromosomal recombination and PCR based (Moradpour and Abdullah 2017) and so on were effectively created in plant transformation. In one Agrobacterium strain, one plasmid harbouring two T-DNAs was marginally adjusted and used to transforme pre-refined rice calli. A pBS vector containing an endo α-1, 3-glucanase gene from Trichdema harzianum alongside actin2 promoter and nos eliminator. The hygromycin phosphotramferase (hpt) gene was utilized for transformation.
In this present investigation, an endo α-1, 3-glucanase gene was identified in Trichdema harzianum through bioinformatics assessment. The gene was confirmed, described and cloned to set up the entire transformation cassette. The rice was transformed utilizing the prepared DNA cassette. The transformed rice lines were developed totally and portrayed. The expression of DNA cassette was confirmed by cloning a gfp gene in the DNA cassette. In this manner, the gene was transformed in rice which demonstrated protection in two progressive generations.
Materials and Methods
Fungal strains, Growth media and Conditions
Rhizoctonia solani (MTCC, Chandigarh, India, Cat#9666) was grown on potato dextrose (Himedia, India, LOT 0000233850) broth at 25 ± 2 °C and 150 RPM in dark for 12 days (Xue et al. 2012). Then the culture was used for bioassays. The potato dextrose agar (0.5% w/v) plate was inoculated with Rhizoctonia solani spore for sub-culturing and preparation of liquid culture.
Trichoderma harzianum (MTCC, Chandigarh, India, Cat#792) was grown on malt extract broth and agar at 28 ± 2 °C and 150 RPM in dark for 7 days.
Agrobacterium tumefaciens (MTCC, Chandigarh, India, Cat#609) was grown on tryptic agar (30 g trypticase soy broth + 15 g agar per litre, pH 7.5) and trypticase soy broth at 30 °C.
Genomic DNA isolation of Trichoderma harzianum and amplification of endo α -1, 3 glucanase gene
Fungal DNA isolation was done through Zymo fungal DNA isolation kit (Zymo Research, U.S.A., Cat#D6005) according to manufacturer protocol. The gene was amplified with specific of the gene ‘endo α-1, 3-glucanase’ forward 5′-TTCGGGATGGTAATCTTTGGCTGCTTC-3′ and reverse 5′-TTCGGGCTAGGACGTCCCAACCCAGC-3′ using thermocycler of 35 cycles. Final product was analysed in 0.8% agarose gel.
Polymerase Chain Reaction
Plasmid DNA of each confirmed clone isolated individually and amplified through PCR followed by PCR purification. The purified product was ligated further and cloned one by one in a single pBS vector to complete the cassette preparation. The confirmed clone again amplified and purified. The amplification of cassette (Forward primer of promoter: 5′-CTTACCAGCTAGCATAGTCGAGGTCAT-3′ and reverse primer of terminator: 5′-TTCGCTCGATCTAGTAACAAAGATGACAC-3′) and only endo α-1, 3-glucanase (Forward primer of gene: 5′-CCCGGGATGGCAATCTTTGGCTGCTTC-3′ and Reverse primer of gene: 5′-CCCGGGCTAGGAGGACCCAACCCAGG-3′) gene was done. The annealing temperature for endo α-1, 3-glucanase was standardized as 57 °C. For characterization purpose, all isolated genomic DNA of each transformed individual was amplified using a set of primer only for the gene. All PCR was performed using slightly modified protocol standardized earlier (Metzker and Caskey 2009) in Mastercycler Pro Eppendorf Vapo Protect Machine.
Cloning of promoter, gene, and terminator in pGEM-T easy vector and pBS vector
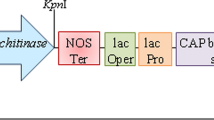
The constitutive promoter actin2 (Forward 5′-GGTACCAGCTAGCATACTCGAGGTCAT-3′ and reverse 5′-GTCGACGATATCCTCGGCTCAGCCA-3′) from rice was directly amplified from isolated genomic DNA of rice variety IR64. Further, the actin2 promoter (1266 bp) was ligated by slightly modified standard protocol mentioned earlier and cloned in pGEM-T easy vector. The nos terminator isolated amplified (Forward 5′-CCCGGGATGGCAATCTTTGGCTGCTTC-3′ and reverse 5′-CCCGGGCTAGGAGGACCCAACCCAGG-3′) and purified from Agrobacterium genomic DNA. Further, the nos terminator (253 bp) was ligated by slightly modified standard protocol mentioned earlier (Xue et al. 2012) and cloned in pGEM-T easy vector. Again purified endo α-1, 3-glucanase gene was ligated and cloned in pGEM-T easy vector. All (promoter, gene and terminator) were cloned in pBS vector sequentially from separate pGEM-T easy vector to make the DNA cassette.
Validation of expression of DNA cassette and its entry
The DNA cassette entry in cells was confirmed by cellular localization experiment. The gfp in DNA cassette just after endo α-1, 3-glucanase gene were cloned in pCAMBIA 1302 vector. Both cassettes were prepared and rice calli, tobacco leaves transformed with them. The explants were kept on wet filter paper with ½ liquid MS medium in sterile plates. Similarly, rice root was also transformed and incubated in the same manner. The images of all explants were taken in fluorescence microscope by preparing slides.
Rice Calli Induction
Mature seeds of Indica rice (IR64) were de-husked by hands; surface sterilized by sequentially washed with sterile water twice, 70% ethanol and finally 4% sodium hypochlorite with 0.1% Tween 20. Surface sterilized seeds were inoculated on MS medium (Murashige and Skoog 1962), supplemented with 30 g/L sucrose, proline 65 mg/L, casein hydrolysate 30 g/L and 2,4-D 2.5 mg/L. Cultures were kept at 28 °C in dark for 1 week then only calli (seed and remaining part removed) sub cultured on the same medium.
Transformation of rice through embryogenic calli and regeneration of transgenic rice plantlets
Agrobacterium tumefaciens strain was transformed with a selectable marker gene (hpt) from pCAMBIA 1301 vector and DNA cassette from pBS vector in a single strain by freeze–thaw method (Chen et al. 2016a, b). The Agrobacterium transformed with hpt gene was grown on LB (Luria–Bertani) agar plate supplied with 50 mg/L Kanamycin. A single, isolated colony was picked and sub-cultured on the same medium. The culture was inoculated in 5 mL LB and plasmid isolated. The plasmid was digested with kpnI-smaI (2.6 kb) and confirmed in agarose gel. Calli induced from mature rice seed on callus induction medium of 1–2 weeks old were used for transformation by Agrobacterium. Agrobacterium strain harbouring two plasmids; one with selectable marker gene hpt and other pBS with DNA cassette was grown on Yeast Extract-mannitol (YEM) medium in dark at 28 °C after reaching growth at the optical density (OD600) of 0.5, the culture centrifuged at 5000 rpm for 10 min at 4 °C. The bacterial pellet was re-suspended in the equal volume of AAM (A protocol for Agrobacterium Mediated transformation) medium (Toriyama and Hinata 1985) supplemented with 20 mg/L acetosyringone. The Agrobacterium culture was incubated at 28 °C and 200 rpm, in the dark for 1 h. Pre-cultured rice calli were immersed in Agrobacterium suspension for 15 min and blotted dry of excess suspension on sterilized filter paper. The blotted rice calli were transferred in 1 mL co-cultivation medium (MS + YEM) for 3 days in the dark at 28 °C. The rice calli were washed thrice with sterile water and then sterile water with 500 mg/L carbenicillin to remove excess Agrobacterium. The blotted rice calli were inoculated on callus induction medium with 500 mg/L carbenicillin and 40 mg/L Hygromycin B for 1 week in dark at 28 °C. Finally proliferating calli were transferred on shoot regeneration medium with 30 mg/L Hygromycin B and incubated at 28 °C in photoperiod of 16:8 h light and dark.
Cell harvesting and DNA isolation
The one gram of leaf sample (any leaf after the third leaf) was collected in 1.5 mL micro centrifuge tube and froze immediately in liquid nitrogen. The samples further crushed with help of liquid nitrogen and dissolved in 400 μL of pre-warmed (65 °C) DNA extraction buffer and kept at the same temperature for 1 h. In between, it was mixed two to three times. Genomic DNA of each rice plant was isolated by CTAB (Cetyltrimethylammonium Bromide) method (Cota-Sánchez et al. 2006) with slight modification. This method was used for extracting unsheared intact genomic DNA of sequencing quality.
The 1 g of rice leaf sample was crushed in 400 μL of DNA extraction buffer (2% CTAB, 100 mM Tris HCl (pH 8), 20 mM EDTA, 1.4 M NaCl, 0.2% β-mercaptoethanol-added just before use). After incubation add an equal volume of chloroform/isoamyl alcohol (24:1) solution followed by a gentle mix and spin 15 min at 14,000 RPM. Carefully transfer the aqueous phase in a clean centrifuge tube. Again repeat the above step to get more purified DNA. Add 1 μL of RNAse to the mixture and keep for 5 min at RT. Add 2/3 volume isopropanol followed by gentle mix. Wash with 70% EtOH twice. Keep for complete disappearance of alcohol smell. Re-suspend the pellet in nuclease-free water or TE (pH 8) and store at − 20 °C.
Bio-assays
The pathogenicity of plant pathogens was checked by simple detached leaf assay (Pettitt et al. 2011). The protocol of bio-assay adopted in this study was modified slightly as mentioned in above reference. The PCR positive rice transgenic lines were exposed to infection of Rhizoctonia solani by the modified bio-assay protocol. To start with, the fungus was grown on agar plate up-to infectious stage. Then spores were collected in clean, sterilized 15 mL centrifuge tube. The collected spore was suspended well by vortexing. The leaf samples of each transformed plant were collected from various panicles randomly and put away in ice quickly. The leaf samples were washed with sterile distilled water in laminar followed by 20% of alcohol for 5 min in shaking condition. The samples were washed with sterile distilled water thrice, dried on filter paper and transferred to water agar plate by covering of wet filter paper in fungal spore suspension. The plates were incubated at 25 °C for 72 h in dark condition. Further, plates were incubated at 25 °C for 7–10 days in greenhouse.
Results
Isolation, cloning and characterization of an endo α-1, 3-glucanase gene from Trichoderma harzianum
It is already mentioned above that no any gene present in rice genome which produces α-1, 3-glucanase enzyme i.e. responsible for degradation of surface α-1, 3 glucan. Trichoderma harzianum having genes for α-1, 3-glucanase enzyme which degrades the excess α-1, 3-glucan after successful infection (Fujikawa et al. 2012). The gene endo α-1, 3-glucanase (1411 bp) was amplified from two different growth stages (conidia and hyphae stage) genomic DNA of Trichoderma harzianum. The amplified product was purified through gel extraction after running in agarose gel (Fig. 1; lane 1 and 2; supplementary data). The promoter and terminator govern the constitutive expression of the gene. A transforming DNA cassette is to be prepared in sequential cloning as a promoter, gene, and terminator pBS vector. The constitutive promoter actin2 from rice and nos terminator from Agrobacterium were cloned individually and sequentially in pGEM-T easy vector (3 kb). Then all individual clones were incorporated into pBS vector to get the complete DNA cassette. The two separate clones of cloned pBS vector were confirmed through repetitive restriction digestions with SmaI (Fig. 2 Supplementary data). The band size (2.9 kb) in agarose gel was observed which indicate the size of DNA cassette. The order and orientation of promoter, gene, and terminator were checked by repetitive restriction digestion with the different combination of restriction endonucleases (Fig. 3 Supplementary data). The size of all fragments of DNA cassette with the different combination of restriction endonucleases was analysed through an online NebCutter tool. All size of fragments was found same as analyzed in above online tool. Lane: 1. Marker; 2. KpnI linearized the clone (5.5 kb); 3. KpnI + SacI cut the clone into three fragments 0.9 + 2.1 + 2.5; 4 KpnI + SmaI cut the clone into two fragments 1.3 + 4.2; 5 XbaI + SacI cut the clone into four fragments 0.5 + 1.0 + 1.5 + 2.5 and 6 XbaI + SacI cut the clone into three fragments 0.5 + 0.75 + 4.25 (Fig. 3 Supplementary data).
Agrobacterium-mediated transformation of DNA cassette in plant cell
With a specific end goal to approve the expression efficiency of the cassette, a cellular localization study was performed in rice calli as well as tobacco whole leaf. Earlier, the gus and gfp gene were used as a reporter gene in the validation of expression of transgene and its localization in plant system. Here, we cloned gfp gene in the DNA cassette after the transgene in pCAMBIA 1302 vector. The pre-cultured rice calli, tobacco leaves and rice seedlings were transformed using above mentioned Agrobacterium tumefaciens mediated transformation method (Akagi et al. 2015). The transformed rice calli, tobacco leaves and rice seedlings were incubated at 25 °C for 48 h with normal photoperiod on wet filter paper with half Murashige and Skoog (MS) medium in aseptic condition. The gfp expression was observed using UV lamp after incubation of 48 h (Fig. 1). The expression of GFP was observed in both rice calli and tobacco leaves. The above findings support the validation of transformation method which is successful and efficient in monocot transformation.

Sub-cellular localization of DNA cassette during transformation. The gfp was cloned in pCAMBIA 1302 (control). The DNA cassette was cloned again in pCAMBIA 1302 by tagging gfp gene with endo α-1, 3-Glucanase gene. a Untransformed rice calli-control and transformed rice calli-gfp. b Untransformed tobacco leaf-control and transformed leaf-gfp. c Untransformed rice seedling-control and transformed rice seedling-gfp
Transformation of rice calli, regeneration and characterization of regenerated plants
For the transformation of rice calli, fresh stock of seed was de-husked by hands. The surface sterilization of de-husked seeds was done as mentioned in methods and materials. After surface sterilization the seeds were dried aseptically and inoculated on callus induction medium. The induced embryogenic calli were sub cultured after 7–10 days of inoculation. The second subculture was done after 14–21 days. The young fragile calli were selected and transformed through Agrobacterium mediated transformation method as mentioned in methods and materials. The transformed rice calli were transferred on selection medium (Fig. 2). Subsequently, viable rice calli were transferred on regeneration medium. The regenerated rice calli were separated carefully and transferred in sterile soil rite. The small plantlets were incubated in plant growth chamber with 90% humidity, 28 °C and a photoperiod of 16 h dark and 8 h light.

Agrobacterium mediated Rice calli transformation and its regeneration in complete plantlets. a Rice calli preparation from seed, first subculture and second subculture. b Rice calli on selection medium after transformation. c Regeneration of transformed rice calli in complete plantlets
To characterize the transformed rice plants, the genomic DNA of each plant was isolated through CTAB method selecting the third/above leaf. The DNA samples were amplified with specific primers using PCR technique as mentioned in methods and materials. The transformed plants were granted as positive by running all amplified DNA sample in agarose gel. The band size of the positive plants was 1.5 kb which corresponding the positive control. In the represented picture of agarose gel, out of 12 transformed plants, only one was negative (Fig. 3a). In T0 generation out of 912 transformed plants, 209 plants were found as pseudo transformant. The positive plantlets were grown further and remaining was discarded. Similarly, a gel picture of 14 DNA samples found as all positive along with positive control in the T1 generation (Fig. 3b). But only 62 plants were also found as false positive out of 873 transformed plants. These observations may be concluded as the inheritance of trans-gene is very high in successive generations.

Screening of transformed plants in T0 and T1 generation through PCR. a In T0 genomic DNA of transformed plants was isolated, amplified in PCR. The trans-gene size 1.5 kb of transformed plant was observed in agarose gel in lane 3–14 which correspond positive control in lane 1. Out of 12 transformed plants, only one was negative in lane 12. Lane 2: marker. b In T1 genomic DNA of transformed plants was isolated, amplified in PCR. The trans-gene size 1.5 kb of transformed plant was observed in agarose gel in lane 1–14 which correspond positive control in lane 16. All 14 transformed plants were positive. Lane 15: marker
In order check the resistance against the sheath blight, bioassay experiment was done. The leaf samples of were collected randomly from each grown plant and cut in equal sizes. The leaf pieces were sterilized by rinsing in 20% alcohol for 5 min followed by washing with sterile water thrice. The explants were treated with fully grown liquid fungal culture of Rhizoctonia solani at the infectious stage by dipping for 50–60 s. The explants were immediately placed on water agar (1%) plates supplied with 2 mg/mL Kinetin. The sheath blight lesions were observed in all explants along with control. The infection in experimental plant leaves was almost absent or very slight which signified the increased resistance level in transformed plants against sheath blight. The same experiment was repeated for both generations [Fig. 4a(i), b(i)]. Another experiment with different spore count was done in both successive generations [Fig. 4a(ii), b(ii)]. We did infect control leaf with a density of 105 fungal spores per mL followed by experimental leaves with 109, 108, 107, 106 and 105 spores per mL respectively. The severity of infection was increased with increasing density of spore. The least infection was noted in case of minimum spore density (105 spores per mL) while control leaf showed maximum infection with the same density of fungal spore. In case of T1 generation, the same experiment was performed with only four spore density 108, 107, 106 and 105. The similar infection pattern was also found in this generation also which favours inefficiency of the developed method.

Bio-assays to check resistance level in transformed rice plants. For T0 generation a (i) bioassay of positive lines with Rhizoctonia solani. (ii) Bioassay with different density of fungal culture. 1 was control plant with 105 spores, 2 with 109 spores, 3 with 108 spores, with 107 spores, with 106 spores, with 105 spores. For T1 generation, b (i) bioassay of positive lines with Rhizoctonia solani. (ii) Bioassay with different density of fungal culture. 1 was control plant with 105 spores, 2 with 108 spores, 3 with 107 spores, with 106 spores, with 105 spores
Discussion
Sheath blight is a devastating disease in rice cultivation after blast around the world. Genetic engineering is successful tool in plant transformation using Agrobacterium. The fungal infection control through fungicide in field is impractical now. In host plants, cuticle layer is the first barrier which senses the fungal infections. Therefore, the fungi are exposed directly to the antifungal enzymes in the apoplast after crossing cuticle layer. Then host PRRs located in cell membrane recognises the fungal PAMPs. Rhizoctonia solani lacking surface α-1, 3-glucan was destroyed by host immunity actions. The destruction of fungal cells is due to the direct contact with the antifungal agents in the apoplast because the plant surfaces are covered by the cuticle wax. Certain fungi developed some mechanism to evoke the immune system of host plant (Fujikawa et al. 2012). It is already reported that bacterial glucanases are able to increase the resistance against blast causing fungus Magnaporthe oryzae (Fujikawa et al. 2012). But the expression of transgene from bacteria may or may not able to stop the entry of pathogenic fungi in the host system. Trichoderma spp. is widely available in the world and able to modify the surrounding environments for their growth, metabolism and sporulation etc. To control the Rhizoctonia solani infection in the field, some extracellular metabolites of Trichoderma spp. were used continuously as a biological fungicide. Their metabolites are volatile and water soluble in nature (Eziashi et al. 2007). The genome of Trichoderma harzianum contains many genes which are responsible for secondary metabolites and enzymes against Rhizoctonia solani. Some of the genes belong to glucanase group. Glucanases having potential to degrade the glucan compound which is abundant in fungal cell wall. These glucans are used as a protective covering of cell wall compounds which act as PAMPs. To overcome such problem an endo α-1, 3-glucanse gene was identified in Trichoderma harzianum which may be able to increase the resistance against the fungal attack. Here, the identified and characterized gene was transformed in embryogenic rice calli through Agrobacterium. For constitutive expression of gene a set of promoter and terminator was cloned to make a complete DNA cassette. After transformation and regeneration of rice plants were checked through different bio-assays protocols (Eziashi et al. 2007). The resistance in transformed plants was increased up-to a remarkable level which support developed method of transformation. This method is validated on the basis of in vitro and in vivo bioassay experiments done. In accordance with these experiments expression of DNA cassette was confirmed by tagging gfp gene in the cassette. The gfp expression was observed in rice calli which to be transformed as well as in tobacco leaves also. The transformation rate was found as 81% in T0 generation which is quite good enough. In T1 generation the transformation rate was 93% which is a very good in plant transformation (Fig. 5a, b). These findings are the main cause to prove the inheritance of trans-gene in successive generations in transformation. The development of resistant variety against sheath blight disease is quite possible in this way. This method may also play a key role in development of transgenic crops on the broad spectrum.

Transformation rate for each generation. a For T0 generation, transformation rate was found as 81%. Total no. of transformed plants was 912. Out of that 209 were false positive. b For T1 generation, transformation rate was found as 93%. Total no. of transformed plants was 873. Out of that 62 were false positive
Abbreviations
- PAMPs:
-
Pathogen associated molecular patterns
- PRRs:
-
Pattern recognition receptors
- hpt :
-
Hygromycin phosphotramferase
- YEM:
-
Yeast extract manitol
- AAM:
-
A protocol for Agrobacterium mediated transformation method medium
References
Akagi A, Jiang CJ, Takatsuji H (2015) Magnaporthe oryzae inoculation of rice seedlings by spraying with a spore suspension. Bio-protocol 5:e1486. https://doi.org/10.21769/BioProtoc.1486
Chen L, Ai P, Zhang J, Deng Q, Wang S, Li S, Zhu J, Li P, Zheng A (2016a) RSIADB, a collective resource for genome and transcriptome analyses in Rhizoctonia solani AG1 IA. Database. https://doi.org/10.1093/database/baw031
Chen L, Wang Q, Chen H, Sun G, Liu H, Wang H (2016b) Agrobacterium tumefaciens-mediated transformation of Botryosphaeria dothidea. World J Microbiol Biotechnol 32:1–5. https://doi.org/10.1007/s11274-016-2045-0
Cota-Sánchez JH, Remarchuk K, Ubayasena K (2006) Ready-to-use DNA extracted with a CTAB method adapted for herbarium specimens and mucilaginous plant tissue. Plant Mol Biol Rep 24:161
de Vetten N, Wolters AM, Raemakers K, van der Meer I, ter Stege R, Heeres E, Visser R (2003) A transformation method for obtaining marker-free plants of a cross-pollinating and vegetatively propagated crop. Nat Biotechnol 21:439–442. https://doi.org/10.1038/nbt801
Eziashi EI, Omamor IB, Dimaro-Oruade EA, Ogunkanmi LA (2007) Control of phytotoxin from Ceratocystis paradoxa using Trichoderma species phytotoxins on oil palm (Elaeis quineensis Jacq.) sprouted seeds. Plant Pathol J 6:324–329. http://www.ansinet.org/ppj
Fujikawa T, Sakaguchi A, Nishizawa Y, Kouzai Y, Minami E, Yano S, Koga H, Meshi T, Nishimura M (2012) Surface α-1, 3-glucan facilitates fungal stealth infection by interfering with innate immunity in plants. PLoS Pathog 8:e1002882. https://doi.org/10.1371/journal.ppat.1002882
Mayo S, Gutierrez S, Malmierca MG, Lorenzana A, Campelo MP, Hermosa R, Casquero PA (2015) Influence of Rhizoctonia solani and Trichoderma spp. in growth of bean (Phaseolus vulgaris L.) and in the induction of plant defense-related genes. Front. Plant Sci 6:685. https://doi.org/10.3389/fpls.2015.00685
Metzker ML, Caskey CT (2009) Polymerase chain reaction (PCR). eLS. https://doi.org/10.1002/9780470015902.a0000998.pub2
Moradpour M, Abdullah SNA (2017) Cisgenesis and Intragenesis as new strategies for crop improvement. In Crop Improvement, pp 191–216. Springer, Cham. https://doi.org/10.3389/fpls.2016.01608
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Pettitt TR, Wainwright MF, Wakeham AJ, White JG (2011) A simple detached leaf assay provides rapid and inexpensive determination of pathogenicity of Pythium isolates to ‘all year round’ (AYR) chrysanthemum roots. Plant Pathol 60:946–956
Scaramelli L, Balestrazzi A, Bonadei M, Piano E, Carbonera D, Confalonieri M (2009) Production of transgenic barrel medic (Medicago truncatula Gaernt.) using the ipt-type MAT vector system and impairment of Recombinase-mediated excision events. Plant Cell Rep 28:197–211. https://doi.org/10.1007/s00299-008-0634-6
Sengupta S, Chakraborti D, Mondal HA, Das S (2010) Selectable antibiotic resistance marker gene-free transgenic rice harbouring the garlic leaf lectin gene exhibits resistance to sap-sucking planthoppers. Plant Cell Rep 29:261–271. https://doi.org/10.1007/s00442-009-1541-4
Svitashev S, Young J, Schwartz C, Gao H, Falco SC, Cigan AM (2015) Targeted mutagenesis, precise gene editing and site-specific gene insertion in maize using Cas9 and guide RNA. Plant Physiol. https://doi.org/10.1104/pp.15.00793
Tang W, Chen H, Xu C, Li X, Lin Y, Zhang Q (2006) Development of insect-resistant transgenic indica rice with a synthetic cry1C gene. Mol Breeding 18:1–10. https://doi.org/10.1007/s11032-006-9002-9
Toriyama K, Hinata K (1985) Cell suspension and protoplast culture in rice. Plant Sci 41:179–183. https://doi.org/10.1016/0168-9452(85)90086-X
Xue M, Yang J, Li Z, Hu S, Yao N, Dean RA, Liu L (2012) Comparative analysis of the genomes of two field isolates of the rice blast fungus Magnaporthe oryzae. PLoS Genet 8:e1002869. https://doi.org/10.1371/journal.pgen.1002869
Acknowledgements
The study was supported by Visargha Agrisciences Pvt. Ltd., TBI, Campus-11, Kalinga Institute of Industrial Technology (KIIT), Deemed to be University, Bhubaneswar-751024, Odisha, India in association with School of Biotechnology, Campus-11, Kalinga Institute of Industrial Technology (KIIT), Deemed to be University, Bhubaneswar-751024, Odisha, India.
Author information
Authors and Affiliations
Contributions
RK designed and did the most of the experiments and wrote the MS. KK, KCH and LK did some part of experiments. BKB edited the MS.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kumar, R., Kumari, K., Hembram, K.C. et al. Expression of an endo α-1, 3-Glucanase gene from Trichoderma harzianum in rice induces resistance against sheath blight. J. Plant Biochem. Biotechnol. 28, 84–90 (2019). https://doi.org/10.1007/s13562-018-0465-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-018-0465-7