
Abstract
Glucokinase-maturity-onset diabetes of the young (GCK-MODY) is an autosomal dominantly inherited disease caused by heterozygous inactivating mutations in the glucokinase gene. It usually presents with mild fasting hyperglycemia. Here, we present an obese patient and her family with GCK-MODY caused by a novel heterozygous p.E51*(c.151.G>T) mutation in the GCK gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glucokinase-maturity-onset diabetes of the young (GCK-MODY, also known as MODY 2) accounts for 2–5% of all diabetes cases [1]. Glucokinase, a key enzyme in glycolysis, is a glucose sensor that maintains glucose homeostasis in pancreatic beta cells [2]. The GCK gene (7p15.3–p15.1) consists of 12 exons and encodes a 465-amino acid protein [3]. GCK-MODY is caused by heterozygous inactivating GCK mutations, which are usually associated with mild hyperglycemia [4]. It is treated only with diet, and complications are extremely rare [5]. Here, we report a novel heterozygous p.E51 mutation encoded by GCK exon 2, which results in the typical GCK-MODY phenotype.
Patient report
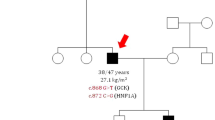
A 17-year-old girl was admitted to our department for evaluation of obesity and hyperglycemia. Despite her obesity (body mass index (BMI), 30.2 kg/m 2), her physical examination had no acanthosis nigricans or other abnormalities. Pubertal assessment revealed Tanner stage V. Blood glucose levels were repeatedly checked and showed mild fasting hyperglycemia as well as an elevated HbA1c level. Standard oral glucose tolerance test with 75 g of glucose was performed showing a fasting glucose of 131 mg/dl, 120-min glucose of 159 mg/dl and normal insulin levels. Serum autoantibodies against glutamic acid decarboxylase, islet cell antibodies, and anti-insulin autoantibodies were negative. Family history revealed that her father, grandmother, and uncle were diagnosed with diabetes (Fig. 1). Considering the clinical features and family history, mutation analysis of the GCK gene was performed. Complete sequencing of coding exons and intron-exon boundaries of the GCK gene identified a heterozygous mutation (c.151.G>T) leading to a stop codon (p.E51*) in the GCK gene.

Family tree of the patient
The father was diagnosed with diabetes at age 30 but he did not go to the doctor visits regularly. The blood glucose levels of both sisters were never tested and the sisters had impaired fasting glucose levels. The patient’s father and two sisters had the same mutation. We performed a 75-g 2-h oral glucose tolerance test (OGTT) (after 12 h of fasting) to assess glycemic/insulin fluctuations in response to a glucose challenge. Clinical and laboratory findings of the patients are given in Tables 1 and 2. The grandmother refused to undergo genetic testing. Unfortunately, we could not describe other family members better as they are living outside the city for a long time.
Patients were treated with appropriate diet and exercise. All patients were screened for diabetes-related microvascular complications. We did not find any diabetes-related complications. Nephropathy was evaluated using microalbuminuria, and retinopathy was assessed by fundoscopic examination. Lipid levels and 24-h urinary albumin excretion rates were normal. None of the patients had diabetic retinopathy.
Mutational analysis
GCK gene sequence analysis was performed by using MiSeq next generation sequencing (NGS) platform, a FDA approved diagnostic system (Illumina, San Diego, CA, USA). Genomic DNA was extracted according to the manufacturer’s standard procedure using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). All coding exons of the GCK gene and their flanking splice site junctions were amplified using PCR primers, designed with PRIMER©—Primer Designer v.2.0 (Scientific & Educational Software programme) software. PCRs were validated by using agarose gel electrophoresis. After PCR amplification, the libraries were prepared with the NexteraXT kit (Illumina Inc.), according to the manufacturer’s instructions. Next-gene sequencing was carried on MiSeq (Illumina Inc.). Sequences were aligned to the hg19 genome within MiSeq Reporter software (Illumina Inc.). Visualization of the data was performed with IGV 2.3 (Broad Institute) software.
In silico analysis was done with Mutation taster software, and it was predicted that variant as a disease causing variant with the probability score 1. As this variant causes a premature stop codon, it causes a truncated non-functional protein.
Discussion
In our patient, a novel heterozygous mutation of c.151.G>T leading to premature stop codon p. E51* was identified in exon 2 of the GCK gene. Glucokinase plays a crucial role in the regulation of insulin secretion and acts as a glucose sensor in pancreatic beta cells. Heterozygous inactivating mutations of GCK cause GCK-MODY characterized by mild fasting hyperglycemia, which is present at birth but often only detected later in life while screening for other reasons [5]. As the result of a change in the required glucose concentration threshold to stimulate insulin secretion, fasting plasma glucose (FPG) ranges between 100 to 153 mg/dl in a majority of patients. Significant worsening of glycemic control occurs with increasing age [6, 7]. In our case, the father’s FPG and HbA1c levels were slightly higher than the children.
Our proband was obese. At the first examination, Type2 diabetes mellitus (T2D) was considered an obese patient with hyperglycemia. But she did not have acanthosis nigricans and metabolic syndrome findings also OGTT showed insulinopenia. Absence of clinical signs such as obesity and the metabolic syndrome in patients with early-onset diabetes favors a diagnosis of MODY [8]. The prevalence of obesity in patients with MODY is as same as normal population. People with GCK-MODY are usually less obese than people with T2D [9]. However, the coexistence of obesity and MODY are more frequently reported because it is likely that obesity is epidemic among teenagers and young adults [8]. The father and other siblings were not obese. We think that our patient’s obesity may have been coincidental. Moreover, a study performed with pediatric-age MODY sufferers observed acanthosis nigricans in 40% of molecularly confirmed cases [10]. Patients with GCK-MODY are also less hyperglycemic than patients with young-onset T2D. The rise of blood sugar level in T2D patients during OGTT is typically above 54 mg/dl [9]. One of the characteristic features of GCK-MODY is a modest increase in the postprandial glucose levels, with a 75-g oral glucose tolerance test increment (120 min glucose minus 0 min glucose) of less than 54 mg/dl in 70% and 83 mg/dl in 95% of patients [11]. We also observed a small increase in glucose levels during OGTT in our patients.
We did not find any diabetes-related complications. Microvascular complications are very rare in individuals with GCK-MODY, because the hyperglycemia is mild, and there is no marked progression [12, 13]. A study consisting of 42 families with GCK-MODY reported that proliferative retinopathy was seen in less than 4% of the patients more than 5 years after initial diagnosis, while peripheral neuropathy and proteinuria rates were 5 and 6%, respectively. Dyslipidemia and hypertension were also reported to have low prevalence [5]. Another study reported a few cases of retinopathy and macrovascular disease [14].
MODY also should be kept in mind in obese patients with mild hyperglycemia, non-obese first degree relatives with early-onset diabetes, and especially lack of insulin resistance, and metabolic syndrome findings. Identification of GCK gene mutations is important for the correct and definitive diagnosis of GCK-MODY and helps the physician predict disease course and initiate appropriate therapy.
Abbreviations
- GCK-MODY:
-
Glucokinase-maturity-onset diabetes of the young
- GCK:
-
Glucokinase
- BMI:
-
Body mass index
- OGTT:
-
Oral glucose tolerance test
- T2D:
-
Type 2 diabetes mellitus
- FPG:
-
Fasting plasma glucose
References
Gat-Yablonski G, Shalitin S, Phillip M. Maturity onset diabetes of the young—review. Pediatr Endocrinol Rev. 2006;3(Suppl 3):514–20. Review
Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL, et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest. 1993;92(5):2092–8.
Iynedjian PB. Mammalian glucokinase and its gene. Biochem J. 1993;293(Pt 1):1–13. Review
Hattersley AT, Turner RC, Permutt MA, Patel P, Tanizawa Y, Chiu KC, O’Rahilly S, Watkins PJ, Wainscoat JS. Linkage of type 2 diabetes to the glucokinase gene. Lancet. 1992;339(8805):1307–10.
Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, Gloyn AL. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30(11):1512–26.
Hattersley AT. Maturity-onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity. Diabet Med. 1998;15:15–24.
Stride A, Vaxillaire M, Tuomi T, et al. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45:427–35.
Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum Mutat. 2013;34(5):669–85.
Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S, Hattersley AT. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care. 2015;38(7):1383–92.
Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, Greenbaum CJ, Imperatore G, Lawrence JM, Marcovina SM, Mayer-Davis E, Rodriguez BL, Steck AK, Williams DE, Hattersley AT. SEARCH for diabetes in youth study group. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for diabetes in youth. J Clin Endocrinol Metab. 2013;98(10):4055–62.
Stride A, Vaxillaire M, Tuomi T, Barbetti F, Njølstad PR, Hansen T, Costa A, Conget I, Pedersen O, Søvik O, Lorini R, Groop L, Froguel P, Hattersley AT. The genetic abnormality in the beta cell determines the response to an oral glucose load. Diabetologia. 2002;45(3):427–35.
Velho G, Blanché H, Vaxillaire M, Bellanné-Chantelot C, Pardini VC, Timsit J, Passa P, Deschamps I, Robert JJ, Weber IT, Marotta D, Pilkis SJ, Lipkind GM, Bell GI, Froguel P. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia. 1997;40(2):217–24.
Stride A, Shields B, Gill-Carey O, Chakera AJ, Colclough K, Ellard S, Hattersley AT. Cross-sectional and longitudinal studies suggest pharmacological treatment used in patients with glucokinase mutations does not alter glycaemia. Diabetologia. 2014;57(1):54–6.
Sagen JV, Bjørkhaug L, Molnes J, Raeder H, Grevle L, Søvik O, Molven A, Njølstad PR. Diagnostic screening of MODY2/GCK mutations in the Norwegian MODY registry. Pediatr Diabetes. 2008;9(5):442–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Funding source
No external funding for this manuscript.
Financial disclosure
The authors have no financial relationships relevant to this article to disclose.
Electronic supplementary material
ESM 1
(DOCX 69 kb)
Rights and permissions
About this article

Cite this article
Ahmet Ucakturk, S., Gunindi, F., Ceylaner, S. et al. A novel genetic mutation in a Turkish family with GCK-MODY. Int J Diabetes Dev Ctries 37, 323–326 (2017). https://doi.org/10.1007/s13410-016-0539-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13410-016-0539-9