
Abstract
Insulin gene (INS) mutations are associated with a rare form of maturity-onset diabetes of the young (MODY). Here, we describe a proband with early-onset diabetes resulting from a heterozygous mutation in the promoter region of INS and summarize the clinical features of INS mutations caused by MODY (INS-MODY) reported in previous studies. The proband presented with proteinuria, mild hyperglycemia, and hypertension at the age of 39 years old; he was negative for the glutamic acid decarboxylase (GAD) antibody and had a family history of diabetes, including his father and aunt. The proband underwent whole exome sequencing, and the mutation in the proband and his father was verified by Sanger sequencing. A literature review was performed to examine all reported cases of INS-MODY to evaluate the clinical characteristics of the probands. A heterozygous INS mutation (c.-332C > G) was detected in the proband and his father, and their phenotypes had unique characteristics. Previous reports have described a total of 26 probands with 16 pathogenic mutations of the INS gene, with clinical features that exhibit great inter- and intrafamilial variability, and onset ages ranging from 2 years, 10 months to 62 years; 88% of patients were diagnosed before 40 years of age. Heterozygous mutations in the promoter region affecting the transcriptional activity of the INS gene may increase the risk of early-onset diabetes in adults, with patients presenting phenotypes that are very similar to type 2 diabetes, and genetic testing is needed to identify these individuals.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The insulin (INS) gene encodes preproinsulin, which comprises the signal peptide, insulin B-chain, C domains, and insulin A-chain. In addition to affecting the transcription and translation of insulin, mutations in the INS gene can also affect all steps of insulin biosynthesis in pancreatic β-cells [1], including endoplasmic reticulum (ER) targeting and translocation of preproinsulin, folding of proinsulin in the ER, trafficking and processing of proinsulin, and the binding of insulin to its receptor. In addition, misfolded proinsulin deposition in the ER can lead to ER stress and pancreatic β-cell apoptosis [2, 3], further promoting the occurrence of diabetes.
Pathogenic mutations of the INS gene are associated with a broad spectrum of clinical manifestations, ranging from severe neonatal onset diabetes to mild adult-onset hyperglycemia, suggesting that the products of different mutant INS alleles behave differently and cause diabetes via different mechanisms [1]. The clinical severity of INS mutations is related to the nature of the specific mutations and the steps of insulin biosynthesis affected by these mutations [4, 5]. In addition to autosomal recessive INS gene mutations that can cause permanent neonatal diabetes mellitus (PNDM), dominant mutations also contribute to the causation of PNDM [6], dominant and recessive INS gene mutations account for approximately 12% of diagnosed PNDM cases [7], and some maturity-onset diabetes of the young (MODY)-type diabetes cases are caused by heterozygous INS gene mutations (INS-MODY or MODY10). A series of cis-sequence elements and their homologous DNA-binding factors in the INS promoter region together ensure cell specificity and the rate of INS transcription [8]. Herein, we report the case of an early-onset diabetes patient carrying a heterozygous variant in the promoter region of the INS gene.
Materials and methods
Case report
The proband was a 49 year-old male who had hyperglycemia and hypertension for 10 years. Details of the laboratory tests were defined as previously described [9]. In 2012, he presented at our hospital with proteinuria and hematuria, with a fasting glucose level of 5.86 mmol/L and HbA1c reading of 6.2% (44.26 mmol/mol). For the next six years, his fasting blood glucose level ranged from 5.18–7.09 mmol/L. In 2018, he was referred to the Department of Endocrinology and Metabolism of Peking University People’s Hospital for elevated fasting glucose levels. No diabetic ketoacidosis was observed. His body mass index (BMI) was 27.8 kg/m2, and the following indices were recorded: fasting glucose 7.3 mmol/L, fasting insulin (Fins) 19.3uU/ml, fasting C-peptide (FCP) 4.21 ng/ml, and HbA1c 7.2% (55.19 mmol/mol). His glutamic acid decarboxylase (GAD) antibody test result was negative. The proband had good glycemia control for several years by diet, and his fasting insulin levels were not low, so even he had only measured the GAD antibody, the proband was diagnosed with type 2 diabetes and started taking SGLT2 inhibitors for proteinuria and diabetes in 2020. His laboratory data for the last decade are detailed in Table 1. The patient’s father and paternal aunt were diagnosed with type 2 diabetes; however, his aunt had died without genetic testing.
The study protocol was approved by the Ethics Committee of Peking University People’s Hospital (China). Written informed consent was obtained from the proband and his father.
Genetic screening
Genetic screening was performed at MyGenostics Inc. (Beijing). All DNA samples were extracted from peripheral blood samples. The GenCap Human Exon V4 capture chip (MyGenostics, China) was used for whole exome sequencing of the proband sample, which was performed using the Illumina HiSeq2500 system. The rare variant of INS was validated by Sanger sequencing in both the proband and his father.
Literatures review
A literature search was performed in the following databases: PubMed, ClinVar, and the Human Gene Mutation Database for mutant INS gene-induced MODY (as of January 2022). The search terms were “INS gene,” “Insulin gene,” “Maturity-onset diabetes of the young 10” and “MODY 10.” All articles published in English reporting patients with the following criteria were included: (1) INS mutations classified as pathogenic or likely pathogenic according to the guidelines recommended by the American College of Medical Genetics and Genomics (ACMG); (2) non-autoimmune diabetes and non-neonatal diabetes; (3) no treatment dependent on insulin and/or measurable C-peptide at least one year after diagnosis of diabetes.
Results
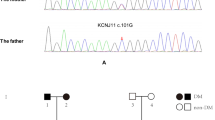
A heterozygous mutation (c.-332C > G) in the promoter region of INS was detected in the proband and his father (Fig. 1). Whole exome sequencing revealed no other mutation of known monogenic diabetes. According to the ACMG guidelines, this mutation can be classified as likely pathogenic (PS3[8], PM2).

DNA sequences of the INS mutation c.-332C > G found in the proband (A) and his father (B). C Indicates pedigrees of the proband. The solid symbol: diabetes status; empty symbol: normoglycemic subject. Arrow indicate the proband. The INS mutation c.-332C > G status is shown under each symbol: NM as heterozygote and NT as not tested. The text below indicates the following: present age, age of onset, treatment, diabetic complications. OHA: oral hypoglycemic agents. DR: diabetic retinopathy. DN: diabetic nephropathy
INS-MODY is a relatively rare type of MODY. The clinical features of the proband found in this study were similar to those of type 2 diabetes, and it is difficult to distinguish him from type 2 diabetes without genetic testing. Therefore, we reviewed the previous literatures to find the clinical characteristics of patients with INS-MODY. A total of 26 probands with 16 pathogenic mutations of INS gene of INS-MODY have been reported in the literature, including one with intronic mutations, two with nonsense mutations, two with frameshift mutations, and 21 with heterozygous mutations (Fig. 2). According to the provided clinical data for probands with pathogenic INS mutations, 92% (24/26) were diagnosed with diabetes, 88% (21/24) of which were diagnosed before 40 years of age, 71% (17/24) of which were diagnosed before 25 years of age, and 63% (15/24) of which were treated with insulin. All of the probands carried heterozygous mutations and the inheritance pattern of their families was autosomal dominant. The clinical features of these cases are summarized in Table 2. There was a significant difference between the different probands. The youngest age at diagnosis was 2 years and 10 months, and the oldest age was 62 years (Supplementary Table 1). Two probands were on a diet to maintain good blood glucose control, and four probands were on insulin but with poor glycemic control [HbA1c > 9% (74.86 mmol/mol)]. Complications of INS-MODY have been reported in only five families; diabetic retinopathy, neuropathy, and microalbuminuria were reported in a proband with a 30-year history of diabetes, while metabolic cataract, nephropathy, and neuropathy were reported in another patient with a frameshift mutation.

A schematic of INS gene structure and the position of the pathogenic mutations of INS-MODY reported in previous studies and this study. Marked in red: the mutation of the proband found in this study (c.-332C>G)
Discussion
This is the first report of a patient with early-onset diabetes due to a heterozygous mutation (c.-332C > G) in the promoter region of the INS gene. The compound heterozygous mutations c.-332C > G and c.-331C > G were first discovered in a pedigree of neonatal diabetes [8].
The most dominant INS mutations are located in the proinsulin domain and disrupt the oxidative folding of the protein, leading to the misfolding of proinsulin in the ER. Misfolded proinsulin accumulates in the ER, disrupts ER protein homeostasis, induces ER chronic stress, and leads to beta cell apoptosis [1, 10]. In addition, misfolded proinsulin can interact abnormally with co-expressed wild type proinsulin through the proinsulin dimerization interface, impairing the folding and ER export of wild type proinsulin, which reduces insulin production and leads to insulin-deficient diabetes [11, 12].
Mutations resulting from the deletion of INS, inactivation of promoters, and deletion of translation initiation can disrupt transcription and translation of the INS gene. Although both INS alleles have been shown to cause neonatal diabetes [8], these recessive INS mutations have also been reported to be associated with early-onset diabetes [13]. The c.-332C > G mutation is located between E1 and A1, and this sequence is conserved in a subpopulation of mammalian species. Multiple base mutations adjacent to this mutation impair INS promoter activity [14]. A previous study reported that the parents of a proband with neonatal diabetes carried the c.-332C > G mutation but did not have diabetes. However, it was not stated in the article whether the parents had performed glucose tolerance tests, and the reported proband had neonatal diabetes, it is possible that his parents were younger and thus not old enough to obtain abnormal glucose tolerance readings. In addition, previous studies have indicated that the mutations c.-331 (C > G, C > A) and c.-332C > G can decrease INS transcriptional activity by up to 90%. The proband in the current study carrying this heterozygous mutation (c.-332C > G) also had early-onset diabetes. Together, these data suggest that the compound heterozygous mutation (c.-332C > G and c.-331C > G) can induce neonatal diabetes, while the c.-332C > G heterozygous mutation on its own can be a pathogenic mutation triggering early-onset diabetes in adults. Recessive INS mutations that cause neonatal diabetes might also function as dominant INS mutations that cause MODY phenotypes.
Previous reported cases have revealed that the clinical features of patients with INS mutations exhibit large inter- and intrafamilial variability, ranging from mild adult-onset hyperglycemia to an onset age of under 3 years old requiring insulin treatment. Even in patients with the same mutations or in the same pedigree, the onset age of diabetes ranged from childhood to middle age, and even some individuals without diabetes at the time of the study (Supplementary Table 1), suggesting that other genetic and environmental factors, including the degree of misfolded proinsulin accumulation and individual differences, may contribute to differences in the clinical features of INS-MODY. The proband of this study was found to have hyperglycemia at the age of 39 years and was overweight, exhibiting microalbuminuria, occasional hyperinsulinemia, hypertriglyceridemia, and low HDL-c levels, suggesting that diabetes in this pedigree results from the INS gene mutation against a background of insulin resistance. In addition, the patient’s father exhibited later onset of diabetes, suggesting that environmental factors, such as age and lifestyle, might have contributed to the development of diabetes in these cases. Because the clinical phenotype of this heterozygous mutation in the promoter region is very similar to that of type 2 diabetes, it is difficult to diagnose such patients if genetic testing is not performed. Thus, screening a larger pool of patients with early-onset diabetes may reveal factors that could serve as indicators whether genetic testing should be performed.
In summary, this study highlights the utility of heterozygous mutations in the promoter region of the INS gene, and suggests that heterozygous mutations affecting the transcriptional activity of INS may increase the risk of early-onset diabetes in adults, indicating that genetic testing is needed in such patients.
Data availability
All datasets are available from the corresponding authors upon reasonable request.
References
Liu M, Sun J, Cui J, Chen W, Guo H, et al. INS-gene mutations: from genetics and beta cell biology to clinical disease. Mol Aspects Med. 2015;42:3–18.
Herbach N, Rathkolb B, Kemter E, Pichl L, Klaften M, et al. Dominant-negative effects of a novel mutated Ins2 allele causes early-onset diabetes and severe beta-cell loss in Munich Ins2C95S mutant mice. Diabetes. 2007;56:1268–76.
Izumi T, Yokota-Hashimoto H, Zhao S, Wang J, Halban PA, et al. Dominant negative pathogenesis by mutant proinsulin in the Akita diabetic mouse. Diabetes. 2003;52:409–16.
Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, et al. Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab. 2010;21:652–9.
Støy J, Steiner DF, Park S-Y, Ye H, Philipson LH, et al. Clinical and molecular genetics of neonatal diabetes due to mutations in the insulin gene. Rev Endocr Metab Disord. 2010;11:205–15.
Gopi S, Gowri P, Panda JK, Sathyanarayana SO, Gupta S, et al. Insulin gene mutations linked to permanent neonatal diabetes mellitus in Indian population. J Diabetes Complications. 2021;35: 108022.
Glaser B. Insulin mutations in diabetes. Diabetes. 2008;57:799–800.
Garin I, Edghill EL, Akerman I, Rubio-Cabezas O, Rica I, et al. Recessive mutations in the INS gene result in neonatal diabetes through reduced insulin biosynthesis. Proc Natl Acad Sci USA. 2010;107:3105–10.
Gong S, Han X, Li M, Cai X, Liu W, et al. Genetics and clinical characteristics of PPARγ variant-induced diabetes in a Chinese Han population. Front Endocrinol. 2021;12: 677130.
Liu M, Hodish I, Rhodes CJ, Arvan P. Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci U S A. 2007;104:15841–6.
Sun J, Xiong Y, Li X, Haataja L, Chen W, et al. Role of proinsulin self-association in mutant INS gene-induced diabetes of youth. Diabetes. 2020;69:954–64.
Yang Y, Shu H, Hu J, Li L, Wang J, et al. A novel nonsense INS mutation causes inefficient preproinsulin translocation into the endoplasmic reticulum. Front Endocrinol. 2022;12: 774634.
Raile K, O’Connell M, Galler A, Werther G, Kühnen P, et al. Diabetes caused by insulin gene (INS) deletion: clinical characteristics of homozygous and heterozygous individuals. Eur J Endocrinol. 2011;165:255–60.
Niu X, Perakakis N, Laubner K, Limbert C, Stahl T, et al. Human Krüppel-like factor 11 inhibits human proinsulin promoter activity in pancreatic beta cells. Diabetologia. 2007;50:1433–41.
Acknowledgements
We thank the study proband and his father for their contributions. We also thank all research staff for their collection of data.This study was supported by grants from National Key Research and Development Program of China (2016YFC1304901), Beijing Municipal Science and Technology Commission Funding (Z141100007414002 andD131100005313008).
Funding
This study was supported by grants from National Key Research and Development Program of China (2016YFC1304901), Beijing Municipal Science and Technology Commission Funding (Z141100007414002 and D131100005313008).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval
The study protocol was approved by the Ethics Committee of Peking University People’s Hospital (China). Written informed consent was obtained from the proband and his father.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Gong, S., Han, X. & Ji, L. A heterozygous mutation at promoter region of insulin gene (INS) accounts for early-onset diabetes: A case report and review of the literature. Int J Diabetes Dev Ctries 44, 145–149 (2024). https://doi.org/10.1007/s13410-023-01205-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13410-023-01205-4