
Abstract
Background
Radiation therapy (RT) is a key anti-cancer treatment that involves using ionizing radiation to kill tumor cells. However, this therapy can lead to short- and long-term adverse effects due to radiation exposure of surrounding normal tissue. The type of DNA damage inflicted by radiation therapy determines its effectiveness. High levels of genotoxic damage can lead to cell cycle arrest, senescence, and cell death, but many tumors can cope with this damage by activating protective mechanisms. Intrinsic and acquired radioresistance are major causes of tumor recurrence, and understanding these mechanisms is crucial for cancer therapy. The mechanisms behind radioresistance involve processes like hypoxia response, cell proliferation, DNA repair, apoptosis inhibition, and autophagy.
Conclusion
Here we briefly review the role of genetic and epigenetic factors involved in the modulation of DNA repair and DNA damage response that promote radioresistance. In addition, leveraging our recent results on the effects of low dose rate (LDR) of ionizing radiation on Drosophila melanogaster we discuss how this model organism can be instrumental in the identification of conserved factors involved in the tumor resistance to RT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Several lines of evidence indicate that cancer cells are heterogeneous in their tumor-initiating properties, which contribute to tumor growth and metastasis development [1, 2]. This is relevant for radiation oncology, as the high inter- and intra-tumoral variability of cancer cells remains the major obstacle to the development of efficient therapies to fight cancer. Radiation therapy (RT) with either photon (conventional radiotherapy) or proton (proton therapy) beams represents one of the key anti-cancer treatment options. The principle of RT is the killing of tumor cells with high-LET (protons) or low-LET (photons) ionizing radiation (IR) while minimizing injuries to normal tissue [3, 4]. It is administered as fractionated or hypofractioned doses over several days or weeks affecting cancer growth by ROS-induced DNA damage and by altering fundamental cell elements such as cell membranes. Around 65% of cancer patients are treated with RT either alone or in combination with other treatments, such as chemotherapy. However short- and long-term adverse effects are often found in cancer patients as a consequence of radiation exposure to healthy tissue that normally surrounds the irradiated tumor [5, 6]. Early clinical data suggest that also low-dose rate (LDR) of radiation (i.e., 0.5-2 Gy per dose) can be exploited as an anti-tumor treatment. LDR has been shown to reprogram the tumor microenvironment (TME), recruit large numbers of effector T cells, thereby inducing tumor vascular normalization, inflammatory microenvironment, increased T cell infiltration, and enhanced anti-tumor effects with extremely reduced toxicity. In addition, these effects can be synergized with immunotherapy, thus rendering tumors more sensitive to immune checkpoint blockade response [7].
Despite the increasing technological advances that have led to the enhancement of this therapeutic approach, many patients experience recurrence and relapse as consequence of tumor radioresistance. The efficacy of IR in eliminating malignant cells can vary among different tumors and depends on either pre-existing mechanism within cancer cells or specific radiation-induced cellular responses. This distinction classifies resistance to RT into two main categories: intrinsic and acquired resistance. Intrinsic resistance is inherent in cancer cells even before the initiation of treatment, and it is closely linked to the inherent characteristics of the tumor. A key player in intrinsic radioresistance is the presence of cancer stem cells (CSCs) within the tumor mass. CSCs constitute a small subset of self-sustaining cells endowed with the unique capacity for self-renewal and the ability to sustain the tumor's growth. If the radiation dosage administered fails to significantly reduce the CSC population, their numbers can increase during treatment, despite visible shrinkage of the tumor. This phenomenon has been documented across various cancer types [2, 8]. Conversely, acquired radioresistance is a process in which cancer cells adapt to the changes induced by irradiation, ultimately leading to resistance against treatment. This adaptation can occur through the activation of specific transcription factors (TFs) triggered by exposure to radiation, enabling cancer cells to elude the lethal effects of IR [9, 10]. Both intrinsic and adaptive radioresistance result from changes in biological processes such as hypoxia response, cell proliferation, epithelial-mesenchymal transition, DNA repair, apoptosis inhibition, and autophagy [11,12,13,14,15].
Furthermore, resistance to radiation treatments could be determined by genetic and epigenetic factors that lead to an alteration of cancer cell metabolism, which extends to tumor microenvironment and ultimately reduces the cytotoxic effect of the therapy [14]. It can be surmised that as RT determines tumor cell death by inducing DNA damage, tumor cells with highly efficient DNA repair are prone to radioresistance, while a defective repair renders these cells radiosensitive.
The RT effect on tumors depends on the type of DNA damage inflicted to IR-exposed tumor tissue. This DNA damage, which consists of either single strand breaks (SSBs) or double strand break (DSB), results from direct DNA ionization, radiolysis of water that produces highly chemically reactive species or from an impairment of mitochondrial functions [4]. Intensive DNA damage that exceeds the DNA repair capacity of tumors will lead tumor cells to cell cycle arrest, senescence and ultimately death. However, a large number of tumors can cope with high levels of genotoxic damage and become radioresistant by activating protective mechanisms against radiation-induced DNA breaks [16].
Here we briefly review the main mechanisms that promote tumor radioresistance focusing on the role of genetic and epigenetic factors involved in the repair of DNA breaks. In addition, we describe how our recent results on the effects of LDR on the model organism Drosophila melanogaster [17] can provide useful insights for the identification of conserved factors involved in tumor resistance to RT.
2 The role of DNA damage response in radioresistance at glance
Exposure to IR induces single-stranded DNA breaks (SSBs), double-stranded DNA breaks (DSBs), base damage, and DNA–protein cross-links in the genomic DNA. Among them, DSBs are the most deleterious lesions that, if not repaired correctly, can lead to genomic instability and cell death [18]. To cope with these genotoxic insults, cells have evolved a sophisticated network of DNA damage response (DDR) pathways.
DSBs repair in normal cells occurs by two major pathways: the non-homologous end joining (NHEJ) and the homologous recombination (HR). NHEJ, which encloses both classical c-NHEJ and alternative alt-EJ/TMEJ, is an error-prone repair system, as when the two DSB ends are recognized, processed and ligated together, often results in the loss or addition of nucleotides at the break site [19]. For this reason, if the damage occurs in a major coding region, the repair will generate a mutation that could adversely affect the functionality of the cell. DSBs recognition during c-NHEJ relies on the main factors Ku70-80, which function as a heterodimer and enable the recruitment of the other proteins critical for promoting the closure of DNA ends. Then, the enzyme DNA-PKcs (DNA-dependent protein kinase, catalytic subunit), which has a high affinity for the DNA-Ku structure, forms the DNA-PK complex [20], that in turn phosphorylates itself and neighboring components. X-ray Repair cross complementing 4 (XRCC4) also plays a role in making this structure stable, and together with Ku, recruits nucleases such as Artemis, PNKP, APLF, WRN, Aprataxin, and the MRN complex (MRE11-RAD50-NBS1), FEN1 and EXO1, that promote resection to ultimately facilitate ligation by the XRCC4-DNA ligase IV complex. In case of a defective or inhibited c-NHEJ pathway, the cell may rely on a back-up pathway, named alt-EJ (also called micro-mediated end-joining; MMEJ, or Theta-mediated end-joining; TMEJ) to repair DSBs [20]. alt-EJ is initiated by PARP-1 recruitment to resected DNA-ends. Following the activation of phosphorylated CtIP, 3′ overhangs are generated by helicases such as the MRN complex. POLQ then binds to long single-stranded DNA (ssDNA) overhangs generated by 5′–3′ resection of DSBs and anneals sequences with 2–6 base pairs of microhomology to use them as primers for DNA synthesis. The stabilized DNA ends are then ligated by LIG3–XRCC1 or LIG1. POLQ also suppresses HR by interacting with RAD51 thereby limiting RAD51-ssDNA nucleofilament formation and suppressing HR activity [21].
In contrast to NHEJ, HR is an error-free DNA repair mechanism that uses the DNA from the sister chromatid as a template for recombination. This implies that this pathway can only be used at certain times in the cell cycle [22]. Damage recognition in HR requires the MRN complex, which favors both short- and long-range DNA end resection by recruiting C-terminal binding protein (CtBP)-interacting protein (CtIP) and exonuclease 1 (EXO1), respectively [23, 24]. DNA end resection results in the generation of a single-stranded DNA tail, which is first stabilized by Replication Protein A (RPA) and then bound by RAD51 that removes RPA. The replacement of RPA on the ssDNA allows RAD51 to promote the invasion of the single-stranded DNA into the homologous double-stranded DNA template, leading to synapsis, novel error-free DNA synthesis, strand dissolution, and repair [25]. This process relies on the activation of the two important protein kinases Ataxia Telangiectasia Mutated (ATM) and Ataxia Telangiectasia and Rad3 related (ATR) by the MRN complex and RPA, respectively.
Differently for DSBs, SSBs are repaired through Base Excision Repair (BER) pathway. BER requires the activation of poly(ADP-ribose) polymerase (PARP) that induces extensive poly-ADP-ribosylation, recruits the Scaffold protein X-ray cross-complementing protein 1 (XRCC1), DNA polymerase B (PolB) and DNA Ligase 3 (Ligase III) to fill the single-nucleotide gap and ligate the newly synthesized DNA strands [26, 27]. SSBs can be also repaired by Nucleotide Excision Repair (NER) and mismatch repair (MMR). Whereas NER removes helix-distorting lesions such as those produced by UV, MMR corrects mismatched bases, insertions and deletions acquired during DNA replication [28,29,30]. Depending on the amount of the DNA damage and on the efficiency of DNA repair, DDR activation can result in cell survival, apoptosis, cell cycle arrest, and senescence [31]. DDR is regulated by the activity of the protein kinases ATM, ATR, and DNA-PKcs. ATM and ATR trigger an activating phosphorylation cascade, which determines the phosphorylation of DDR mediators (e.g., p53-binding protein 1 (53BP1), H2A histone family member X at Serine 139 (H2AXSer139 or H2AX), breast cancer type 1 susceptibility protein (BRCA1), and downstream kinases checkpoint kinases 1/2 (Chk1/Chk2). Chk1 and Chk2 then in turn phosphorylate downstream effectors such as p53. The activation of DNA-PKcs induces the formation of a complex with the Ku heterodimer (Ku70/Ku80) that is required for NHEJ (see below).
How cells choose either NHEJ or HR to repair DSBs, it depends on several factors that include cell cycle phase, chromatin quality (euchromatin vs heterochromatin), and amount of resection [32]. HR is generally favored in the S/G2 phases of the cell cycle, when the sister chromatid is available as a donor template, although both HR and NHEJ can take place throughout the cell cycle [24]. In mammalian cells, the DNA repair pathway choice relies on a regulatory circuit that involves 53BP1-RIF1 and BRCA1-CtIP [33]. During G1, RIF1, which is recruited to the DSB site by phosphorylated 53BP1, prevents end resection thus promoting NHEJ. In the S/G2 phases, CtIP is phosphorylated by CDK2 and, associated with BRCA1, removes 53BP1 and RIF1 from the DSB sites. Consequently, the MRN complex is recruited on the same sites and initiates end resection.
The loss of 53BP1 rescued the severe genomic instability of BRCA1 mutants and the sensitivity of BRCA1-mutant cells to PARP inhibitors [34, 35]. 53BP1 depletion in BRCA1-deficient cells restores, to some degree, HR in a manner that depends on the activation of end resection. This phenotypic reversal relies on a functional antagonism between BRCA1 and 53BP1, which also compete for accumulation at DNA damage sites [33, 36,37,38]. These findings indicate that initiating end resection is a key decision point in DSB repair pathway choice, with a direct impact on the therapeutic efficacy of PARP inhibitors. Interestingly, depleting any single subunit of shieldin (that consists of REV7, a known 53BP1-pathway component, and the three hitherto uncharacterized proteins C20orf196/SHLD1, FAM35A/SHLD2, and CTC-534A2.2/SHLD3 [39]), in various BRCA1-deficient cell lines suppressed their sensitivity to PARPi to a degree comparable to that of 53BP1 depletion. This suggests that the shieldin complex, like 53BP1, suppresses DNA end resection and supports c-NHEJ [40]. The evidence that shieldin shares the same functions as 53BP1 and that it can act genetically as part of 53BP1-RIF1 pathway, lead to hypothesize that shieldin might represent the 53BP1 effector during DSBs repair pathway choice [40].
3 Tumor Radioresistance to RT results from dysregulation of DNA repair factors
Several findings have highlighted that modulation of key factors involved in DNA repair and cell cycle regulation described above can account for promoting resistance to RT of several tumors. For example, radioresistant populations of breast cancer cells derived from IR elicit increased activation of ATM and upregulation of E-box binding homeobox 1 (ZEB1), an EMT-inducing transcription factor. ZEB1 in turns enhances the stabilization of CHK1 and HR-dependent DNA repair thus leading to radioresistance [41]. Recent works have also shown that the recurrent glioblastoma (GBM) cells that are generated from resistant residual cells, undergo a shift from activation of ATM to ATR mediated repair pathway, escaping the ATM inhibition and leading to better survival of these cells following RT [42]. Furthermore, ectopic expression of 14–3-3σ, a family member of 14–3-3 proteins (14–3-3β, ϵ, θ/τ, ζ, σ, γ and η) in humans implicated in the development of cancer, has been reported to enhance NHEJ in different tumors upregulating Chk2 and by increasing PARP-1 expression thereby promoting radioresistance [43]. PARP-1 increased expression has been also shown to promote radioresistance in prostate cancer and in renal cell carcinoma [44, 45]. In the latter, this hyperactivation is caused by the loss of DOC-2/DAB2 interactive protein (DAB2IP), a potent tumor suppressor, which is frequently lost in Renal Cell Carcinoma (RCC). Indeed, DAB2IP directly interacts with PARP-1 and affects its turnover by recruiting E3-ligases (e.g., RanBP2, TRIP12, and RNF40). Loss of DAB2IP results in extended PARP-1 protein expression and ultimately enhances its role in DNA repair [44]. Radioresistance in lung cancer has been recently correlated to the upregulation of DNA protein Rad17 and MRN complex both in vitro and in vivo. In this case a Rad17-dependent recruitment of the MRN complex was determined by the overexpression of IQGAP3 GTPase-activating protein 3 that physically interacts with Rad17 [46]. RNA-seq data from large cohorts of patients from the TCGA project revealed that expression levels of RAD51 were significantly elevated in advanced-stage lung adenocarcinoma tumors compared with normal tissues and were associated with poor survival [47]. Moreover, NSCLC cells that express the KRAS mutant form A549 and LU99A are more radioresistant than wild-type cells because mutant KRAS can upregulate RAD51 expression through oncogene MYC, thus favoring a more efficient DNA damage repair and thereby cell survival [47]. Overexpression of RAD51 was also observed in CDC133 + lung cancer cells to improve, along with Exo1, repair proficiency of A549 cell lines [48]. Increased levels of DNA-PKc and MRN complex have been found in radioresistant cervical cancer cells [49] that also overexpressed the Rho GTPase RhoC highlighting the role for ChoC-ROCK2 signaling in DNA repair.
Chromatin factors that normally bind to both DNA and histones are also involved in mediating radioresistance. Expression of High-mobility group box 1 (HMGB1), a structural protein of chromatin that regulates several biological processing including DNA repair [50] has been associated with radioresistance in bladder cancer, Esophageal squamous-cell carcinoma (ESCC) tumor and breast cancer. Recent findings revealed that increased expression of HMGB1 in these tumors influences DNA damage pathways, apoptosis, and autophagy [51,52,53]. Diverse tumors aberrantly express Cancer/Testes (CT) Antigens, whose normal distribution is generally restricted to germ cells. Some CT antigens are involved in DNA repair and suggested to play a role in radioresistance. For instance, the CT melanoma antigen-A4 (MAGE-A4) interacts with the ubiquitin ligase RAD18 that activates Trans-lesion synthesis (TLS), which represents a crucial DNA-damage tolerance mechanism and allows DNA synthesis in cells where DNA damage has occurred [54]. Moreover, the CT antigen HORMAD1 has been proposed to promote radioresistance in Lung adenocarcinoma cells by enhancing HR in response to IR [55].
The massive involvement of DDR pathways in inducing tumor radioresistance has driven to exploit the targeting key DNA damage repair (DDR) factors as an important tool to improve the efficacy of RT. Several strategies are being employed to overcome radioresistance: (a) The development of DDR Inhibitors, small molecules and drugs that specifically target DDR factors, such as proteins involved in DNA repair pathways like ATM [56], DNA PKcs [57], CHK1 [58, 59], or WEE1 [60]. These inhibitors disrupt the repair machinery, making it difficult for tumor cells to efficiently mend radiation-induced DNA damage; (b) Radiosensitizing agents, compounds that enhance the sensitivity of tumor cells to radiation. They consist of small molecules (such as oxygen mimics and halogenate base analogs [61, 62]), macromolecules (such as antibodies and short peptides that have high affinity with antigens and receptors overexpressed on the surface of tumor cells [63, 64]) and nanomaterials (in particular, heavy-metal nanomaterials with high atomic number (Z) as they can absorb, scatter, and emit radiation energy [65, 66]); (c) Synthetic Lethality that implies the simultaneous targeting of DDR factors in combination with radiation leading cell death. For example, PARP inhibitors have shown success in targeting BRCA-mutated cancer cells, inducing synthetic lethality when combined with radiotherapy [67]. Combining immunotherapy with radiotherapy can also sensitize tumor cells by promoting immunogenic cell death, activating the immune system to target cancer cells. Immune checkpoint inhibitors, like anti-PD-1 or anti-CTLA-4, can further enhance this effect [68]. These strategies hold great promise in addressing radioresistance and improving the outcomes of radiotherapy.
4 Changes in the epigenome influence DNA repair in cancer cells after radiation
A growing number of works have highlighted that changes in the epigenome can promote radioresistant phenotypes of cancer cells. DNA methylation, post-translational modifications of histones and/or chromatin remodeling, altered expression of ncRNAs are implicated in changing the expression of specific genes to avoid the cytotoxic effects of radiation treatments [69]. In cancer cells, DNA methylation that occurs in specific genomic sites, can indeed play a role mainly in determining intrinsic radioresistance as it confers tolerance to the effect of RT. Moreover, following radiotherapy, cancer methylome can be reprogrammed on genes associated with radioresistance. Interestingly, while hypermethylation has been identified in promoters of tumor suppressor genes, hypomethylation was identified in specific genes, including oncogenes [70].
DNA methylation
Recent findings suggest that DNA methylation regulates the expression of specific transcriptional factors, which in turn control the expression of radioresistance gene. In small cell lung cancer (SCLC) tumors, radiation increases methylation in the promoter of hTERT gene and this modification ultimately leads to the overexpression of hTERT [71]. In vitro experiments revealed that methylation and upregulation of hTERT conferred radioresistance of SCLC cells promoting overexpression of the histone-lysine N-methyltransferase EZH2, the enzymatic catalytic subunit of Polycomb Repressive Complex 2 (PRC2). Increased levels of this enzyme, which is the catalytic subunit of the polycomb repressor complex 2 (PRC2), resulted in the increase of H3K27 trimethylation (H3K27me3) that correlates with SCLC cell survival following irradiation. The dynamic methylome changes induced by RT can also results from differential expression of DNA methyltransferases and demethylases. Indeed, the overexpression of DNMT3B has been recently associated with increased radioresistance in nasopharyngeal cancer. Depletion of DNMT3B determined demethylation of p53 and p21 promoters leading to apoptosis and cell cycle arrest that reverted radioresistance both in vivo and in vitro [72]. In addition, recent studies have shown that DNMT3A and DNMT3B, are also overexpressed in Rhabdomyosarcoma (RMS) compared to normal muscle tissue. Indeed, a combination of DNMT3a or DNMT3B silencing with IR resulted in increased radiosensitivity in RMS. In particular, the silencing of DNMT3A induced cellular senescence through the upregulation of p16 and p21, while DNMT3B depletion caused significant DNA damage and impaired DNA repair mechanisms (ATM, DNA-PKcs, and Rad51) increasing sensitivity to irradiation [73]. Thus, targeting DNMT3A and DNMT3B may be a promising strategy to sensitize RMS cells to RT, especially in cases of metastatic or recurrent RMS tumors.
Interestingly, methylation marks associated with radioresistance of breast cancer cells can be also transmitted to the daughter cells even after a long recovery period post-irradiation suggesting that this heritable epigenetic mechanism underlying radioreristance could enable cell migration and invasion of subsequent cell progeny [74]. As demethylation of the specific tumor suppressor genes reverts radioresistance, DNMT encoding genes might represent potential therapeutic targets for the development of epigenetic drugs. However, as the inhibition of DNMTs might determine several unwanted effects in the methylome, great attention should be paid when considering the employment of demethylating agents (such as 5-aza-20-deoxycytidine) in combination with RT.
Post-translational modifications of histones and/or chromatin remodeling
The response to RT is also influenced by histone post-translational alterations or by modifications of histone modifiers enzymes [69]. For instance, alterations of histone methylation have been also invoked as mechanisms responsible for radioresistance. Reduction of the levels of RIZ1/KMT8 histone methyltransferase was associated with radioresistance of cervical cancer cells. Moreover, whereas H3K36me2 and H3K4me2 marks were shown to facilitate efficient NHEJ of glioblastoma cells and therefore to promote survival of radiation-resistant residual cells [75], inhibition of EZH2 reduced H3K27me3 and favored euchromatin and DNA transcription in the A7 glioblastoma cells. JARID1B (KDM5B)-mediated lysine demethylation is involved in the stemness maintenance in radioresistant cancer cells. Increased expression of histone demethylase JARID1B (KDM5B) has been indeed found in radioresistant OSCC cells and considered as the favorite mechanism that cancer cells use to avoid the cytotoxic effects of RT. Histone acetylation is also involved in triggering radioresistance. Histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate histone acetylation level by transferring or removing an acetyl group from acetyl CoA to lysine residues, respectively. In general, histone lysine acetylation stimulates gene expression, whereas deacetylation is associated with gene silencing. HDACs have been found overexpressed in several tumor types, such as RMS, where they play a crucial role in cancer onset, progression, and resistance to therapies, including RT. For instance, in vitro experiments revealed that the pharmaceutical inhibition of Class I HDACs radiosensitized the two major molecular RMS subtypes such as fusion-positive (FP)-RMS expressing PAX3-FOXO1 and fusion-negative (FN)-RMS [76,77,78]. Moreover, expression of histone deacetylases HDAC4 and HDAC6 negatively correlates with survival of patients with glioblastoma tumors, after RT [79]. Acquired radioresistance was also found associated with an increase of H3K9me3 foci in lung cancers. Interestingly, the treatment of these cancer cells with HDAC-pan inhibitors reduced the number of H3K9me3 foci and promoted radiosensitivity [80]. Radioresistance in breast cancer cells was found correlated with decreased levels of H3K9ac, H3K27ac, and H3S10pK14ac that promoted chromatin compaction, suggesting that an imbalance of HDAC-HAT activity and histone phospho-acetylation could also be implicated in acquired radioresistance [81]. Despite the large number of evidence of the involvement of HDAC in establishing radioresistance, little is known about the role of HATs.
ncRNAs
Several findings have proven that also ncRNAs play a role in determining cancer cell radioresistance [82]. ncRNAs are considered as non-traditional epigenetic regulators of gene expression and indirectly they may regulate heterochromatin formation, histone modification and DNA methylation. Among these, miRNAs have attracted more attention due to their interplay with other epigenetic effectors that are known to affect the RT response. In particular, the crosstalk between miRNAs and DNA methylation has been shown to modulate the radiosensitivity of cancer cells in response to radiation in some tumors. For example, the hypermethylation of the promoters of miRNA tumor suppressors, such as miR-9, miR-24 e miR-21 is associated with radioresistance of glioblastoma multiforme, lung cancer cells and nasopharyngeal carcinoma [83]. This suggests that the identification of miRNAs regulated by methylation might represent a double target to revert radioresistance phenotypes in cancer. lncRNAs, that recruit chromatin-modifying complexes, interact with DNA regulatory sequences or regulated miRNAs, can influence both innate and acquired radioresistance of cancer cells [84, 85]. The lncRNA-mediated regulation of miRNAs has been described as the main mechanism that preserves CSCs after RT in different tumors by modulating the epithelial/mesenchymal transition (EMT) and/or improving DNA repair induced by radiation [86,87,88,89]. As a consequence, knocking down specific lncRNAs can revert radioresistance in certain tumors. Indeed, depletion of NEAT1_2, a lncRNA overexpressed in hepatocellular carcinoma and cervical cancer that acts as a sponge for mirR-101-3p and miR-193b-3p, respectively, radiosensitizes tumor cells allowing both miRNA to suppress the expression of genes that evade radiotherapy effects [90, 91]. In addition to miRNA regulations, lncRNAs can act as scaffold for complexes involved in histone post-translational modifications (PTMs) and this function can affect the tumor cell response to RT. One example is provided by lncRNA PVT1 that recruits the histone acetyltransferase KAT2A and the histone methyltransferase WDRC5 to promote the acetylation of H3K9 [92]. This histone PTM is responsible for the binding of the transcription factor TIF1b and for the induction of NF90 transcription, which has been associated with nasopharyngeal tumor growth in mice after RT. Tumor survival after RT has been also found in colorectal cancer cells as result of concomitant overexpression of lncRNA LINC00630 and EZH2. High levels of both factors lead to gene methylation and (therefore to suppression of expression) of BEX1 determining a poor radiation response, which can be reverted by 5-azacytidine treatment and EZH2 depletion [93].
5 DNA repair and CSC radioresistance: a short overview
A low level of DNA lesions triggers DNA repair mechanisms and DNA damage checkpoints, which arrest cell cycle progression in the presence of DNA damage and allow cells to repair DNA before returning to the proliferative pool. Accumulating preclinical evidence indicates that many of these protective mechanisms are activated in cancer stem cells (CSCs) populations possibly resulting in treatment resistance. CSCs are a subpopulation of cells that exhibit distinctive self-renewal, proliferation, and differentiation features that play a critical role in cancer initiation, maintenance, and progression. It has been proposed that CSCs could be in large part responsible for the inherent radioresistance of a different number of tumors. Furthermore, genetic diversity, epigenetics, and tumor microenvironment are known to control cancer cell stemness, thereby influencing patient response to therapy [94]. Self-defense mechanisms that CSCs use to elude RT include but are not limited to amplification of alternative oncogenes, increased expression of ATP binding cassette (ABC) membrane transporters, overexpression of anti-apoptotic proteins [95]. Particularly relevant is the finding that patient-derived as well as cultured CSCs exhibit a robust DDR as compared to relatively more differentiated malignant cells [8]. After irradiation, resistance correlates with less DNA damage in glioma and breast CSCs. Moreover, enhanced activation of ATM and ATR, as well as their two downstream checkpoint kinase Chk1 and Chk2 has been shown to mediate radioresistance of glioblastoma. CD133+ glioma stem cells elicited also altered expression of NBS1, a component of the MRN complex involved in DSBs sensor activity [96]. Increased expression of DDR factors and other DNA repair genes were also observed in CD133+ CSCs from lung carcinoma, breast carcinoma-inducing cells and pancreatic CSCs. It is believed that enhanced DDR could result from a general adaptation to high levels of DNA replication stress and increased ROS defense which are already active in untreated cells and that would cause and increase activation of DDR after irradiation thereby contributing to tumor radioresistance [97, 98].
Increasing evidence suggests that CSCs radioresistance can be promoted also by dysregulation of miRNAs through constitutive activation of carcinogenic signaling pathways [99, 100]. A high-throughput screen of prostate CSCs revealed that miR-139 and miR-183 could alter the pre-metastatic niche. The expression of miR-20b-5p and miR- 125a-5p, is known to promote the development of radioresistant esophageal carcinomas [101]. Furthermore, miRNAs belonging to the Let-7 family, have been associated to the process of tumorigenesis and resistance [102]. Similarly, some circular RNAs act as regulators of the process of tumor initiation, carcinogenesis, and resistance to radiation. circ_0008344, for example, has been shown to interact with miR-433-3p, regulating the response to radiation in glioma cells [103].
6 Animal models in radioresistance studies
The elucidation of radioresistance mechanisms in cancer pathogenesis has been hindered by limited access to patient samples, tumor heterogeneity, and the difficulty to modeling their biology in appropriate model systems. Performing radiobiology experiments on organisms resistant to radiation consents to appreciate also small variations of molecular pathways altered by radiation. Usually in humans very high exposures as 10–20 Gy can lead to death, thus not always is possible to observe long-term effects [104]. By contrast, using a radioresistant organism the time to observe the IR-induced effects is longer. Studies on analytical structural analysis coupled with next generation sequencing and proteomic approaches of the prokaryotic radiation resistant thermophiles could provide fundamental and conserved molecular insights that will help develop strategies to improve radiation therapies [105]. In addition, these organisms can produce natural compounds that, if isolated and purified, can provide potential therapeutics including anticancer drugs, anti-oxidant etc. [105, 106]. Radiation therapy can also benefit the genomic and proteomic studies on radiation tolerance of tardigrades, tiny invertebrates belonging to the phylum Tardigrada, that possess unique radioresistance features. Exposures of IR at 0.5 or 1 kGy do not affect the survival rate of these animals both in the hydrate and dehydrate states [107], and stress-related tardigrade genes may be transfected to human cells providing increased tolerance to osmotic stress and IR. Thus, the understanding of molecular mechanisms that underlie the radioresistance of tardigrades could be of a pivotal importance for cancer research.
Radiobiology studies also on well-established model organisms, such as the nematode C. elegans and the fruit fly D. melanogaster, have provided useful insights in the identification of factors involved in determining radioresistance. The availability of complete genome sequences and sophisticated genetic/molecular tools, the high level of gene and protein identity with humans, the possibility to conduct genetic screens looking for genetic and molecular interactions render both systems suitable models for most aspect of human medical research including cancer biology [108]. In addition, unique and intrinsic properties elicited by these organisms can provide even more revealing information on important biological mechanisms involved in radioresistance. For example, meiotic pachytene nuclei in C. elegans gonads are hyper-resistant to X-ray irradiation as consequence of high level of expression of enzymes involved in homologous recombination such as the product of RAD51, DMCI (LIM15) homolog 1 (Ce-rad-51), rather than apoptotic exclusion [109]. Interestingly, genome wide RNAi looking for modifiers of hyper resistance of pachytene cells has led to the identification of conserved pathways involved in this response that can provide useful insights in the RT research [110].
Drosophila, like other insects, is more resistant to radiation compared to mammals. Indeed, it has demonstrated that exposure with radiation doses from 10 to 30 Gy does not reduce the survival until adulthood [111]. However, Drosophila is more radiosensitive during development rather than in the adult stage [111, 112]. It has been found that exposure with 40 Gy during the late third-instar larvae enhances the adult mortality of flies compared to unirradiated control [111]. Indeed, although in Drosophila IR exposure eradicates more than half of larval cells, it allows a complete regeneration to produce an adult fly of normal size and pattern [113]. Drosophila is also a well-established animal model to investigate basic principles of tumorigenesis in the developing or ageing organism [114,115,116,117]. The establishment of different brain tumor models also has demonstrated the existence of a complex heterogeneity in the cellular composition of tumor tissues that could shed light on the mechanisms of tumor relapse [118, 119]. In addition, flies have been used to demonstrate that single gene inactivation perturbing the asymmetric divisions of neural stem cells (NSCs), called neuroblasts (NBs) in Drosophila, during development, can rapidly cause NB amplification and aggressive malignant tumors in transplantation assays [120, 121]. Systemic gene targeting in flies bearing tumors can also potentially be exploited in a curative context. Very recently, the ubiquitous loss of the transcription factor SPT5 that physically interacts with MYC oncoproteins and is essential for efficient transcriptional activation of MYC targets, has been demonstrated to extend the lifespan of flies with brain tumors, pointing Spt5 as a potential drug target for human tumors [122].
Drosophila larval imaginal discs, consisting of single-cell layered sacs of epithelial cells that develop into the adult appendages such as eyes and wings, also are frequently used as genetic models for growth control and tumor development. Maintenance of apical-basal polarity of epithelial cells is critical for suppression of neoplastic tumor development [123,124,125]. Disc cell clones can be genetically manipulated in vivo using genetic mosaic techniques. Clones of cells expressing a constitutively activated form of Ras (RasV12) form benign tumors, which develop into malignant tumors if mutations in the apico-basal polarity gene such as scribble (scrib) or discs large (dlg) are present. Interestingly, irradiation of the imaginal disc bearing RasV12 clones, has revealed that RasV12 cells with higher p53 expression support survival of neighboring RasV12 cells with lower p53 expression by inducing the Drosophila JAK/STAT ligands Unpaired1-3Upd, suggesting that cooperation between oncogenic Ras and wild-type p53 stimulates STAT non-cell autonomously to promote tumor radioresistance in a non-cell autonomous manner [126]).
7 The molecular characterization of Radio-adaptive Response (RAR) in Drosophila as a model to investigate radioresistance
The use of Drosophila as a model organism to study DNA repair has been proven to be very valuable to decipher the cellular and molecular mechanisms that control and coordinate the different DNA damage induced responses such as cell cycle arrest, DNA repair, senescence, apoptosis, and radioresistance [127, 128]. Drosophila appear to use the major repair pathways as other organisms and most of the key DNA repair factors are conserved between flies and humans (see Table 1), although in some cases genes that are critical for certain repair processes in other model organisms are not found in Drosophila [129].
We propose that the genetic and molecular characterization of the biological effects of LDR exposures on fruit flies could provide useful insights for understanding the tumor radioresistance in human cells. LDR can induce radio-adaptive response (RAR) that is a protective phenomenon where a small initial low radiation dose (priming) reduces the response to a subsequent high radiation dose (challenging). RAR relies on an efficient DNA repair activity and increased level of antioxidant enzymes [130,131,132]. First discovered in cultured human lymphocytes [133], RAR has been observed in several mammalian systems as well as in zebrafish and Drosophila using various end points such as cell death, chromosome breakage, mutation rate and DNA repair [17, 130, 131]. Moreover, the effects of RAR depend on several factors such as the cell, tissue, animal types, as well as the genetic background. The dose, dose rate, and time period between priming and challenging dose may be crucial for a cell to induce RAR. It is still unclear what dose and dose rate might trigger RAR in humans [132, 134]. This is also complicated by the high degree of inter-individual variation, which in turn depends on the radio-sensitivity of an individual. Thus, the evaluation of whether low radiation doses or dose rates represent a risk estimate factor for radiation protection purposes requires a better understanding of the RAR.
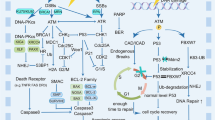
We recently found that the chronic treatment with a priming γ-radiation dose of 0.4 Gy (at dose rate of about 2.5 mGy/h, herein LDR) strongly reduces (~ 50%) the frequency of chromosome breaks occurring at least in S-G2 in third instar larval brains indicating that the LDR triggers a RAR that renders Drosophila mitotic cells more resistant than untreated cells to DNA damage [17]. Our results also indicated that this RAR does not rely on a modulation of apoptosis and that it depends on post-replicative repair mechanisms. Genetic analyses revealed that mutants in DNA damage repair master genes failed to elicit RAR indicating that radioresistance required a canonical repair and DDR. We have demonstrated that radio-adapted cells showed increased levels of Nbs1 and Rad50 proteins as well as an accelerated dynamic of the recruitment of γH2AV (the fly counterpart of mammal γH2AX). Collectively, our data suggest that RAR depends on a more efficient DNA damage repair than to the fact that the chromatin is less sensitive to damage [17]. Finally, by RNA-Seq analysis we found that RAR was associated to a modulation of genes mainly involved in the regulation of non-coding RNAs demonstrating that this response is mediated by epigenetic mechanisms. We found that radioresistant Drosophila mitotic cells exhibit a reduction in the expression of loqs-RD, which encodes an isoform of the fly ortholog of the mammalian Dicer partners TARBP/PACT. The ds-RNA binding protein Loqs-PD interacts with Dicer-2 stimulating this helicase during the nucleolytic processing of hairpin-derived endo-siRNA, cis-Natural Antisense Transcript (NAT)-derived endo-siRNA, as well as exo-siRNAs generated from an inverted repeat transgene [135, 136]. The Dicer2-LoqsPD complex is then required to process esiRNA precursor hairpins with long stems, which results in the production of AGO2-associated small RNAs. DSBs are known to recruit locus-specific siRNAs in different organisms, including flies, to facilitate HR-dependent DNA repair [137, 138]. DNA damage-induced siRNAs in flies are generated upon transcription, which is limited in a region between a transcription start site and the end of the broken DNA. DNA ends at the break act as transcription initiation site that could lead to the generation of antisense transcripts, which ultimately give rise to dsRNA and then siRNAs [139]. However, giving that DNA repair in fruit flies appears independent on siRNA biogenesis [140] and that DNA repair in loqs mutants is not impaired, it can be envisaged that endogenous siRNAs derived from cis-antisense transcripts as well as from other dsRNA sources that are normally converted to siRNA by Loqs-PD, are not directly involved in repairing DSBs in flies. We propose that reduced levels of Loqs-PD lead to the accumulation of esiRNA precursors that could result in a more efficient DNA repair or a more proficient DDR (Fig. 1). A class of esiRNA precursors involved in this effect is represented by cis-NATs as long antisense transcripts originating from a DSB are known to template DSB repair through an RNA:DNA hybrid intermediate [141, 142]. Moreover, cis-NATs can induce chromatin and DNA epigenetic changes and promote chromatin remodeling [143] (Fig. 1). Thus, permanence of cis-NATs would likely favor a more competent activity of DNA repair machinery of either LDR-primed or loqs mutant cells, compared to unprimed cells, thus resulting in a reduction of chromosome breaks following γ-ray acute irradiation (Fig. 1). However, given that Loqs proteins are necessary for the regulation of miRNAs pathways involved in self-renewal of Drosophila stem cells [144, 145], our data could also suggest that RAR is also associated with a modulation of pre-microRNA processing during stem cell maintenance.

Model that explains why Loqs PD depletion protects Drosophila chromosomes from excessive damage. We can speculate that following IR-induced lncRNA transcription (i.e. sense and antisense transcription), the resulting long dsRNAs are processed by a canonical RNAi machinery into Ago2-LoqsPD loaded esiRNA which are able to silence the cognate transcripts. However, as discussed in the text, the contribution of these siRNAs in DNA repair is very limited. Although lncRNA transcription at chromosome breaks in flies results mainly from RNA polymerase II (RNA pol II), the contribution of other RNA polymerase is still debated (RNA pol?). Our recent results indicate that depletion of Loqs PD either as a consequence of LDR treatment or by strong hypomorphic and nulla mutations in Loqs encoding gene, could affect the formation of esiRNAs and in turn determine accumulation of dsRNA precursors which ultimately favor a more efficient DNA repair activity (see text for further details)
Our analysis on LDR treated flies reveals that the radio-adaptive response shares common features with that observed in tumor cells and in CSCs that are resistant to IR. First, in both cases the radioresistance results from an adaptation of cells to stress already in the untreated state. Second, resistance correlates with significantly less DNA damage in both radio-adapted Drosophila neuroblasts and cancer cells [135, 146,147,148,149], which depends on increased activation of the DNA damage response after irradiation rather than on an intrinsic reluctance of chromatin to break. Third, resistance of Drosophila cells and of tumor and/or CSCs manifests during the S phase by HR and involves the accumulation of DDR factors including the components of the MRN complex, which are well conserved between mammals and fruit flies [129] (Table 1). Thus, we are convinced that data obtained from the analysis of radio-adaptive response in flies could provide useful insights for the study of tumor radioresistance and its link to the DNA damage response.
In order to confirm the importance of Loqs-PD in human cancers and its involvement in tumor radioresistance, we firstly analyzed dependency of human cancer cell lines from Loqs-PD human orthologs, PRKRA and TARBP2, using DepMap portal (https://depmap.org/portal/). Our preliminary Chronos score results revealed that although no dependency has been observed for TARBP2 (Data not shown), a large number of cancers (Anaplastic Thyroid cancer, Adenosquamous Pancreatic tumor, Leiomyosarcoma, Liposarcoma, Lung Neuroendocrine tumor, Undifferentiated Pleomorphic Sarcoma and Rhabdoid cancer) exhibited a strong dependency on PRKRA (Fig. 2). The double stranded RNA-activated protein kinase PKR is a mediator of the effects of interferon. It is mainly required for the activation of ElF2AK2/PKR in the absence of dsRNA leading to phosphorylation of ElF2S1/EFl2-alpha and inhibition of translation and induction of apoptosis. It is also involved in the production of siRNAs for post-transcriptional gene silencing. PRKRA mutation is associated with dystonia, but evidence of its involvement in tumors is very limited and only refers to its overexpression [150,151,152]. We then analyzed PRKRA gene expression levels in tumor biopsies from the TCGA human pan-cancer dataset using Ualcan web tool (https://ualcan.path.uab.edu/index.html). Our analysis revealed that, whereas PRKRA expression varies among the tumors analyzed, 7 tumors showed PRKRA down-regulation compared to the normal counterpart, suggesting a putative tumor suppressor function in this subset of human cancers that should be further explored. Interestingly, these tumors included Bladder Cancer [153], Thyroid Carcinoma [154], Kidney Chromophobe Carcinoma [155] and Prostate adenocarcinoma [156], which are well known radioresistant tumors (Fig. 2B). Accordingly, mutational analysis of alterations, obtained with cBioPortal (https://www.cbioportal.org/) (Fig. 2C) in either PRKRA gene or protein, showed that PRKRA is poorly affected by genomic alterations such as mutations and deletions while mRNA downregulation represents a very frequent alteration for this gene in the Prostate adenocarcinoma, Uveal Melanoma, Low-grade glioma, Pheochromocytoma, Bladder Cancer, Thyroid Carcinoma and Kidney Carcinoma tumors that have been also in this case described to be resistant to RT treatments [153,154,155,156,157,158,159]. It is worth noting that low levels of PRKRA mRNA are the most prominent feature of the Kidney Chromophobe Carcinoma tumor, a subtype or radioresistant RCC. Altogether, we think that these preliminary results suggest a potential association between loss of PRKRA and tumor radioresistance. Whether this association can explain the radioresistance of these tumors, it is a task we are currently tackling.

Impact of PRKRA expression on human cancer cell lines and tumor tissue. A Analysis of the dependency from PRKRA expression on 1095 human cancer cell lines performed using DepMap portal (https://depmap.org/portal/). Dependency is reported as Chronos score. B In-depth analysis of PRKRA expression in human cancer biopsies. All data were obtained from TGCA pan-cancer atlas via UALCAN web tool. The statistical analysis performed was One-Way ANOVA. P-values of less than 0.05 were considered significant (***p < 0.001; **p < 0.005; *p < 0.05). C Mutational Profile of PRKRA on human cancer biopsies. Alteration frequency: percentage of patients carrying the indicated alteration. All data were obtained from pan-cancer TGCA atlas using cBioPortal (https://www.cbioportal.org/). BLCA: Bladder urothelial carcinoma; BRCA: Breast invasive carcinoma; CESC: Cervical squamous cell carcinoma; CHOL: Cholangiocarcinoma; COAD: Colon adenocarcinoma; ESCA: Esophageal carcinoma; GBM: Glioblastoma multiforme; HNSC: Head and Neck squamous cell carcinoma; KICH: Kidney chromophobe; KIRC: Kidney renal clear cell carcinoma; KIRP: Kidney renal papillary cell carcinoma; LIHC: Liver hepatocellular carcinoma; LUAD: Lung adenocarcinoma; LUSC: Lung squamous cell carcinoma; PAAD: Pancreatic adenocarcinoma; PRAD: Prostate adenocarcinoma; PCPG: Pheochromocytoma and Paraganglioma; READ: Rectum adenocarcinoma; SARC: Sarcoma; SKCM: Skin cutaneous melanoma; THCA: Thyroid carcinoma; THYM: Thymoma; STAD: Stomach adenocarcinomna; UCEC: Uterine corpus endometrial carcinoma
8 Conclusions
Despite several improvements in cancer treatment, tumor relapse continues to occur in a high proportion of patients after radiation therapy. We have reviewed here the genetic and epigenetic players involved in the enhanced DNA repair of different cancer cells following irradiation. However, for most tumors the events that lead to genome maintenance strategies are not quite clear thus limiting the development of radio-sensitizing approach that could overcome radioresistance. Nevertheless, we proposed that understanding of the genetic and molecular bases that underlie radioresistance in the model organism Drosophila melanogaster, could provide useful insights for the identification of new conserved factors/pathways involved in resistance to radiation. Indeed, our recent characterization of radio-adaptive response in flies has revealed that the RNA binding protein Loqs/PRKRA could be bona fide considered an unprecedented factor involved in tumor radioresistance [17]. These results, along also with our recent observations [160], add to the very large set of evidence that underscore the importance of Drosophila as an attractive invertebrate model organism for human cancer biology [114, 161, 162]. Finally, as the radioadaptive response is part of the non-(DNA)-targeted effects that can be maintained even for long period and are continuously monitored for assessing the risk associated with low dose/low dose rate during human daily life, our results, combined also with our observations on the effects of reduced background radiations [163,164,165] can also provide useful insights for studying the environmental and occupational adaptive response [131, 132, 134].
Data Availability
Not applicable.
References
E. Abad, D. Graifer, A. Lyakhovich, Cancer Lett. 474, 106–117 (2020). https://doi.org/10.1016/j.canlet.2020.01.008
M. Baumann, M. Krause, R. Hill, Nat. Rev. Cancer 8, 545–554 (2008). https://doi.org/10.1038/nrc2419
R. Mohan, D. Grosshans, Adv. Drug Deliv. Rev. 109, 26–44 (2017). https://doi.org/10.1016/j.addr.2016.11.006
D. Schaue, W.H. McBride, Nat. Rev. Clin. Oncol. 12, 527–540 (2015). https://doi.org/10.1038/nrclinonc.2015.120
R.A. Chandra, F.K. Keane, F.E.M. Voncken, C.R. Thomas Jr., Lancet 398, 171–184 (2021). https://doi.org/10.1016/S0140-6736(21)00233-6
R. Yahyapour, E. Motevaseli, A. Rezaeyan, H. Abdollahi, B. Farhood, M. Cheki, S. Rezapoor, D. Shabeeb, A.E. Musa, M. Najafi, V. Villa, Clin. Transl. Oncol. 20, 975–988 (2018). https://doi.org/10.1007/s12094-017-1828-6
F.G. Herrera, P. Romero, G. Coukos, Trends Immunol. 43, 173–179 (2022). https://doi.org/10.1016/j.it.2022.01.006
A. Schulz, F. Meyer, A. Dubrovska, K. Borgmann, Cancers (Basel) 11, (2019). https://doi.org/10.3390/cancers11060862
J.C. Alamilla-Presuel, A.M. Burgos-Molina, A. Gonzalez-Vidal, F. Sendra-Portero, M.J. Ruiz-Gomez, Int. J. Radiat. Biol. 98, 1301–1315 (2022). https://doi.org/10.1080/09553002.2022.2047825
C. Galeaz, C. Totis, A. Bisio, Front. Oncol. 11, 662840 (2021). https://doi.org/10.3389/fonc.2021.662840
H.E. Barker, J.T. Paget, A.A. Khan, K.J. Harrington, Nat. Rev. Cancer 15, 409–425 (2015). https://doi.org/10.1038/nrc3958
K.J. McKelvey, A.L. Hudson, M. Back, T. Eade, C.I. Diakos, Mamm. Genome 29, 843–865 (2018). https://doi.org/10.1007/s00335-018-9777-0
D. Nantajit, D. Lin, J.J. Li, J. Cancer Res. Clin. Oncol. 141, 1697–1713 (2015). https://doi.org/10.1007/s00432-014-1840-y
A. Mittal, M. Nenwani, I. Sarangi, A. Achreja, T.S. Lawrence, D. Nagrath, Trends Cancer 8, 855–869 (2022). https://doi.org/10.1016/j.trecan.2022.05.005
C.M. West, S.E. Davidson, S.A. Elyan, R. Swindell, S.A. Roberts, C.J. Orton, C.A. Coyle, H. Valentine, D.P. Wilks, R.D. Hunter, J.H. Hendry, Int. J. Radiat. Biol. 73, 409–413 (1998). https://doi.org/10.1080/095530098142248
T. Helleday, E. Petermann, C. Lundin, B. Hodgson, R.A. Sharma, Nat. Rev. Cancer 8, 193–204 (2008). https://doi.org/10.1038/nrc2342
A. Porrazzo, F. Cipressa, A. De Gregorio, C. De Pitta, G. Sales, L. Ciapponi, P. Morciano, G. Esposito, M.A. Tabocchini, G. Cenci, Commun. Biol. 5, 905 (2022). https://doi.org/10.1038/s42003-022-03885-w
J. Vignard, G. Mirey, B. Salles, Radiother. Oncol. 108, 362–369 (2013). https://doi.org/10.1016/j.radonc.2013.06.013
B. Zhao, E. Rothenberg, D.A. Ramsden, M.R. Lieber, Nat. Rev. Mol. Cell Biol. 21, 765–781 (2020). https://doi.org/10.1038/s41580-020-00297-8
H.H.Y. Chang, N.R. Pannunzio, N. Adachi, M.R. Lieber, Nat. Rev. Mol. Cell Biol. 18, 495–506 (2017). https://doi.org/10.1038/nrm.2017.48
R. Ceccaldi, J.C. Liu, R. Amunugama, I. Hajdu, B. Primack, M.I. Petalcorin, K.W. O’Connor, P.A. Konstantinopoulos, S.J. Elledge, S.J. Boulton, T. Yusufzai, A.D. D’Andrea, Nature 518, 258–262 (2015). https://doi.org/10.1038/nature14184
W.D. Wright, S.S. Shah, W.D. Heyer, J. Biol. Chem. 293, 10524–10535 (2018). https://doi.org/10.1074/jbc.TM118.000372
M.E. Moynahan, M. Jasin, Nat. Rev. Mol. Cell Biol. 11, 196–207 (2010). https://doi.org/10.1038/nrm2851
L.S. Symington, J. Gautier, Annu. Rev. Genet. 45, 247–271 (2011). https://doi.org/10.1146/annurev-genet-110410-132435
P. Sung, L. Krejci, S. Van Komen, M.G. Sehorn, J. Biol. Chem. 278, 42729–42732 (2003). https://doi.org/10.1074/jbc.R300027200
S. Zhao, S. Tadesse, D. Kidane, Int. Rev. Cell Mol. Biol. 364, 163–193 (2021). https://doi.org/10.1016/bs.ircmb.2021.05.002
D.J. Biechele-Speziale, T.B. Sutton, S. Delaney, DNA Repair (Amst) 116, 103345 (2022). https://doi.org/10.1016/j.dnarep.2022.103345
K.W. Caldecott, Trends Cell Biol. 32, 733–745 (2022). https://doi.org/10.1016/j.tcb.2022.04.010
I. Kamileri, I. Karakasilioti, G.A. Garinis, Trends Genet. 28, 566–573 (2012). https://doi.org/10.1016/j.tig.2012.06.004
J. Jiricny, Cold Spring Harb. Perspect. Biol. 5, a012633 (2013). https://doi.org/10.1101/cshperspect.a012633
G. Sulli, R. Di Micco, F. d’Adda di Fagagna, Nat. Rev. Cancer 12, 709–720 (2012). https://doi.org/10.1038/nrc3344
R. Scully, A. Panday, R. Elango, N.A. Willis, Nat. Rev. Mol. Cell Biol. 20, 698–714 (2019). https://doi.org/10.1038/s41580-019-0152-0
C. Escribano-Diaz, A. Orthwein, A. Fradet-Turcotte, M. Xing, J.T. Young, J. Tkac, M.A. Cook, A.P. Rosebrock, M. Munro, M.D. Canny, D. Xu, D. Durocher, Mol. Cell 49, 872–883 (2013). https://doi.org/10.1016/j.molcel.2013.01.001
P. Bouwman, A. Aly, J.M. Escandell, M. Pieterse, J. Bartkova, H. van der Gulden, S. Hiddingh, M. Thanasoula, A. Kulkarni, Q. Yang, B.G. Haffty, J. Tommiska, C. Blomqvist, R. Drapkin, D.J. Adams, H. Nevanlinna, J. Bartek, M. Tarsounas, S. Ganesan, J. Jonkers, Nat. Struct. Mol. Biol. 17, 688–695 (2010). https://doi.org/10.1038/nsmb.1831
J.E. Jaspers, A. Kersbergen, U. Boon, W. Sol, L. van Deemter, S.A. Zander, R. Drost, E. Wientjens, J. Ji, A. Aly, J.H. Doroshow, A. Cranston, N.M. Martin, A. Lau, M.J. O’Connor, S. Ganesan, P. Borst, J. Jonkers, S. Rottenberg, Cancer Discov. 3, 68–81 (2013). https://doi.org/10.1158/2159-8290.CD-12-0049
J.R. Chapman, P. Barral, J.B. Vannier, V. Borel, M. Steger, A. Tomas-Loba, A.A. Sartori, I.R. Adams, F.D. Batista, S.J. Boulton, Mol. Cell 49, 858–871 (2013). https://doi.org/10.1016/j.molcel.2013.01.002
J.R. Chapman, A.J. Sossick, S.J. Boulton, S.P. Jackson, J. Cell Sci. 125, 3529–3534 (2012). https://doi.org/10.1242/jcs.105353
M. Zimmermann, F. Lottersberger, S.B. Buonomo, A. Sfeir, T. de Lange, Science (New York, N.Y.) 339, 700–704 (2013). https://doi.org/10.1126/science.1231573
S.M. Noordermeer, S. Adam, D. Setiaputra, M. Barazas, S.J. Pettitt, A.K. Ling, M. Olivieri, A. Alvarez-Quilon, N. Moatti, M. Zimmermann, S. Annunziato, D.B. Krastev, F. Song, I. Brandsma, J. Frankum, R. Brough, A. Sherker, S. Landry, R.K. Szilard, M.M. Munro, A. McEwan, T. Goullet de Rugy, Z.Y. Lin, T. Hart, J. Moffat, A.C. Gingras, A. Martin, H. van Attikum, J. Jonkers, C.J. Lord, S. Rottenberg, D. Durocher, Nature 560, 117–121 (2018). https://doi.org/10.1038/s41586-018-0340-7
D. Setiaputra, D. Durocher, EMBO Rep 20, (2019). https://doi.org/10.15252/embr.201847560
P. Zhang, Y. Wei, L. Wang, B.G. Debeb, Y. Yuan, J. Zhang, J. Yuan, M. Wang, D. Chen, Y. Sun, W.A. Woodward, Y. Liu, D.C. Dean, H. Liang, Y. Hu, K.K. Ang, M.C. Hung, J. Chen, L. Ma, Nat. Cell Biol. 16, 864–875 (2014). https://doi.org/10.1038/ncb3013
E. Kaur, M. Ketkar, S. Dutt, Med. Oncol. 39, 50 (2022). https://doi.org/10.1007/s12032-022-01657-4
Y. Chen, Z. Li, Z. Dong, J. Beebe, K. Yang, L. Fu, J.T. Zhang, Mol. Cancer Res. 15, 418–428 (2017). https://doi.org/10.1158/1541-7786.MCR-16-0366
E.J. Yun, C.J. Lin, A. Dang, E. Hernandez, J. Guo, W.M. Chen, J. Allison, N. Kim, P. Kapur, J. Brugarolas, K. Wu, D. He, C.H. Lai, H. Lin, D. Saha, S.T. Baek, B.P.C. Chen, J.T. Hsieh, Clin. Cancer Res. 25, 4542–4551 (2019). https://doi.org/10.1158/1078-0432.CCR-18-3004
Z. Kong, D. Xie, T. Boike, P. Raghavan, S. Burma, D.J. Chen, A.A. Habib, A. Chakraborty, J.T. Hsieh, D. Saha, Cancer Res. 70, 2829–2839 (2010). https://doi.org/10.1158/0008-5472.CAN-09-2919
Y. Zeng, X. Jie, B. Wu, G. Wu, L. Liu, S. Xu, Cancer Lett. 493, 254–265 (2020). https://doi.org/10.1016/j.canlet.2020.08.042
J. Hu, Z. Zhang, L. Zhao, L. Li, W. Zuo, L. Han, BMB Rep. 52, 151–156 (2019). https://doi.org/10.5483/BMBRep.2019.52.2.213
A. Desai, B. Webb, S.L. Gerson, Radiother. Oncol. 110, 538–545 (2014). https://doi.org/10.1016/j.radonc.2013.10.040
A. Pranatharthi, P. Thomas, A.H. Udayashankar, C. Bhavani, S.B. Suresh, S. Krishna, J. Thatte, N. Srikantia, C.R. Ross, S. Srivastava, J. Exp. Clin. Cancer Res. 38, 392 (2019). https://doi.org/10.1186/s13046-019-1385-7
D. Tang, R. Kang, H.J. Zeh, M.T. Lotze, Nat. Rev. Immunol. (2023). https://doi.org/10.1038/s41577-023-00894-6
H. Ma, S. Zheng, X. Zhang, T. Gong, X. Lv, S. Fu, S. Zhang, X. Yin, J. Hao, C. Shan, S. Huang, Cell Death Dis. 10, 136 (2019). https://doi.org/10.1038/s41419-019-1355-1
S. Ke, F. Zhou, H. Yang, Y. Wei, J. Gong, Z. Mei, L. Wu, H. Yu, Y. Zhou, Int. J. Oncol. 46, 1051–1058 (2015). https://doi.org/10.3892/ijo.2014.2793
S. Shrivastava, J.J. Mansure, W. Almajed, F. Cury, G. Ferbeyre, M. Popovic, J. Seuntjens, W. Kassouf, Mol. Cancer Ther. 15, 471–479 (2016). https://doi.org/10.1158/1535-7163.MCT-15-0581
Y. Gao, E. Mutter-Rottmayer, A.M. Greenwalt, D. Goldfarb, F. Yan, Y. Yang, R.C. Martinez-Chacin, K.H. Pearce, S. Tateishi, M.B. Major, C. Vaziri, Nat. Commun. 7, 12105 (2016). https://doi.org/10.1038/ncomms12105
Y. Gao, J. Kardos, Y. Yang, T.Y. Tamir, E. Mutter-Rottmayer, B. Weissman, M.B. Major, W.Y. Kim, C. Vaziri, Sci. Rep. 8, 15304 (2018). https://doi.org/10.1038/s41598-018-33601-w
I. Hickson, Y. Zhao, C.J. Richardson, S.J. Green, N.M. Martin, A.I. Orr, P.M. Reaper, S.P. Jackson, N.J. Curtin, G.C. Smith, Cancer Res. 64, 9152–9159 (2004). https://doi.org/10.1158/0008-5472.CAN-04-2727
J.H.L. Fok, A. Ramos-Montoya, M. Vazquez-Chantada, P.W.G. Wijnhoven, V. Follia, N. James, P.M. Farrington, A. Karmokar, S.E. Willis, J. Cairns, J. Nikkila, D. Beattie, G.M. Lamont, M.R.V. Finlay, J. Wilson, A. Smith, L.O. O’Connor, S. Ling, S.E. Fawell, M.J. O’Connor, S.J. Hollingsworth, E. Dean, F.W. Goldberg, B.R. Davies, E.B. Cadogan, Nat. Commun. 10, 5065 (2019). https://doi.org/10.1038/s41467-019-12836-9
P.T. Ferrao, E.P. Bukczynska, R.W. Johnstone, G.A. McArthur, Oncogene 31, 1661–1672 (2012). https://doi.org/10.1038/onc.2011.358
K. Brooks, V. Oakes, B. Edwards, M. Ranall, P. Leo, S. Pavey, A. Pinder, H. Beamish, P. Mukhopadhyay, D. Lambie, B. Gabrielli, Oncogene 32, 788–796 (2013). https://doi.org/10.1038/onc.2012.72
S.X. Pfister, E. Markkanen, Y. Jiang, S. Sarkar, M. Woodcock, G. Orlando, I. Mavrommati, C.C. Pai, L.P. Zalmas, N. Drobnitzky, G.L. Dianov, C. Verrill, V.M. Macaulay, S. Ying, N.B. La Thangue, V. D’Angiolella, A.J. Ryan, T.C. Humphrey, Cancer Cell 28, 557–568 (2015). https://doi.org/10.1016/j.ccell.2015.09.015
S. Rey, L. Schito, M. Koritzinsky, B.G. Wouters, Adv. Drug Deliv. Rev. 109, 45–62 (2017). https://doi.org/10.1016/j.addr.2016.10.002
S. Wang, P. Zhao, C. Zhang, Y. Bu, J. Phys. Chem. B 120, 2649–2657 (2016). https://doi.org/10.1021/acs.jpcb.5b11432
C. Lhuillier, N.P. Rudqvist, O. Elemento, S.C. Formenti, S. Demaria, Genome Med 11, 40 (2019). https://doi.org/10.1186/s13073-019-0653-7
L. Bourillon, C. Bourgier, N. Gaborit, V. Garambois, E. Lles, A. Zampieri, C. Ogier, M. Jarlier, N. Radosevic-Robin, B. Orsetti, H. Delpech, C. Theillet, P.E. Colombo, D. Azria, A. Pelegrin, C. Larbouret, T. Chardes, Int. J. Cancer 145, 1838–1851 (2019). https://doi.org/10.1002/ijc.32273
Y. Mi, Z. Shao, J. Vang, O. Kaidar-Person, A.Z. Wang, Cancer Nanotechnol 7, 11 (2016). https://doi.org/10.1186/s12645-016-0024-7
M. Babaei, M. Ganjalikhani, Bioimpacts 4, 15–20 (2014). https://doi.org/10.5681/bi.2014.003
H.E. Bryant, N. Schultz, H.D. Thomas, K.M. Parker, D. Flower, E. Lopez, S. Kyle, M. Meuth, N.J. Curtin, T. Helleday, Nature 434, 913–917 (2005). https://doi.org/10.1038/nature03443
Z.I. Hu, A.Y. Ho, H.L. McArthur, Int. J. Radiat. Oncol. Biol. Phys. 99, 153–164 (2017). https://doi.org/10.1016/j.ijrobp.2017.05.029
K.M. Smits, V. Melotte, H.E. Niessen, L. Dubois, C. Oberije, E.G. Troost, M.H. Starmans, P.C. Boutros, M. Vooijs, M. van Engeland, P. Lambin, Radiother. Oncol. 111, 168–177 (2014). https://doi.org/10.1016/j.radonc.2014.05.001
E.H. Kim, A.K. Park, S.M. Dong, J.H. Ahn, W.Y. Park, Oncogene 29, 4725–4731 (2010). https://doi.org/10.1038/onc.2010.223
G. Zhai, J. Li, J. Zheng, P. An, X. Chen, X. Wang, C. Li, J. Radiat. Res. 61, 674–683 (2020). https://doi.org/10.1093/jrr/rraa052
C. Wu, E. Guo, J. Ming, W. Sun, X. Nie, L. Sun, S. Peng, M. Luo, D. Liu, L. Zhang, Q. Mei, G. Long, G. Hu, G. Hu, Mol. Ther. Oncolytics 17, 306–319 (2020). https://doi.org/10.1016/j.omto.2020.04.007
S. Camero, G. Vitali, P. Pontecorvi, S. Ceccarelli, E. Anastasiadou, F. Cicchetti, E. Flex, S. Pomella, M. Cassandri, R. Rota, F. Marampon, C. Marchese, A. Schiavetti, F. Megiorni, Cells 10, (2021). https://doi.org/10.3390/cells10112956
S.P. Zielske, J. Cell. Biochem. 116, 212–217 (2015). https://doi.org/10.1002/jcb.24959
E. Kaur, J. Nair, A. Ghorai, S.V. Mishra, A. Achareker, M. Ketkar, D. Sarkar, S. Salunkhe, J. Rajendra, N. Gardi, S. Desai, P. Iyer, R. Thorat, A. Dutt, A. Moiyadi, S. Dutt, Neuro Oncol. 22, 1785–1796 (2020). https://doi.org/10.1093/neuonc/noaa128
A. Rossetti, F. Petragnano, L. Milazzo, F. Vulcano, G. Macioce, S. Codenotti, M. Cassandri, S. Pomella, F. Cicchetti, I. Fasciani, C. Antinozzi, L. Di Luigi, C. Festuccia, F. De Felice, M. Vergine, A. Fanzani, R. Rota, R. Maggio, A. Polimeni, V. Tombolini, G.L. Gravina, F. Marampon, Int. J. Radiat. Biol. 97, 943–957 (2021). https://doi.org/10.1080/09553002.2021.1928786
F. Marampon, V. Di Nisio, I. Pietrantoni, F. Petragnano, I. Fasciani, B.M. Scicchitano, C. Ciccarelli, G.L. Gravina, C. Festuccia, A. Del Fattore, M. Tombolini, F. De Felice, D. Musio, S. Cecconi, P. Tini, M. Maddalo, S. Codenotti, A. Fanzani, A. Polimeni, R. Maggio, V. Tombolini, Cancer Lett. 461, 90–101 (2019). https://doi.org/10.1016/j.canlet.2019.07.009
M. Cassandri, S. Pomella, A. Rossetti, F. Petragnano, L. Milazzo, F. Vulcano, S. Camero, S. Codenotti, F. Cicchetti, R. Maggio, C. Festuccia, G.L. Gravina, A. Fanzani, F. Megiorni, M. Catanoso, C. Marchese, V. Tombolini, F. Locatelli, R. Rota, F. Marampon, Int J Mol Sci 22, (2021). https://doi.org/10.3390/ijms221910671
F. Marampon, F. Megiorni, S. Camero, C. Crescioli, H.P. McDowell, R. Sferra, A. Vetuschi, S. Pompili, L. Ventura, F. De Felice, V. Tombolini, C. Dominici, R. Maggio, C. Festuccia, G.L. Gravina, Cancer Lett. 397, 1–11 (2017). https://doi.org/10.1016/j.canlet.2017.03.028
P. Wang, D. Yuan, F. Guo, X. Chen, L. Zhu, H. Zhang, C. Wang, C. Shao, Oncotarget 8, 52823–52836 (2017). https://doi.org/10.18632/oncotarget.17275
A. Sharda, M. Rashid, S.G. Shah, A.K. Sharma, S.R. Singh, P. Gera, M.K. Chilkapati, S. Gupta, Clin. Epigenetics 12, 4 (2020). https://doi.org/10.1186/s13148-019-0800-4
M. Podralska, S. Ciesielska, J. Kluiver, A. van den Berg, A. Dzikiewicz-Krawczyk, I. Slezak-Prochazka, Cancers (Basel) 12, (2020). https://doi.org/10.3390/cancers12061662
S. Wang, R. Zhang, F.X. Claret, H. Yang, Mol. Cancer Ther. 13, 3163–3174 (2014). https://doi.org/10.1158/1535-7163.MCT-14-0317
B. Ricciuti, C. Mencaroni, L. Paglialunga, F. Paciullo, L. Crino, R. Chiari, G. Metro, Med. Oncol. 33, 18 (2016). https://doi.org/10.1007/s12032-016-0731-2
J. Kozlowska-Maslon, K. Guglas, A. Paszkowska, T. Kolenda, M. Podralska, A. Teresiak, R. Blizniak, K. Lamperska, J Pers Med 12, (2022). https://doi.org/10.3390/jpm12101605
L. Bie, S. Luo, D. Li, Y. Wei, Y. Mu, X. Chen, S. Wang, P. Guo, X. Lu, Curr. Cancer Drug Targets 20, 700–709 (2020). https://doi.org/10.2174/1568009620666200504114000
Z. Guo, Y.H. Wang, H. Xu, C.S. Yuan, H.H. Zhou, W.H. Huang, H. Wang, W. Zhang, Cell Death Dis. 12, 69 (2021). https://doi.org/10.1038/s41419-020-03302-2
L. Qian, Q. Fei, H. Zhang, M. Qiu, B. Zhang, Q. Wang, Y. Yu, C. Guo, Y. Ren, M. Mei, L. Zhang, Y. Zhu, B. Yang, DNA Cell Biol. (2020). https://doi.org/10.1089/dna.2020.5771
Y.H. Wang, Z. Guo, L. An, Y. Zhou, H. Xu, J. Xiong, Z.Q. Liu, X.P. Chen, H.H. Zhou, X. Li, T. Liu, W.H. Huang, W. Zhang, Cell Death Dis. 12, 454 (2021). https://doi.org/10.1038/s41419-021-03728-2
C. Klec, F. Prinz, M. Pichler, Mol. Oncol. 13, 46–60 (2019). https://doi.org/10.1002/1878-0261.12404
H. Zhou, Y. Wang, Z. Liu, Z. Zhang, L. Xiong, Y. Wen, Acta Biochim. Biophys. Sin. (Shanghai) 54, 153–162 (2022). https://doi.org/10.3724/abbs.2021022
Y. Wang, W. Chen, J. Lian, H. Zhang, B. Yu, M. Zhang, F. Wei, J. Wu, J. Jiang, Y. Jia, F. Mo, S. Zhang, X. Liang, X. Mou, J. Tang, Cell Death Differ. 27, 695–710 (2020). https://doi.org/10.1038/s41418-019-0381-y
F. Liu, W. Huang, J. Hong, C. Cai, W. Zhang, J. Zhang, Z. Kang, IUBMB Life 72, 1404–1414 (2020). https://doi.org/10.1002/iub.2263
P.L. Bedard, A.R. Hansen, M.J. Ratain, L.L. Siu, Nature 501, 355–364 (2013). https://doi.org/10.1038/nature12627
H. Zahreddine, K.L. Borden, Front. Pharmacol. 4, 28 (2013). https://doi.org/10.3389/fphar.2013.00028
L. Cheng, Q. Wu, Z. Huang, O.A. Guryanova, Q. Huang, W. Shou, J.N. Rich, S. Bao, EMBO J. 30, 800–813 (2011). https://doi.org/10.1038/emboj.2011.10
R.D. Carruthers, S.U. Ahmed, S. Ramachandran, K. Strathdee, K.M. Kurian, A. Hedley, N. Gomez-Roman, G. Kalna, M. Neilson, L. Gilmour, K.H. Stevenson, E.M. Hammond, A.J. Chalmers, Cancer Res. 78, 5060–5071 (2018). https://doi.org/10.1158/0008-5472.CAN-18-0569
M. Diehn, R.W. Cho, N.A. Lobo, T. Kalisky, M.J. Dorie, A.N. Kulp, D. Qian, J.S. Lam, L.E. Ailles, M. Wong, B. Joshua, M.J. Kaplan, I. Wapnir, F.M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S.K. Lau, S.R. Quake, J.M. Brown, I.L. Weissman, M.F. Clarke, Nature 458, 780–783 (2009). https://doi.org/10.1038/nature07733
Y. Zhao, D. Wei, Y. Zhang, J. Ji, Essays Biochem. 66, 345–358 (2022). https://doi.org/10.1042/EBC20220007
K. Yoshida, Y. Yamamoto, T. Ochiya, Regen. Ther. 17, 1–7 (2021). https://doi.org/10.1016/j.reth.2021.01.004
D. Chen, H. Su, Y. Li, X. Wu, Y. Li, C. Wei, D. Shi, Y. Gao, Q. Zhou, Q. Wang, X. Jin, C. Xie, Aging (Albany NY) 13, 9566–9581 (2021). https://doi.org/10.18632/aging.202690
Y. Ma, N. Shen, M.S. Wicha, M. Luo, Cells 10, (2021). https://doi.org/10.3390/cells10092415
L. Di, X. Zhao, J. Ding, J. Biosci. 46, (2021). https://doi.org/10.1007/s12038-021-00198-8
K. Kamiya, K. Ozasa, S. Akiba, O. Niwa, K. Kodama, N. Takamura, E.K. Zaharieva, Y. Kimura, R. Wakeford, Lancet 386, 469–478 (2015). https://doi.org/10.1016/S0140-6736(15)61167-9
A. Pavlopoulou, G.D. Savva, M. Louka, P.G. Bagos, C.E. Vorgias, I. Michalopoulos, A.G. Georgakilas, Mutat. Res. Rev. Mutat. Res. 767, 92–107 (2016). https://doi.org/10.1016/j.mrrev.2015.10.001
O.V. Singh, P. Gabani, J. Appl. Microbiol. 110, 851–861 (2011). https://doi.org/10.1111/j.1365-2672.2011.04971.x
K.I. Jonsson, I. Holm, H. Tassidis, Results Probl. Cell Differ. 68, 231–249 (2019). https://doi.org/10.1007/978-3-030-23459-1_10
C.H. Stuelten, C.A. Parent, D.J. Montell, Nat. Rev. Cancer 18, 296–312 (2018). https://doi.org/10.1038/nrc.2018.15
T. Takanami, A. Mori, H. Takahashi, A. Higashitani, Nucleic Acids Res. 28, 4232–4236 (2000). https://doi.org/10.1093/nar/28.21.4232
G. van Haaften, R. Romeijn, J. Pothof, W. Koole, L.H. Mullenders, A. Pastink, R.H. Plasterk, M. Tijsterman, Curr. Biol. 16, 1344–1350 (2006). https://doi.org/10.1016/j.cub.2006.05.047
L.J. Sudmeier, S.P. Howard, B. Ganetzky, Dis. Model. Mech. 8, 669–677 (2015). https://doi.org/10.1242/dmm.019786
M. Vaisnav, C. Xing, H.C. Ku, D. Hwang, S. Stojadinovic, A. Pertsemlidis, J.M. Abrams, PLoS ONE 9, e104858 (2014). https://doi.org/10.1371/journal.pone.0104858
T.T. Su, Adv. Exp. Med. Biol. 1167, 225–236 (2019). https://doi.org/10.1007/978-3-030-23629-8_13
C. Gonzalez, Nat. Rev. Cancer 13, 172–183 (2013). https://doi.org/10.1038/nrc3461
K. Siudeja, S. Nassari, L. Gervais, P. Skorski, S. Lameiras, D. Stolfa, M. Zande, V. Bernard, T.R. Frio, A.J. Bardin, Cell Stem Cell 17, 663–674 (2015). https://doi.org/10.1016/j.stem.2015.09.016
S.R. Singh, P. Aggarwal, S.X. Hou, Adv. Exp. Med. Biol. 1167, 175–190 (2019). https://doi.org/10.1007/978-3-030-23629-8_10
S. Li, H. Wang, C. Groth, Biosci. Rep. 34, (2014).https://doi.org/10.1042/BSR20140008
S. Genovese, R. Clement, C. Gaultier, F. Besse, K. Narbonne-Reveau, F. Daian, S. Foppolo, N.M. Luis, C. Maurange, Elife 8, (2019). https://doi.org/10.7554/eLife.50375
K. Narbonne-Reveau, E. Lanet, C. Dillard, S. Foppolo, C.H. Chen, H. Parrinello, S. Rialle, N.S. Sokol, C. Maurange, Elife 5, (2016). https://doi.org/10.7554/eLife.13463
J.A. Knoblich, Nat. Rev. Mol. Cell Biol. 11, 849–860 (2010). https://doi.org/10.1038/nrm3010
E. Caussinus, C. Gonzalez, Nat. Genet. 37, 1125–1129 (2005). https://doi.org/10.1038/ng1632
J. Hofstetter, A. Ogunleye, A. Kutschke, L.M. Buchholz, E. Wolf, T. Raabe, P. Gallant, Life Sci. Alliance 7, (2024). https://doi.org/10.26508/lsa.202302130
I. Elsum, L. Yates, P.O. Humbert, H.E. Richardson, Essays Biochem. 53, 141–168 (2012). https://doi.org/10.1042/bse0530141
D.T. Bergstralh, D. St Johnston, Essays Biochem. 53, 129–140 (2012). https://doi.org/10.1042/bse0530129
F. Martin-Belmonte, M. Perez-Moreno, Nat. Rev. Cancer 12, 23–38 (2011). https://doi.org/10.1038/nrc3169
Y.L. Dong, G.P. Vadla, J.J. Lu, V. Ahmad, T.J. Klein, L.F. Liu, P.M. Glazer, T. Xu, C.Y. Chabu, Commun. Biol. 4, 374 (2021). https://doi.org/10.1038/s42003-021-01898-5
C. Khan, S. Muliyil, B.J. Rao, Int. Rev. Cell Mol. Biol. 345, 173–224 (2019). https://doi.org/10.1016/bs.ircmb.2018.12.001
Y.H. Song, Mol. Cells 19, 167–179 (2005)
J. Sekelsky, Genetics 205, 471–490 (2017). https://doi.org/10.1534/genetics.116.186759
G. Esposito, A. Campa, M. Pinto, G. Simone, M.A. Tabocchini, M. Belli, Radiat. Prot. Dosimetry. 143, 320–324 (2011). https://doi.org/10.1093/rpd/ncq474
M. Nenoi, B. Wang, G. Vares, Hum. Exp. Toxicol. 34, 272–283 (2015). https://doi.org/10.1177/0960327114537537
S. Tapio, V. Jacob, Radiat. Environ. Biophys. 46, 1–12 (2007). https://doi.org/10.1007/s00411-006-0078-8
G. Olivieri, J. Bodycote, S. Wolff, Science (New York, N.Y.) 223, 594–597 (1984). https://doi.org/10.1126/science.6695170
Y. Shibamoto, H. Nakamura, Int. J. Mol. Sci. 19, (2018). https://doi.org/10.3390/ijms19082387
R. Fukunaga, B.W. Han, J.H. Hung, J. Xu, Z. Weng, P.D. Zamore, Cell 151, 912 (2012). https://doi.org/10.1016/j.cell.2012.10.029
J.V. Hartig, K. Forstemann, Nucleic Acids Res. 39, 3836–3851 (2011). https://doi.org/10.1093/nar/gkq1324
W. Wei, Z. Ba, M. Gao, Y. Wu, Y. Ma, S. Amiard, C.I. White, J.M. Rendtlew Danielsen, Y.G. Yang, Y. Qi, Cell 149, 101–112 (2012). https://doi.org/10.1016/j.cell.2012.03.002
M. Gao, W. Wei, M.M. Li, Y.S. Wu, Z. Ba, K.X. Jin, M.M. Li, Y.Q. Liao, S. Adhikari, Z. Chong, T. Zhang, C.X. Guo, T.S. Tang, B.T. Zhu, X.Z. Xu, N. Mailand, Y.G. Yang, Y. Qi, J.M. Rendtlew Danielsen, Cell Res 24, 532–541 (2014). https://doi.org/10.1038/cr.2014.36
K.M. Michalik, R. Bottcher, K. Forstemann, Nucleic Acids Res. 40, 9596–9603 (2012). https://doi.org/10.1093/nar/gks711
C.D. Malone, G.J. Hannon, Cell 136, 656–668 (2009). https://doi.org/10.1016/j.cell.2009.01.045
H. Keskin, Y. Shen, F. Huang, M. Patel, T. Yang, K. Ashley, A.V. Mazin, F. Storici, Nature 515, 436–439 (2014). https://doi.org/10.1038/nature13682
H. Keskin, C. Meers, F. Storici, RNA Biol. 13, 157–165 (2016). https://doi.org/10.1080/15476286.2015.1116676
M.A. Faghihi, C. Wahlestedt, Nat. Rev. Mol. Cell Biol. 10, 637–643 (2009). https://doi.org/10.1038/nrm2738
J.K. Park, X. Liu, T.J. Strauss, D.M. McKearin, Q. Liu, Curr. Biol. 17, 533–538 (2007). https://doi.org/10.1016/j.cub.2007.01.060
K. Forstemann, Y. Tomari, T. Du, V.V. Vagin, A.M. Denli, D.P. Bratu, C. Klattenhoff, W.E. Theurkauf, P.D. Zamore, PLoS Biol. 3, e236 (2005). https://doi.org/10.1371/journal.pbio.0030236
S. Bao, Q. Wu, R.E. McLendon, Y. Hao, Q. Shi, A.B. Hjelmeland, M.W. Dewhirst, D.D. Bigner, J.N. Rich, Nature 444, 756–760 (2006). https://doi.org/10.1038/nature05236
I. Vitale, G. Kroemer, Cell Res. 27, 720–721 (2017). https://doi.org/10.1038/cr.2017.43
I. Vitale, G. Manic, R. De Maria, G. Kroemer, L. Galluzzi, Mol. Cell 66, 306–319 (2017). https://doi.org/10.1016/j.molcel.2017.04.006
T.M. Phillips, W.H. McBride, F. Pajonk, J. Natl. Cancer Inst. 98, 1777–1785 (2006). https://doi.org/10.1093/jnci/djj495
F. Wang, L. Zhu, Q. Xue, C. Tang, W. Tang, N. Zhang, C. Dai, Z. Chen, Oxid. Med. Cell. Longev. 2022, 1125932 (2022). https://doi.org/10.1155/2022/1125932
T. Hisamatsu, M. McGuire, S.Y. Wu, R. Rupaimoole, S. Pradeep, E. Bayraktar, K. Noh, W. Hu, J.M. Hansen, Y. Lyons, K.M. Gharpure, A.S. Nagaraja, L.S. Mangala, T. Mitamura, C. Rodriguez-Aguayo, Y.G. Eun, J. Rose, G. Bartholomeusz, C. Ivan, J.S. Lee, K. Matsuo, M. Frumovitz, K.K. Wong, G. Lopez-Berestein, A.K. Sood, Mol. Cancer Ther. 18, 162–172 (2019). https://doi.org/10.1158/1535-7163.MCT-17-1050
J. Qiu, M. Feng, G. Yang, D. Su, F. Zhao, Y. Liu, J. Tao, W. Luo, T. Zhang, Heliyon 9, e17194 (2023). https://doi.org/10.1016/j.heliyon.2023.e17194
A. Mehmandar-Oskuie, K. Jahankhani, A. Rostamlou, S. Arabi, Z. Sadat Razavi, A. Mardi, Biomed. Pharmacother. 165, 115242 (2023). https://doi.org/10.1016/j.biopha.2023.115242
R. Robb, L. Yang, C. Shen, A.R. Wolfe, A. Webb, X. Zhang, M. Vedaie, M. Saji, S. Jhiang, M.D. Ringel, T.M. Williams, Clin. Cancer Res. 25, 4749–4760 (2019). https://doi.org/10.1158/1078-0432.CCR-18-3625
W.F. Mourad, J. Dutcher, R.D. Ennis, Am. J. Clin. Oncol. 37, 498–505 (2014). https://doi.org/10.1097/COC.0b013e31825d5522
L. Chaiswing, H.L. Weiss, R.D. Jayswal, D.K.S. Clair, N. Kyprianou, Crit. Rev. Oncog. 23, 39–67 (2018). https://doi.org/10.1615/CritRevOncog.2018025946
S. Grynberg, R. Stoff, N. Asher, R. Shapira-Frommer, J. Schachter, O. Haisraely, Y. Lawrence, G. Ben-Betzalel, Ther. Adv. Med. Oncol. 14, 17588359221131520 (2022). https://doi.org/10.1177/17588359221131521
W. Lin, Z. Huang, Y. Xu, X. Chen, T. Chen, Y. Ye, J. Ding, Z. Chen, L. Chen, X. Qiu, S. Qiu, Aging (Albany NY) 12, 9188–9204 (2020). https://doi.org/10.18632/aging.103189
V. Seifert, S. Richter, N. Bechmann, M. Bachmann, C.G. Ziegler, J. Pietzsch, M. Ullrich, Cancers (Basel) 13, (2021). https://doi.org/10.3390/cancers13030385
G. Bosso, F. Cipressa, L. Tullo, G. Cenci, Cell Death Discov. 9, 317 (2023). https://doi.org/10.1038/s41420-023-01598-5
D. Chatterjee, W.M. Deng, Adv. Exp. Med. Biol. 1167, 1–14 (2019). https://doi.org/10.1007/978-3-030-23629-8_1
M. Sonoshita, R.L. Cagan, Curr. Top. Dev. Biol. 121, 287–309 (2017). https://doi.org/10.1016/bs.ctdb.2016.07.008
P. Morciano, F. Cipressa, A. Porrazzo, G. Esposito, M.A. Tabocchini, G. Cenci, Radiat. Res. 190, 217–225 (2018). https://doi.org/10.1667/RR15083.1
P. Morciano, R. Iorio, D. Iovino, F. Cipressa, G. Esposito, A. Porrazzo, L. Satta, E. Alesse, M.A. Tabocchini, G. Cenci, J. Cell. Physiol. 233, 23–29 (2018). https://doi.org/10.1002/jcp.25889
A. Porrazzo, G. Esposito, D. Grifoni, G. Cenci, P. Morciano, M.A. Tabocchini, Int. J. Mol. Sci. 23, (2022). https://doi.org/10.3390/ijms23105472
Author information
Authors and Affiliations
Contributions
A.P., M.C., wrote part of the main manuscript text and prepared Figure 2 A.D. prepared Figure 1 and Table 1 All authors reviewed the manuscript
Corresponding author
Ethics declarations
This manuscript is a review article and does not involve a research protocol requiring approval by the relevant institutional review board or ethics committee.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Porrazzo, A., Cassandri, M., D’Alessandro, A. et al. DNA repair in tumor radioresistance: insights from fruit flies genetics. Cell Oncol. 47, 717–732 (2024). https://doi.org/10.1007/s13402-023-00906-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-023-00906-6