
Abstract
Canola (Brassica napus L.), an agro-economically important crop in the world, is sensitive to many fungal pathogens. One strategy to combat fungal diseases is genetic engineering through transferring genes encoding the pathogenesis-related (PR) proteins such as chitinase which cause the chitin degradation of fungal cell wall. Chitinase Chit42 from Trichoderma atroviride (PTCC5220) plays an important role in biocontrol and has high antifungal activity against a wide range of phytopathogenic fungi. This enzyme lacks a chitin binding domain (ChBD) which is involved in binding activity to insoluble chitin. In the present study, we investigated the effect of chitin binding domain fused to Chit42 when compared with native Chit42. These genes were over-expressed under the CaMV35S promoter in B. napus, R line Hyola 308. Transformation of cotyledonary petioles was achieved by pBISM2 and pBIKE1 constructs containing chimeric and native Chit42 genes respectively, via Agrobacterium method. The insertion of transgenes in T0 generation was verified through polymerase chain reaction (PCR) and Southern blot analysis. Antifungal activity of expressed chitinase in transgenic plants was also investigated by bioassays. The transgenic canola expressing chimeric chitinase showed stronger inhibition against phytopathogenic fungi that indicates the role of chitin binding domain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Canola provides oils for industrial lubricants, human consumption, animal feeds and also can be used as green fertilizer and composting crops (Dixon 2006; Seberry et al. 2009). The productivity of canola is limited by several abiotic and biotic stress factors (Grover and Pental 2003; Dutta et al. 2005; Ullah et al. 2012). Fungal diseases have served as a major cause of yield losses (Oerke et al. 1994). Agricultural operations, utilization of chemical compounds and using resistant varieties are three common procedures for controlling fungal diseases (Barone and Frusciante 2007). Since the agrochemicals and conventional breeding approaches often have certain limitations, it is advisable to develop fungus-resistant plants through genetic engineering (Chang et al. 2002).
Chitinases are member of pathogenesis-related proteins which are often acid soluble, extracellular and protease resistant. Chitinases from Trichoderma sp. have been shown to have strong antifungal activities. They catalyze hydrolysis of Chitin that is a major component of many fungal cell walls, insect exoskeletons and crustacean shells and the second most abundant polysaccharide in nature (Collinge et al. 1993; Graham and Sticklen 1994; Neuhaus 1999).
Chitinase Chit42 is an endo-chitinase of T. atroviride PTCC5220 which plays a key role in biocontrol activities against phytopathogenic fungi (Limon et al. 2004; Harighi et al. 2006). Chit42 lacks a chitin-binding domain (ChBD) (Kojima et al. 2005). Chitin-binding domain linked to the catalytic site of some chitinases via a linker region (Arakane et al. 2003; Limon et al. 2004). This domain is a tunnel-like structure which permits tight interaction with the polymeric substrate and facilitates chitinase binding, thus allowing efficient degradation of chitin (Van Aalten et al. 2001; Hardt and Laine 2004). Serratia marcescens is one of the most extensively studied chitinolytic bacteria producing multiple chitinases (Felse and Panda 1999; Zakariassen et al. 2009).
In current study to investigate the role of chitin binding domain from S. marcescens chitinase B on enzyme activity of T. atroviride Chit42, two constructs including Chit42 and chimeric chitinase with fused ChBD at C-terminal site were transformed to canola plants via Agrobacterium method. The evaluation of the antifungal activity of the putative transgenic plants was carried out to study the effect of ChBD in the chimeric chitinase.
Materials and methods
Plant, fungal and bacterial materials
The seeds of canola (B. napus L.) R line Hyola 308, as a genes receptor, was provided by the Oilseed and Development Company, Tehran, Iran. Sclerotinia sclerotiorum, Rhizoctonia solani, Fusarium oxysporum, Verticillium dahlia and Alternaria solani were provided by Dr. H. Afshari-Azad from Iranian Research Institute of Plant Protection. The fungi were proliferated on PDA (Potato dextrose agar) medium and sub-cultured as needed.
Agrobacterium tumefaciens strain LBA4404 was used for plant transformation and Escherichia coli strain DH5α (Cinnagen, Iran) was performed in all molecular biological experiments. The bacteria were grown in LB (Luria-Bertani) medium at appropriate temperatures (37 °C for E. coli and 28 °C for A. tumefaciens) along with shaking in 180 rpm.
Preparation of explants and the bacterial strain for transformation
Seeds were sterilized using 70 % ethanol for 2 min and 0.1 % HgCl2 for 10 min. Then they were washed several times with sterilized water, plated on ½MS medium (Murashige and Skoog 1962) and incubated in the presence of light for 5 days. After germination, the cotyledonary petioles were cut and pre-cultured on CM solid medium (MS with 3.5 mg/L of Benzylaminopurine). After 2 days, the explants were subjected for transformation process. Single colonies of A. tumefaciens contaning pBIKE1 (chit42 gene) and pBISM2 (chimeric chitinase gene) were used to incubate in LB medium including 50 mg/L of kanamycin and were grown overnight at 28 °C with constant shaking (180 rpm) to mid-log phase. The bacterial culture was transferred to fresh medium and cultivated until the optical density (OD 600) of 0.4 was achieved. Then the bacterial cells were collected by centrifugation and re-suspended in fresh ½MS medium for the subsequent inoculation step.
Transformation and selection procedure
Explants were submerged in a bacterial suspension for 5 min with constant shaking and then removed excessive moisture using sterile filter paper and put them on CM medium in Petri dishes for co-cultivation at 25 °C for 3 days.
After co-cultivation, due to inhibit the growth of A. tumefaciens attached to the explants, they were rinsed with sterile water including 200 mg/L cephatoxim and then transferred to MS medium containing 3.5 mg/L of BAP (Benzylaminopurine), 8 mg/L of kanamycin and 200 mg/L of cephatoxim. The explants were transferred to MS medium with 15 mg/L of kanamycin and 200 mg/L of cephatoxim, after shoot initiation. The regenerated shoots (about 3 cm in length) were separated from the explants and transferred to MS medium with 2 mg/L of 3-Indolebutyric acid (IBA) and 200 mg/L of cephatoxime for rooting and formation the complete plants. All the above media contained 3 % (w/v) of sucrose and 8 g/L of agar, pH 5.8. The explants were cultured at 23 ± 2 °C and under 16/8 h photoperiod with light intensity of 2000 Lux.
Molecular analysis of the transgenic canola
The leaves from the untransgenic and transgenic canola were collected, lyophilized and minced into a fine powder for extraction of genomic DNA (Doyle and Doyle 1991). PCR analysis was used for the first evidence of the transgene presence in the assumed transgenic plants. DNA fragment harboring of chimeric and native Chit42 genes was amplified by PCR using the genomic DNA and (C42F4–5´-CGTTCCCGCAAGCAAGATCG- 3´/NOSR- 5´-CCAGTGAATTCCCGATCTAGTAAC-3´) primers. PCR was carried out as explained: an initial denaturation at 94 °C for 15 min followed by 40 cycles of denaturation at 94 °C for 1 min, annealing at 58 °C for 1 min, extension at 72 °C for 1 min and a final extension at 72 °C for 10 min. The resulting PCR products were detected by electrophoresis on 1 % (w/v) agarose gel.
Enzyme assay
Young leaves from putative transformants and untransformed canola plants were frozen in liquid nitrogen and were ground to make fine powder. The soluble proteins were also extracted in 50 mM sodium acetate buffer (pH 7.0). In this assay chitinase activity was generally measured in the reaction mixture (total 500 ml) containing colloidal chitin as a substrate (3.8 mg) and the crud of enzymes from transgenic plants containing chitinases (200 μg/ml). The reaction was performed at 37 °C for 1 h and then centrifuged at 6000×g for 5 min. The supernatant was boiled along with potassium tetra borate buffer (100 μl) for 3 min. In following 3 ml of DMAB reagent [10 g of Di-methyl amino benzaldehyde in 100 ml of 10 N chloridric acid (12.5 % v/v) and glacial acetic acid (87.5 % v/v)] was added to the reaction, incubated at 37 °C for 20 min and the amount of GLcNAc produced in the reaction was calculated using the defined method by Zeilinger et al. (1999) according to the standard diagram of GLcNAc. One unit of enzyme activity was described as the amount of enzyme that catalyses the release of 1 μmol GLcNAc in 60 min at 37 °C. The assay for each sample was performed in three biological replicates. Total soluble protein concentration was determined according to Bradford (1976), using BSA (Bovine serum albumin) diagram as standard.
Southern blot analysis
Genomic DNA was extracted from fresh leaves of nontransgenic plants as the control and putative transgenic ones using the CTAB method (Doyle and Doyle 1991). PCR positive plants with high enzyme activity and untransformed control plants were analyzed by Southern blot assay to confirm the integration of the transgenes.
The extracted genomic DNA (20 μg) was digested using HindIII enzyme. The digested DNAs were seperated on 0.8 % (w/v) agarose gel, transferred onto a nylon membrane (Amersham Hybond N+; Amersham International Plc, Amersham, UK) and then hybridised to the Dig-dUTP labelled CaMV 35S probe. A partial internal fragment (631 bp in size) was achieved from PCR amplification of the CaMV35S fragment using CaMV35SF (5´- GGACTAACTGCATCAAGAACACAG- 3´)/CaMV35SR (5´- GAAGGATAGTGGGATTGTGCGTC- 3´) primers and plasmid pBI121 containing the native and chimeric chitinase as template, subjected to DIG DNA labeling (Roche Applied Science GmbH, Mannheim, Germany) and used as a probe in hybridization experiments.
Bioassay of the transgenic canola plants
The antifungal activity of the extracted proteins from transgenic plants was tested using biological assays as mentioned in below. Canola leaf materials (0.5 g) were grinded to a powder in liquid nitrogen by a pestle and mortar. The soluble proteins were then extracted in 50 mM Tris–HCl (pH 8.8). The extracts were shaken for 1 h at 4 °C and subsequently centrifuged at 13,000 g for 35 min at 4 °C (Wang et al. 2006). The resulting supernatants were stored at 4 °C. The protein content was determined against BSA according the Bradford assay (Bradford 1976).
Radial diffusion assay (Broglie et al. 1991)
For detection of chitinase antifungal activity of Chit42 and chimeric chitinase, fungal growth inhibitory assay was used. The zone of inhibition assay for antifungal activity was determined using 100 × 15 mm plates including 25 ml of PDA. After development of the mycelia, 5 mm holes were made around and at a distance of 1 cm away from the rim of the mycelia colony. Equal aliquots (40–45 μg) of crude proteins from transgenic plants contaning Chit42 and the chimeric chitinase were added to the holes. The plates were incubated at 28 °C for 24 h until mycelial growth had enveloped peripheral hole containing the negative control the extraction buffer of protein and the crude of proteins from untransformed plant. The fungal species included S. sclerotiorum, R. solani, F. oxysporum and V. dahlia. The assay for each sample was performed three replicates. Inhibition area was measured using Image Tools Software.
Disc diffusion assay (Nweze and Mukherjee 2010)
At first some fungal species such as F. oxysporum, V. dahlia, A. solani were grown on PDA plate at 28 °C for 5 days. The spores were washed with sterile water and then were collected and counted by Thoma Lam. Based on a modified method of Nweze, sterile paper disks (6 mm in diameter) were placed on the PDA medium. Then 1 μl from 2 × 107 cell/ml dilution of spore suspension of F. oxysporum, V. dahlia and A. solani was added to each disk. Following the same concentration of total proteins from leaf of transgenic canola containing chimeric and native Chit42 were added to the disks. The protein from untransgenic plants and protein extracting buffer were served as control. Plates were incubated at 28 °C until spore germination and mycelia growth was seen in negative control disks. Finally, the inhibitory effect of Chit42 and chimeric chitinase enzymes on the growth of these fungi were compared relative to each other and to the controls. The assay for each sample was carried out in three replicates. Diameter of growth zones were measured using Image Tools Software.
Spore germination assay (Broekaert et al. 1997)
Spore suspension of A. alternate, F. oxysporum and V.dahlia (2 × 104 cell/ml) in half-strength PDB (Potato dextrose broth) containing the crude protein of transgenic canola with native or chimeric chitinase were incubated at 28 °C with shaking (150 rpm) for 48 h. Crude protein from untransgenic canola plant was used as a control. After 48 h incubation, the growth of the fungi was determined by measuring OD-values at 595 nm. The percentage of growth inhibition was calculated as 100 × the ratio of the A595 of the control minus the A595 of the sample over the A595 of the control. The experiments were conducted three times.
Light microscopy assay (Kronland and Stanghellini 1988 and Benhamou et al. 1993)
This assay used for studying the effect of Chit42 enzyme of transgenic canola plants on morphological changes and the cell wall of mycelium in R. solani. The crude protein (70 μg) of transgenic canola was added to 2 days grown R. solani on a slide covered with a thin layer of PDA. Crude protein from untransgenic canola plant was used as a control. After 3, 6 and 24 h of incubation at 37 °C, the slide was examined for morphological changes and degradation of fungi cell wall under a light microscope.
Statistical analysis
The statistical differences were assessed based on the ANOVA (analysis of variance) using SPSS Software (Ver. 15, USA). Differences were considered significant at a probability level of P < 0.05. Mean values were compared using least significant different and Duncan tests.
Results and discussion
The main objective of this work was to obtain transgenic canola (B. napus) plants harboring a novel chitinase with chitin binding domain (chimeric chitinase) and native chitinase (chit42) genes and to evaluate the effect of this domain in antifungal activity. The ChBD from soil bacteria S. marcescens was selected to join to chitinase (Chit42) of T. atroviride.
Transformation and selection of transgenic plant
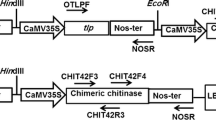
The constructs pBIKE1 containing Chit42 gene and pBISM2 containing chimeric chitinase gene were used for over expression of these genes in canola. They are under the control of CaMV35S promoter and the nopaline synthase as terminator in the plant expression vector pBI121. Selectable marker gene in this vector is nptII (kanamycin resistance gene) (Supplementary Fig. 1).
These constructs were mobilized into A. tumefaciens and subsequently used for B. napus, R line Hyola 308 transformation. The explants were co-cultivated with A. tumefaciens and then transferred to selection medium containing kanamycin and cefotaxime. The independent transgenic canola lines were successfully rooted (Supplementary Fig. 2).
The success of Agrobacterium-mediated plant transformation is a biological progress which depends on various factors such as; species genotype, strain of Agrobacterium, selectable marker, regeneration capacity of target cells and accessibility of the bacterium to the regenerable cells. Also, CaMV35S promoter used to ensure high levels of gene expression in all plant tissues. Agrobacterium mediated method for transformation and use of CaMV35S promoter have been reported by several researchers as impressive parameters (Cardoza and Stewart 2003; Kahrizi et al. 2007; Liu et al. 2010).
The experiment was performed with a total of 400 and 451 explants of canola using the pBIKE1 and pBISM2 constructs respectively. The numbers of regenerated shoots obtained at 8 mg/L kanamycin were 280 and 225 for these transgenes. Shoots were transferred to a medium containing 15 mg/L kamamycin and only 129 and 133 green shoots were obtained. Then, 65 and 59-rooted plantlets were transferred to pots (Table 1). PCR analysis confirmed the presence of the Chit42 in 20 and chimeric chitinase in 21 kanamycin-resistant putative transgenic plants (Table 1). Successful introduction of transformation efficiency of Chit42 and chimeric chitinase were calculated as 5 % and 4.65 % respectively (Table 1). The observed satisfactory transformation rates were similar to various reports in B. napus (Wang et al. 2005; Liu et al. 2011) and other Brassica species, B. rapa and B. juncea (Cho et al. 2001; Das et al. 2006). The transformants were phenotypically analyzed and compared with the controls and any abnormalities in the growth, size or reproduction did not show in transgenic lines.
Molecular analysis of regenerated plants
The ka namycin- resistant clones were subjected to PCR analysis for integration confirm of the transgenes chimeric chitinase and Chit42. The corresponding fragment, 700 bp of the Chit42 gene, was amplified using specific primers (C42F4/NOSR) (Supplementary Fig. 3a). The lines containing chimeric chitinase were also analyzed by PCR using the same primers and the expected fragment (925 bp) was amplified from these transgenic lines (Supplementary Fig. 3b). The Chit42 and chimeric chitinase specific primers did not amplify the corresponding fragments in untransformed plants.
A set of virG primers (virGf/virGr) was used to detect Agrobacterium contamination that might have escaped the selection. PCR detection under different conditions showed no bands using transgenic line DNA as template. A 738 bp band was found using Agrobacterium DNA as control (data not shown).
Enzyme activity assay
Chitinase activity from leaf tissues of the wild type and PCR positive transgenic plants, containing native and chimeric chitinase, was assayed in the presence of colloidal chitin. The specific enzyme activity of different transgenic plants varied from 0.72 ± 0.24 to 4.02 ± 0.39 U/μg for the lines containing (Chit42) and 0.79 ± 0.23 to 6.48 ± 0.52 U/μg for the lines harboring (chimeric chitinase). There was a significant difference between enzyme activities in transgenic lines (harboring chimeric and native Chit42) with control plant (wild type). Among all transgenic lines, T18, T7, T21, T13 and T2 harboring Chit42 and T21, T9, T3, T2 and T1 with chimeric chitinase showed the highest specific enzyme activity. Furthermore, chimeric chitinase transformants (6.48 ± 0.52 to 2.46 ± 0.60 U/μg) in general demonstrated higher enzyme activity compared to transgenic lines harboring Chit42 (4.02 ± 0.39 to 3.66 ± 0.15 U/μg) (Table 2). In previous study, the results revealed that the fusion of ChBD to chitinase improved the enzyme affinity to crystalline and colloidal chitin (Matroodi et al. 2013). The effect of ChBD on binding chitinase to chitin substrate was also described by Hashimoto et al. (2000) where they showed that the ChBD deletion from chitinase A1 greatly decreased the efficiency of chitin degradation. The enzyme activities of all T0 transformants having Chimeric and native Chit42 showed various levels due to their different copy number and localization of integrated heterologous transgenes (Yamamoto et al. 2000; Iyer et al. 2000; Matzke et al. 2000). The transgenic lines harboring chimeric chitinase (T21, T9, T3, T2 and T1) and native Chit42 (T18, T7, T21, T13 and T2) with high enzyme activity were selected for Southern blot analysis in order to define the number of integrated T-DNAs.
Southern blot analysis
Southern blot analysis was performed in PCR positive plants demonstrating high chitinase activity and the results showed that the transgenes were integrated into the transformed plants. Amplified CaMV35S promoter was labeled as probe. Genomic DNA from transformed lines contaning chimeric chitinase (T21, T9, T3, T2 and T1) and Chit42 (T21, T18, T13, T7 and T2) were digested with HindIII (Supplementary Fig. 1). Each hybridization band corresponded to one separate transgens copy, because the integrated genes were cut ones by HindIII enzyme while the other HindIII site was localized in the plant genome. Whereas each integration event occurs independently and by chance, transgenic plants exhibited unique hybridization profiles. The transgenes copy number was appraised as one in T9, T3 and T1 lines with chimeric chitinase and T18 and T2 lines harboring Chit42 genes. Two bands were detected in T2 (chimeric chitinase), T21 and T7 harboring Chit42. In the rest of the lines, T21 and T13 containing chimeric and native Chit42 respectively, three bands were indicated. The transgenic lines carried one to three copies of these genes which are according to the results of Moloney et al. (1989), who reported single or multiple copy insertion into the canola genome. The size of hybridization bands from all tested transgenic plants were more than 2.5 kb for chimeric chitinase and 2.4 kb for Chit42. The HindIII digested pBIKE1 plasmid was used as positive control. No transgene insertion was detected in untransformed plant (Supplementary Fig. 4).
Antifungal activity
In order to study antifungal activity of transgenes, the crude proteins were extracted from single copy transformant leaves containing chimeric (T9, T3 and T1) and native Chit42 (T18 and T2) genes. The inhibitory effect was evaluated in vitro against five major phytopathogenic fungi. According to disk diffusion assay results, hyphal growth of F. oxysporum, V. dahlia and A. solani substantially decreased using total proteins of all transgenic lines. Moreover, germinated spores in the presence of chimeric chitinase were lower than lines with Chit42 in all cases. No inhibition was observed in the case of control plant (Fig. 1).

Inhibitory activity of the chimeric chiinase and Chit42 toward a A. solani, b V. dahlia, c F. oxysporum and d Growth zone diameter of pathogenic fungal growth. T18 and T2 (Chit42); T9, T3 and T1 (chimeric chitinase); Buff: Extraction buffer of protein as negative control; WT: the crude of proteins from wild type or untransformed canola plant as negative control. Results represent the average and standard deviation of three experiments
The Inhibition rate was also calculated using spore germination assay. Significant differences were found between transgenic and negative controls (wild type plant and protein extraction buffer). These results showed that the restriction of hyphal growth in A. solani, F. oxysporum and V. dahlia fungi using crude enzymes containing chimeric chitinase with inhibition rate of 55 %, 39 % and 36 % was stronger, when compared to those with Chit42 with inhibition rate of 33 %, 16 % and 18 % respectively (Fig. 2).

The growth inhibition (%) of Chit42 and chimeric chitinase on spore germination. Growth of A. solani, F. oxysporum and V. dahlia in crude protein extracts from transgenic plant containing chimeric chitinase (T9, T3 and T1) and Chit42 (T18 and T2) and non-transformed (WT) canola leaves. Absorbance of the reaction mixture (crude protein extract + spore suspension + potato dextrose broth) after 48 h incubation was measured at 595 nm. Bars represent means and standard errors and those with different lower case letters are significantly different at (p > 0.05) by least significant difference (LSD) test. Results represent the average and standard deviation of three experiments
As shown in Fig. 3, the extracted total proteins (45 μg) from transgenic lines exhibited antifungal activity against R. solani, S. sclerotiorum, F. oxysporum and V. dahlia based on radial diffusion assay. According to this result chitinase enzyme exhibited stronger inhibition against S. sclerotiorum and R. solani (Fig. 3a, b and e) than F. oxysporum and V. dahlia fungi (Fig. 3c, d and e). This may be due to the intrinsic variability of chitin in fungal cell wall, which exists in several forms (Van de Velde and Kiekens 2004; Aranaz et al. 2009). In addition, the antifungal activity of chimeric chitinase was significantly higher than Chit42 (Fig. 3e). This finding seems to result from the subsite structure in the binding cleft of this enzyme (Sasaki et al. 2002). The same results have also been reported by Matroodi et al. (2013). Protein extraction buffer and crude proteins from wild type plants, as negative control, showed no or negligible inhibition against four fungal pathogens tested.

Inhibitory activity of the chimeric chitinase and Chit42 toward a S. sclerotiorum, b R. solani, c F. oxysporum and d V. dahlia; e Inhibition zone diameter of pathogenic fungal growth. T18 (Chit42); T9 (chimeric chitinase); Extraction buffer of protein as negative control (Buff); the crude of proteins from wild type or untransformed canola plant as negative control (WT). In diagram the bars represent means and standard errors for chimeric Chit42 (T9, T3 and T1) and Chit42 (T18 and T2) and those with different lower case letters are significantly different at (p > 0.05) by least significant difference (LSD) test. Results represent the average and standard deviation of three experiments
Microscopic observations revealed that the morphology and the cell wall of R. solani were changed and degraded after exposure to crude protein extracts from transgenic plants. Hyphal samples growing in PDA medium deposited on microscopic slides were studied at 3, 6 and 24 h using light microscopy (Fig. 4). Observation of control sample exposed to crude proteins of untransformed plant showed the presence of a dense and regular mycelium. They were described by their hyphal natural diameter and regular shape (WT in Fig. 4). Morphological changes, mainly localized at the hyphal tips, were recognizable within 3 h after exposure of hyphal samples to the crude proteins of transgenic plants. The apical zone of most hyphae displayed distinct swelling (Fig. 4a, arrows). The transgenic plants containing either of enzymes are able to degrade the cell wall of R. solani mycelium (Fig. 4). After 6 h exposure, the R. solani hyphae were shown abnormal growth with noticeable swelling, branching and distortion (Fig. 4b). Then after 24 h enzyme exposure, hyphae was noticeably reduced in size and the degradation of cell wall was occurred (Fig. 4c, arrows). Similar to our findings, a number of other studies have implicated the chitinase enzyme to be responsible for degradation of the cell wall of phytopathogenic fungi (Harighi et al. 2006).

Morphological changes induced in Rhizoctonia solani hyphae after exposure to the chitinase enzyme. a Mycelium of R. solani, 3 h after addition of proteins containing chimeric chitinase and native Chit42. The apical zone of young, emerging branches shows pronounced swelling (arrows). b Mycelium of R. solani, 6 h after addition of chitinase enzyme. Emerging branches are abnormally shaped and markedly swollen. c Mycelium of R. solani, 24 h after addition of chitinase. Emerging degradation of R. solani cell wall shows pronounced swelling (arrows). All showed events including single copy of transferred gene with high chitinas activity. WT; Control hyphae exposed to crude proteins of untransformed plant showing a regularly mycelium. Scale bar =20 μm
In conclusion, the addition of a ChBD increases enzyme activities and antifungal properties of chitinase, so the transgenic canola expressing chimeric chitinase showed stronger inhibition against phytopathogenic fungi. Transformation of genes with enhanced antifungal activity would allow us to develop fungal resistant crop plants and is likely to reduce dependence on chemical fungicides. Meanwhile, this work demonstrates that, the expression of these genes had no deleterious phenotypic effects on the transgenic plants.
Abbreviations
- DMAB:
-
Di-methyl amino benzaldehyde
- CTAB:
-
Cetyl Trimethyl Ammonium Bromide
- GLcNAc:
-
N-acetylglucosamine
References
Arakane Y, Zhu Q, Matsumiya M, Muthukrishnan S, Kramer KJ (2003) Properties of catalytic, linker and chitin-binding domains of insect chitinase. Insect Biochem Mol Biol 33:631–648
Aranaz I, Mengibar M, Harris R, Panos I, Miralles B, Acosta N, Galed G, Heras A (2009) Functional characterization of chitin and chitosan. Curr Chem Biol 3:203–230
Barone A, Frusciante L (2007) Molecular marker-assisted selection for resistance to pathogens in tomato, Marker-Assisted Selection, Current status and future perspectives in crops, livestock, forestryand fish. pp 153–164
Benhamou N, Brogl K, Brogl R (1993) Antifungal effect of bean endochitinase on Rhizoctonia solani: ultrastructural changes and cytochemical aspects of chitin breakdown. Can J Microbiol 39:318–328
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254
Broekaert WF, Cammue BPA, De Bolle MFC, K T, GW d S, RW O (1997) Antimicrobial peptides from plants. Crit Rev Plant Sci 16:297–323
Broglie K, Chet I, Holliday M, Cressman R, Biddle P, Knowlton S, Mauvais CJ, Broglie R (1991) Transgenic plants with enhanced resistance to the fungal pathogen Rhizoctonia solani. Science 254:1194–1197
Cardoza V, Stewart CN (2003) Increased Agrobacterium-mediated transformation and rooting efficiencies in canola (Brassica napus L.) from hypocotyls segment explants. Plant Cell Rep 21:599–604
Chang M, Culley D, Choi J, Hadwiger L (2002) Agrobacterium mediated co transformation of a pea β-1,3 glucanase and chitinase genes in potato (Solanum tuberosum L. c. v. Russet Burbank) using a single selectable marker. Plant Sci 163:83–89
Cho HS, Cao J, Ren JP, Earle ED (2001) Control of Lepidopteran insect pests in transgenic Chinese cabbage (Brassica rapa ssp ekinensis) transformed with a synthetic Bacillus thuringiensis cry1C gene. Plant Cell Rep 20:1–7
Collinge DB, Kragh KM, Mikkelsen JD, Nielsen KK, Rasmussen U (1993) Plant Chitinases. Plant J 3:31–40
Das B, Goswami L, Ray S, Ghosh S, Bhattacharyya S, Das S, Majumder AL (2006) Agrobacterium-mediated transformation of Brassica juncea with a cyanobacterial (Synechocystis PCC6803) delta-6 desaturase gene leads to production of gamma-linolenic acid. Plant Cell Tissue Organ Cult 86:219–231
Dixon GR (2006) Vegetable Brassicas and related crucifers- volume 14 of crop production science in horticulture. CAB International, Glascow
Doyle JJ, Doyle JL (1991) Isolation of plant DNA from fresh tissue plant. Plant Mol Biol 9:340–342
Dutta I, Majumder P, Saha P, Ray K, Das S (2005) Constitutive and phloem specific expression of Allium sativum leaf agglutinin (ASAL) to engineer aphid (Lipaphis erysimi) resistance in transgenic Indian mustard (Brassica juncea). Plant Sci 169:996–1007
Felse PA, Panda T (1999) Regulation and cloning of microbial chitinase genes. Appl Microbiol Biotechnol 51:141–151
Graham LS, Sticklen MB (1994) Plant Chitinases. Can J Bot 72:1057–1083
Grover A, Pental D (2003) Breeding objectives and requirements for producing transgenics for major field crops of India. Curr Sci 84:310–320
Hardt M, Laine RA (2004) Mutation of active site residues in the chitin-binding domain ChBDChiA1 from chitinase A1 of Bacillus circulans alters substrate specificity: use of a green fluorescent protein binding assay. Arch Biochem Biophys 426:286–297
Harighi MJ, Motallebi M, Zamani MR (2006) Antifungal activity of heterologous expressed chitinase 42 (Chit42) from Trichoderma atroviride PTCC5220. Iran J Biotech 4:95–103
Hashimoto M, Ikegami T, Seino S, Ohuchi N, Fukada H, Sugiyama J, Shirakawa M, Watanabe T (2000) Expression and characterization of the chitin-binding domain of chitinase A1 from Bacillus circulans WL-12. J Bacteriol 182:3045–3054
Iyer LM, Kumpatla SP, Chandrasekharan MB, Hall TC (2000) Transgene silencing in monocots. Plant Mol Biol 43:323–346
Kahrizi D, Salmanian AH, Afshari A, Moieni A, Mousavi A (2007) Simultaneous substitution of Gly96 to Ala and Ala183 to Thr in 5-enolpyruvylshikimate-3-phosphate synthase gene of E. coli (K12) and transformation of rapeseed (Brassica napus L.) in order to make tolerance to glyphosate. Plant Cell Rep 26:95–104
Kojima M, Yoshikawa T, Ueda M, Nonomura T, Matsuda Y, Toyoda H, Miyatake K, Arai M, Fukamizo T (2005) Family 19 chitinase from Aeromonas sp. No. 10S-24: role of chitin-binding domain in the enzymatic activity. J Biochem 137:235–242
Kronland WC, Stanghellini ME (1988) Clean slide technique for the observation of anastomosis and nuclear condition of Rhizoctonia solani. Phytopathology 78: 820–822
Limon MC, Chacon MR, Mejias R, Delgado-Jarana J, Rincon AM, Codon AC, Benitez T (2004) Increased antifungal and chitinase specific activities of Trichoderma harzianum CECT 2413 by addition of a cellulose binding domain. Appl Microbiol Biot 64:675–685
Liu T, Liu L, Jiang X, Hou J, Fu K, Zhou F, Chen J (2010) Agrobacterium-mediated transformation as a useful tool for the molecular genetics study of the phytopathogen Curvularia lunata. Eur J Plant Pathol 126:363–371
Liu H, Guo X, Naeem MS, Liu D, Xu L, Zhang W, Tang G, Zhou W (2011) Transgenic Brassica napus L. lines carrying a two gene construct demonstrate enhanced resistance against Plutella xylostella and Sclerotinia sclerotiorum. Plant Cell Tiss Organ Cult 106:143–151
Matroodi S, Motallebi M, MR Z, Moradyar M (2013) Designing a new chitinase with more chitin binding and antifungal activity. World J Microbiol Biotechnol 29:1517–1523
Matzke MA, Mette MF, Matzke AJM (2000) Transgene silencing by the host genome defense: implications for the evolution of epigenetic control mechanism in plant and vertebrates. Plant Mol Biol 43:401–415
Moloney MM, Walker JM, Sharma KK (1989) High efficiency transformation of Brassica napus using Agrobacterium vectors. Plant Cell Rep 8:238–242
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Neuhaus JM (1999) Plant chitinases (PR-3, PR-4, PR-8, PR11). In: Datta SK, Muthukrishnan S (eds) Pathogenesisrelated proteins in plants. CRC Press, Florida, pp. 77–105
Nweze E, Mukherjee P (2010) Agar-based disk diffusion assay for susceptibility testing of dermatophytes. J Clin Microbiol 10:3750–3752
Oerke E, Dehne H, Schonbeck F, Weber A (1994) Crop production and crop protection-estimated losses in major food and cash crops. Elsevier Science, Amsterdam
Sasaki C, Yokoyama A, Itoh Y, Hashimoto M, Watanabe T, Fukamizo T (2002) Comparative study of the reaction mechanism of family 18 chitinases from plants and microbes. J Biochem 131:557–564
Seberry DE, Mailer RJ, Parker PA (2009) Quality of Australian canola. AOF (Australian Oilseeds Federation) Publishing physicsWeb.http://www.australianoilseeds.com/data/assets/pdf_file/0004/6592/2009_Book.pdf. Accessed 2009
Ullah F, Banu A, Nosheen A (2012) Effects of plant growth regulators on growth and oil quality of canola (Brassica napus L.) under drought stress. Pak J Bot 44:1873–1880
Van Aalten DMF, Komander D, Synstad B, Gaseidnes S, Peter MG, Eijsink VGH (2001) Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Pnas 98:8979–8984
Van de Velde K, Kiekens P (2004) Structure analysis and degree of substitution of chitin, chitosan and dibutyrylchitin by FT-IR spectroscopy and solid state 13C NMR. Carbohydr Polym 58:409–416
Wang J, Chen Z, Du J, Sun Y, Liang A (2005) Novel insect resistance in Brassica napus developed by transformation of chitinase and scorpion toxin genes. Plant Cell Rep 24:549–555
Wang J, Zheng L, Wu J, Tan R (2006) Involvement of nitric oxide in oxidative burst, phenylalanine ammonia-lyase activation and Taxol production induced by low-energy ultrasound in Taxus yunnanensis cell suspension cultures. Nitric Oxide 15:351–358
Yamamoto T, Iketani H, Ieki H, Nishizawa Y, Notsuka K, Hibi T, Hayashi T, Matsuta N (2000) Transgenic grapevine plants expressing a rice chitinase with enhanced resistance to fungal pathogens. Plant Cell Rep 19:639–646
Zakariassen H, Aam BB, Horn SJ, Varum KM, Sorlie M, Eijsink VGH (2009) Aromatic residues in the catalytic center of chitinase A from Serratia marcescens affects processivity, enzyme activity, and biomass-converting efficiency. J Biol Chem 284:10610–10617
Zeilinger S, Galhaup C, Payer K, Woo SL, Mach RL, Fekete C, Lorito M, Kubicek CP (1999) Chitinase gene expression during mycoparasitic interaction of Trichoderma harzianum with its host. Fungal Genet Biol 26:131–140
Acknowledgments
This research was financially supported by the National Institute of Genetic Engineering and Biotechnology (NIGEB) of I. R. Iran.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they do not have any conflict of interest.
Electronic supplementary material
Supplementary Fig. 1
Schematic representation of the T-DNA region of the transformation vector a pBIKE1 (containing of chit42 gene) and b pBISM1 (containing of chimeric chitinase). Nos- pro- nopaline synthase promoter; NPTII- gene for neomycin phosphotransferase; Nos- ter- terminator of nopaline synthase; 35 s-Pro- 35S promoter of cauliflower mosaic virus (JPEG 611 kb)
Supplementary Fig. 2
Transformation and regeneration of transgenic canola plants: a Germinated seeds, b Cotyledons cultivated on co- culture medium, c Regenerated shoots in growth and selective medium, d Regenerated plantlets with well developed roots and leaves, e Covered regenerated plantlets in pots, f Transformed canola plants in pots acclimated to non-aseptic environment (JPEG 5125 kb)
Supplementary Fig. 3
PCR analysis of different putative transgenic lines: a Expected 700 bp fragment amplified from putative transgenic plants containing Chit42 using C42F4/NOSR primers. Ctrl+, pBIKE1 plasmid DNA template as positive control; WT, untransformed sample; M, DNA ladder Mix (Fermentas, CA). b Expected 925 bp fragment amplified by PCR from the DNA isolated from putative transgenic plants containing chimeric chitinase using C42F4/NOSR primers. Ctrl+, pBISM1 plasmid DNA template as positive control; WT, untransformed sample; M, DNA ladder Mix (Fermentas, CA) (JPEG 2194 kb)
Supplementary Fig. 4
Southern blot analysis of the transgenic canola plants transformed with the Chit42 and chimeric chitinase encoding genes. Genomic DNA from the canola plant was digested with HindIII and hybridized with the digoxigenin-labelled 631 bp partial internal fragment of the CaMV35S as a probe, to show integration of the DNA into the plant genome and the number of integrations. The numbers identify each independent transgenic plant tested. Lane 1, HindIII digested DNA from transgenic plant containing chimeric chitinase (T21); lane 2 (T9); lane 3 (T2); lane 4 (T1); lane 5 (T3); lane 6, HindIII digested DNA from transgenic plant containing Chit42 (T18); lane 7 (T13); lane 8 (T7); lane 9 (T2); lane 10 (T21); Untransformed canola genomic DNA digested with HindIII is shown in lane WT. ctrl+; pBIKE1 plasmid as positive control. M; molecular marker. The size of hybridization bands from all tested transgenic plants were more than 2.5 kb for chimeric chitinase and 2.4 kb for Chit42. (JPEG 2095 kb)
Rights and permissions
About this article

Cite this article
Ziaei, M., Motallebi, M., Zamani, M.R. et al. A comparative study of transgenic canola (Brassica napus L.) harboring either chimeric or native Chit42 genes against phytopathogenic fungi. J. Plant Biochem. Biotechnol. 25, 358–366 (2016). https://doi.org/10.1007/s13562-015-0347-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13562-015-0347-1