
Abstract
Purpose
Human head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide. Currently, surgical resection plus a combination of chemotherapy and radiotherapy is the standard treatment for HNSCC, and the 5-year survival rate of patients with HNSCC remains very low because of the higher incidence of metastasis with consequent recurrence. Here, we aimed to investigate the potential role of DNA N6-methyladenine (6mA) demethylase ALKBH1 in tumor cell proliferation in HNSCC.
Methods
The expression of ALKBH1 in 10 pairs of HNSCC/normal tissues and 3 HNSCC cell lines were measured by qRT‒PCR and western blotting. Colony formation, flow cytometry, patient-derived HNSCC organoid assays were used to assess the role of ALKBH1 in HNSCC cell proliferation in cell lines and human HNSCC patients. MeDIP-seq, RNA sequencing, Dot blotting and western blotting were used to evaluate the regulatory effect of ALKBH1 on the expression of DEAD-box RNA helicase DDX18. A dual-luciferase reporter assay was used to assess the putative effect of DNA 6mA levels on DDX18 transcription.
Results
ALKBH1 was highly expressed in HNSCC cells and patient tissues. Functional experiments revealed that ALKBH1 knockdown in SCC9, SCC25, and CAL27 cells inhibited their proliferation in vitro. Using patient-derived HNSCC organoid assay, we found that knockdown of ALKBH1 inhibited the proliferation and colony formation of HNSCC patients-derived organoids. Moreover, we found that ALKBH1 can enhance DDX18 expression by erasing DNA 6mA level and regulating its promoter activity. ALKBH1 deficiency blocked tumor cell proliferation by inhibiting DDX18 expression. Exogenous overexpression of DDX18 rescued the cell proliferation arrest caused by ALKBH1 knockdown.
Conclusion
Our data reveal the important role of ALKBH1 in regulating proliferation of HNSCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Head and neck squamous cell carcinoma (HNSCC) is the most common malignancy that arises from the transformed mucosal epithelium in the oral cavity, pharynx, and larynx [1]. Surgical resection, chemotherapy, and radiotherapy are the standard treatments for patients with HNSCC in clinic [2,3,4]. However, high incidence of metastasis associated with recurrence frequently occurs, and the 5-year survival rate of patients with HNSCC remains low [2, 5,6,7]. Thus, it is critical to explore the underlying mechanisms for HNSCC metastasis, which might contribute to developing potential strategies to block the metastasis and HNSCC treatment.
AlkB members are known as the 2-oxoglutarate (2OG) and Fe(II)-dependent dioxygenase superfamily [8], and nine homologs including ALKBH 1–8 and FTO (fat mass and obesity-associated protein) are encoding in human genome [9]. They involve in various biological processes, such as DNA repair, RNA metabolism, histone demethylation, or fatty acid metabolism [10]. Overexpression of different ALKBH members has been observed in various cancer types [11,12,13,14,15,16]. For instance, higher expression of ALKBH2 is detected in bladder cancer, and its knockdown suppresses tumor development. Interestingly, ALKBH2 downregulation also sensitizes glioma cells to alkylating agents [17, 18]. Our previous work reveals a key role of ALKBH5 in acute myeloid leukemia [19]. Recent works demonstrate that ALKBH1 functions as a demethylase of DNA N6-methyladenine (6mA) [20, 21], and involves in many physiological and pathological conditions [22]. ALKBH1-demethylated DNA 6mA modification triggers vascular calcification via osteogenic reprogramming in chronic kidney disease [23]. ALKBH1 also promotes adiponeic differentiation via regulation of HIF-1 signaling [24]. However, the roles of ALKBH1 in HNSCC remains elusive. In this study, we observe high expression of ALKBH1 in patients with HNSCC, and find that ALKBH1 promotes HNSCC proliferation by regulating the expression of DEAD-box RNA helicase DDX18 in a DNA 6mA-dependent manner. Overall, this work uncovers a new role of ALBKH1 in HNSCC.
2 Materials and Methods
2.1 Human HNSCC samples and cell lines
Human HNSCC tumors and normal oral mucosal tissues were obtained from the Hospital of Stomatology of Wuhan University. All experiments involving human samples were conducted in compliance with all relevant ethical regulations, and were approved by the Medical Ethics Committees of the School of Medicine, Wuhan University.
HNSCC patient-derived cell lines SCC25, SCC9, and CAL27 were maintained in DMEM containing 10% FBS and antibiotics (streptomycin and penicillin) at 37 °C in a 5% CO2 atmosphere.
2.2 Patient-derived HNSCC organoids culture
Patient-derived HNSCC organoids culture was performed as previously described with minor modifications [25].Patient material was collected from pathology material in Advanced DMEM +/+/+ medium (Advanced DMEM/F12, supplemented with 1×GlutaMAX, 1% Penicillin–streptomycin, and 10 mmol/L HEPES). For collection of patient material, 100 µg/mL Primocin was added to the Advanced DMEM +/+/+ medium. For normal tissue samples, excess fat or muscle tissue was removed to enrich for epithelial cells, and tissue was cut into small fragments. Fragments were incubated at 37 °C in 0.125% Trypsin in the Advanced DMEM +/+/+ medium until digested. Every 10 min, the tissue suspension was sheared using 1 mL pipette. Digestion was monitored closely to prevent excess incubation in trypsin. Incubation was performed for a maximum of 60 min. When complete, Trypsin was diluted by addition of 10 mL the Advanced DMEM +/+/+ medium. Suspension was strained over a 100 µ m Easy Strainer filter and centrifuged at 1,000 rpm. The resulting pellet was resuspended in ice-cold 70% 10 mg/mL cold Cultrex growth factor–reduced BME type 2 in human organoid medium (1 × B27 supplement, 1.25 mmol/L N-acetyl-l-cysteine, 10 mmol/L Nicotinamide, 50 ng/mL human EGF, 500 nM A83-01, 10 ng/mL human FGF10, 5 ng/mL human FGF2, 1 µM Prostaglandin E2, 0.3 µM CHIR 99,021, 1 µM Forskolin, 20% R-spondin and Noggin conditioned medium). Droplets of approximately 10 µL were plated on the bottom of preheated suspension culture plates. After plating, plates were inverted and put at 37 °C for 30 min to let the BME solidify. Subsequently, pre-warmed organoid medium was added to the plate. For the first week, 10 µM Rho-associated kinase (ROCK) inhibitor Y-27,632 was added to the medium to aid outgrowth of organoids for the primary tissue.
Organoids were split between 7 and 14 days after initial plating. For passaging, organoids were collected from the plate by disrupting the BME droplets with a P1000, collecting and washing in 10 mL +/+/+. Pellet was resuspended in 1 mL of TrypLE Express and incubated at 37 °C. Digestion was closely monitored and suspension was pipetted up and down every 5 min to aid disruption of the organoids. TrypLE digestion was stopped when organoids were disrupted into single cells by adding 10 mL +/+/+. Cells were subsequently re-suspended in ice-cold 70% BME in organoid medium and plated at suitable ratios (1:5 to 1:20) to allow efficient outgrowth of new organoids. After splitting, 10 µ mol/L Y-27,632 was always added to aid outgrowth of organoids from single cells. Medium was changed every 2–3 days and organoids were split once every 1–2 weeks.
2.3 R-Spondin and noggin conditioned medium
Thaw a vial of 293T-HA-Rspol-Fc cells from liquid nitrogen storage. Divide one vial of frozen cells into 1 x T75 flasks containing 15ml of growth medium (no Zeocin or Puromycin). After 3–4 days, when cells are 80% confluent, split 1:5. 4 x T75 flasks cells were cryopreserved and 1 x T75 flasks cell was cultured with 12ml of growth medium (DMEM + 10% FBS + 1%P/S + Zeocin (300ug/ml)). After 3–4 days, when cells are 80% confluent, split 1:5. Add 12ml media containing zeocin to 1 x T75 flask and 12 ml media without zeocin to 4 x T75 flasks. Culture for another 2–3 days until 80% confluent. Trypsinize 4 x T75 flasks without Zeocin with 1:3 diluted Trypsin-EDTA in PBS (from 0.25% Trypsin-EDTA, at room temperature for approximately 30 s or until cell start detach from the flask) neutralize with DMEM + 10% FBS and centrifuge at 200 g (1,338 rpm). Remove the trypsin and medium, divide the cell pellet into 10 x T75 flasks each containing 20ml medium for making conditioned medium (Advanced DMEM/F12, supplemented with 1×GlutaMAX, 1% Penicillin–streptomycin, and 10 mM HEPES). Harvest after one week. Pool medium from all flasks into centrifuge tube, centrifuge at 1000 g(2,990 rpm) for 10 min and filter supernatant through a 0.22 μm filter. Noggin Conditioned medium acquisition is similar, except that antibiotics are changed from Zeocin to Puromycin.
2.4 ALKBH1 demethylase assays
Demethylation assays was performed as previously described with minor modifications [26]. Briefly, the reactions were performed in a 50 uL demethylation reaction buffer containing 50pmol DNA oligos, 500ng recombinant ALKBH1 protein, 50 μm HEPES (pH = 7.0), 50 μm KCl, 1mM MgCl2, 2mM ascorbic acid, 1mM a-KG, and1mM (NH4)2Fe(SO4)2·6H2O. Reactions were carried out for 1 h at 37ºC and stopped with 5 mM EDTA followed by heating at 95ºC for 10 min. Then 2 mL of a reaction product was used for dot blotting.
2.5 Plasmids and lentivirus production
Lentivirus pLKO.1 and pHKO.23 vectors were used. All the target sequences for shRNA are listed in the supplemental Table 1. Short hairpin RNA constructs against ALKBH1 were designed and cloned into pLKO.1 according to the instructions. For the ALKBH1 rescue experiment, human ALKBH1 cDNA was cloned into a vector following shALKBH1#3’, which targets the 3’UTR of ALKBH1. For the TWIST rescue experiment, short hairpin RNA constructs against TWIST were cloned into vectors following shALKBH1#1 and shALKBH1#2. For the DDX18 rescue experiment, human DDX18 cDNA was cloned into a vector following shALKBH1. Lentiviruses were produced in HEK293T cells transfected using PEI with viral packaging constructs pMD2. G and pSPAX2. Viral supernatants were harvested at 48 and 72 h after transfection and filtered through a 0.45 μm filter with low protein binding membrane (Millipore).
2.6 6mA dot blot
For 6mA dot blot, DNA samples were denatured at 99ºC for 10 min, cooled down on ice for 3 min, neutralized with 10% vol of 6.6 M ammonium acetate. Samples were UV crosslinked to the membrane, and membrane was blocked with 5% nonfat dry milk (in 1X TBS) for 1–2 h and incubated with a specific anti-6mA antibody (1:2000 dilution, Synaptic Systems, 202,003) overnight at 4ºC. HRP-conjugated secondary antibodies was added to the blots for 1 h at room temperature and the membrane was developed with ECL Western Blotting Substrate (Bio-Rad) and exposure with X-Ray Super RX Films (Fujifilm).
2.7 Purification of recombinant proteins
Purification of recombinant proteins was performed as previously described with minor modifications [27]. ALKBH1 full length protein was cloned into pGEX-4P‐1 expression vector(addgene,28,060), which provides a N-terminal GST tag for purification and a Prescission protease site for removal of the GST tag after purification. BL21(DE3) competent cells were cultured in 2X YT broth at 37°C at 220 rpm until optical density at 600 nm reached 0.6–0.9. Subsequently, expression was induced in 0.5 mM IPTG for 13 h at 25ºC. The culture was harvested by centrifuging at 6000 rpm for 15 min at 4ºC. The pellets from 1 L of bacterial culture were re‐suspended with 30 mL of lysis buffer (50mM Tris HCl, pH 7.5, 5mM EDTA, 200mM NaCl, freshly added 5 mg/mL Lysozyme, 1mM DTT, PMSF, protease inhibitor cocktail) and incubated on ice for 30 min. The lysate was sonicated at 90% amplitude (30 s pulse-on and 90 s pulse‐off). The sonicated samples were centrifuged at 13,000 rpm for 30 min at 4°C. The supernatant was filtered and incubated with Glutathione Agarose beads at 4ºC. The beads were washed with high–‐salt wash buffer (50mM Tris HCl, pH 7.5, 500mM NaCl) three times and washed once again with Prescission protease cleavage buffer (50mM Tris HCl, pH 7.5, 1mM EDTA, 150 mM NaCl, 1mM DTT). The beads were incubated with Prescission protease (GE Healthcare Life Sciences) added to the cleavage buffer overnight at 4°C, or GST tagged proteins were eluted with elution buffer (Reduced glutathione, 50mM Tris HCL, pH8.0).
2.8 Cell proliferation and in vitro colony-forming assay
For HNSCC cell proliferation assays, human HNSCC cells (SCC9, SCC25 and CAL27) were transduced with lentivirus and selected with 2 mg/ml puromycin for 2 days. After selection, cells were seeded into 96-well plates at the concentration of 5000 cells per well in triplicates. Cell proliferation was assessed every 48 h using Colorimetric CellTiter 96 AQueous One Solution Cell Proliferation assay (Promega, Madison, WI). Experiments were performed in triplicate. For colony-forming assay, 2,500 cells were plated into 6 well plates in triplicate and cultured for 10–14 days in DMEM supplemented with 10% FBS. Cells were fixed with 95% ethanol and stained with 0.5% crystal violet.
For patient-derived HNSCC organoids, organoids were dissociated into single cells by first triturating them in media through a fire-polished glass pipette, and then by enzymatic dissociation with 2 mg/mL dispase dissolved in TrypLE (Life Technologies), until the organoids appeared as single cells under the microscope. Cells were counted, and diluted to 10 cells/mL in a mixture of complete media, Rho Kinase inhibitor Y-27,632, and Cultrex growth factor–reduced BME type 2. 100 µL of this mixture (1000 cells per well) was plated in 96-well plates (Nunc), whose wells had been previously coated with a bed of GFR-Matrigel to prevent attachment of the cells to the bottom of the plate. Cell viability was measured every 48 h using the CellTiter-Glo assay (Promega). All cell viability experiments were conducted in quadruplicate and standard deviations were reported.
For colony formation assay, organoids were dissociated to single cells as described above. Cells were sorted into organoid medium and were resuspended in Matrigel and cultured in 24-well plates with Rho Kinase inhibitor Y-27,632, for 10–14 days. The number of colonies formed in each well was assessed using a bright-field microscope.
2.9 MeDIP-qPCR, MeDIP-seq and data analysis of MeDIP–seq
MeDIP experiments were performed as previously described with minor modifications [26, 28, 29]. Genomic DNA was purified with DNeasy kit (QIAGEN). DNA samples were fragmented into 200-300 bp fragments by fragmentation buffer with Bioruptor. Then, adaptors were ligated to genomic DNA fragments following the Illumina protocol. The ligated DNA fragments were denatured at 95ºC for 5 min. Then, the single-stranded DNA fragments were immunoprecipitated with 6mA antibodies (Abcam, ab151230) in IP buffer (150mMNaCl, 0.1% NP-40,10mM Tris-HCl, pH 7.9, 2mM EDTA) overnight at 4 degrees. The mixture was then immunoprecipitated by incubation with protein-A/G beads (Thermo Fisher) at 4ºC for an additional 2 h. The beads were then separated and washed with 1x IP buffer three times before eluted with 6mA elution buffer for two times. DNA was further purified using PCR Purification Kit (QIAGEN) and analysed by PCR on a QuantStudio 7 Flex Real Time PCR System (Applied Biosystem). PCR parameters were 95 °C for 2 min and 40 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 15 s, followed by 72 °C for 1 min. All the ChIP–qPCR data presented were at least three biological replicates. Primer sequences are in Table 1. Error bars represent standard deviation (three biological replicates).
For MeDIP–seq, IP DNA and input DNA were PCR amplified with Illumina indexing primers. Sequencing was carried out on Illumina HiSeq X Ten. Reads from input and IP sequencing libraries were quality controlled with fastqc (v0.11.5) and mapped to the human genome reference (GRCh38/hg38) using bowtie2 (v2.2.9) with default parameters or bowtie (v1.2.2) with parameters -v2 -k1–best. Bam files were generated with samtools 1.3.1, which was followed by making bigwig files with deeptools (v/3.0.2) (binsize 10). Reads were normalized to Reads Per Genome Coverage (RPGC) with deeptools (v/3.0.2) bamCoverage function. MACS2 was also used for peak calling, which generated similar results as SICER. For visualizing 6mA peaks, .bigwig files were loaded in IGV (v2.4) and adjusted to the same scale.
2.10 RNA-seq and data analysis
For RNA-seq, total RNA was isolated using TRIzol reagent (Thermo Fisher). Poly(A) mRNA was subsequently purified from 1 µg of total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module. NEBNext Ultra Directional RNA Library Prep Kit (NEB, #E7760) was used for library preparation. RNA libraries were sequenced on an Illumina HiSeq X Ten platform with paired-end reads (150-bp read length). RNA-seq data were quantified with Kallisto (v0.43.1) and summarized to the gene level by tximport (v1.10.1). EdgeR (v3.24.3) was employed for data normalization and differential expression analysis of RNA-seq counts.
2.11 Quantitative RT-PCR
Total RNA from HNSCC cells was purified using TRIzol (Life Technologies) according to the manufacturer’s instructions. One microgram of purified total RNA was reverse transcribed using the ReverTra Ace qPCR RT Kit (TOYOBO). The levels of specific RNAs were measured using a Bio-Rad real-time PCR machine and Fast SybrGreen PCR master mix according to the manufacturer’s instructions. Primer sequences are listed in Supplemental Table 1. The 2−ΔΔCt method was used to normalize expression to GAPDH for cell lines.
2.12 Western blot analysis
Cells were lysed in RIPA buffer with a protease inhibitor cocktail (Roche), and the total cell lysates were resolved with SDS-PAGE gels. Membranes were blocked for 30 min at room temperature in TBS supplemented with 5% nonfat dried milk and incubated overnight at 4 °C with primary antibody diluted in the same blocking buffer. After three washes in TBST, membranes were incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies diluted in blocking buffer and then washed an additional three times with TBST before incubation with ECL Western Blotting Substrate (Bio-Rad) and exposure with X-ray Super RX Films (Fujifilm).
2.13 Luciferase reporter assay
HEK293T and SCC9 cells seeded in 24-well plates were transfected with the pMIR-REPORT luciferase vector fused with or without the promoter. Transfection efficiency was quantified by co-transfection with an actin promoter-driven Renilla luciferase reporter. The activities of firefly luciferase and Renilla luciferase in each well were calculated by a dual-luciferase reporter assay system (Promega). The ratios between the promoter-reporter and Renilla control were determined 48 h after shRNA treatment. The relative luciferase activities were further normalized to those in cells transfected with the firefly luciferase vector control under the same treatment conditions.
2.14 Statistical analysis
Student’s t test was used for significance testing. The log-rank test was used to compare survival curves. P values of less than 0.05 were considered statistically significant. Statistical analyses were performed using GraphPad Prism 7.0 or the R statistical environment. In the figures, asterisks indicate *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
3 Results
3.1 ALKBH1 is highly expressed in human HNSCC
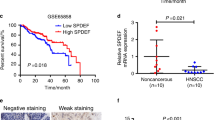
We first assessed the expression level of ALKBH1 in human HNSCC. We interrogated RNA sequencing data of 44 normal head and neck epithelial tissues and 278 HNSCC tissues from the TCGA database. Interestingly, compared to healthy control, high expression level of ALKBH1 was observed in most HNSCC patients (Fig. 1A). A similar result was also observed in different HNSCC subtypes, including atypical, basal, classical and mesenchymal subtypes (Fig. 1B). We further validated high expression of ALKBH1 using 10 human HNSCC samples compared to normal adjacent tissues (Fig. 1C). Consistently, we also detected higher expression of ALKBH1 protein in patient-derived primary HNSCCs (Fig. 1D). In addition, high expression of ALKBH1 at both mRNA and protein level was also observed in different patient-derived HNSCC cell lines (SCC9, SCC25, CAL27) (Fig. 1E and F). Next, we investigated the prognostic value of ALKBH1 in HNSCC. As expected, elevated expression of ALKBH1 correlated with poor overall survival of AML patients (Fig. 1G). Collectively, these findings imply that ALKBH1 might play an important role in human HNSCC.

ALKBH1 is upregulated in human HNSCC. (A)ALKBH1 expression pattern investigated using the TCGA database (278 cases of HNSCC tissues and 44 cases of normal tissues). The expression values were log2 transformed. (B) Comparison of the expression levels of ALKBH1 in different subtypes of HNSCC patients with normal controls based on TCGA datasets. The expression values were log2 transformed. (C)ALKBH1 levels detected in 10 HNSCC tumor tissues and paired non-tumor tissues using qRT-PCR. (D) Immunoblots showing ALKBH1 expression in normal controls (n = 3) and HNSCC patient samples (n = 4). (E). ALKBH1 expression in HNSCC cell lines (SCC9, SCC25 and CAL27) and DOK cells analyzed by qRT-PCR; GAPDH was used as a housekeeping gene(F) Immunoblots showing ALKBH1 expression in normal controls and HNSCC cell lines (SCC9, SCC25 and CAL27) using TUBULIN as a loading control. (G) Kaplan-Meier plots of overall survival in TCGA cohorts for HNSCC patients, stratified on the basis of ALKBH1 expression above (ALKBH1high) or below (ALKBH1low) the median.
3.2 ALKBH1 promotes the proliferation of HNSCC cells
To investigate the biological function of ALKBH1 in HNSCC, we abrogated ALKBH1 expression in HNSCC cells (SCC9, SCC25, and CAL27) by short hairpin RNA (shRNA)-mediated knockdown (KD). Compared with the scramble control, the shRNAs targeting different regions of ALKBH1 (including the coding region and 3’UTR) markedly decreased its expression level in different HNSCC cells (Fig. 2A). Interestingly, we found that knockdown of ALKBH1 significantly inhibited the growth of HNSCC cells (Fig. 2B). Further, we found that ALKBH1 knockdown obviously inhibited the cycling stage and increased the quiescence stage in the cell cycle of HSNCC cells (Fig. 2C and D). Surprisingly, ALKBH1 knockdown did not significantly affect apoptosis (Fig. 2E and F). Thus, these data suggest that ALKBH1 is essential for promoting HNSCC proliferation.
To rule out the possibility of off-target effects, we restored ALKBH1 expression by inserting its cDNA that is resistant to shALKBH1 targeting the 3’UTR and can be detected by primers for CDS region but not 3’UTR region (Fig. 2.G). As expected, we found that reintroduction of ALKBH1 substantially rescued the defects in the cellular growth and clonogenic ability of HNSCC cells caused by ALKBH1 deficiency (Fig. 2H). Together, these results indicate that ALKBH1 is required for the proliferation of human HNSCC cells.

ALKBH1 promotes the proliferation of HNSCC cells. (A) Immunoblots showing the knockdown efficiency of ALKBH1 in SCC9, SCC25 and CAL27 cells. (B) Growth curves of patient-derived HNSCC SCC9, SCC25 and CAL27 cells after knockdown of ALKBH1. (C and D) Cell cycle analysis of HNSCC cells after ALKBH1 knockdown. (E and F) Apoptosis analysis of HNSCC cells after ALKBH1 knockdown. (G) qRT-PCR analysis showing endogenous and exogenous ALKBH1 expression level after transduction with the indicated lentiviruses. Primer pair for ALKBH1 3’UTR and CDS region were used for detecting endogenous and exogenous ALKBH1 respectively. (H) Colony-forming unit assay of patient-derived HNSCC cells after knockdown of ALKBH1
3.3 ALKBH1 regulates proliferation of patient-derived primary HNSCC cells
We next investigated the function of ALKBH1 in HNSCC patients. To do this, we successfully established HNSCC patient-derived organoid system (Fig. 3A and B). Consistently, when compared with the control group, ALKBH1 knockdown significantly inhibited proliferation and reduced colony formation of patient-derived HNSCC organoids (Fig. 3C and D). We also found that deletion of ALKBH1 blocked the cycling and arrested cells in quiescence stage of HNSCC, but did not significantly affect apoptosis (Fig. 3E-H). Thus, these data indicate that ALKBH1 regulates proliferation of patient-derived primary HNSCC cells.

ALKBH1 regulates proliferation of HNSCC patient-derived organoids. (A and B) Schematic outline for the patient-derived HNSCC organoid system. (C) Growth curves of patient-derived HNSCC organoids after ALKBH1 knockdown. (D) Colony-forming unit assay using patient-derived HNSCC organoids from three individual patients after knockdown of ALKBH1. (E and F) Cell cycle distribution of HNSCC patient-derived primary cells after ALKBH1 knockdown. (G and H) Apoptosis analysis of HNSCC patient-derived primary cells after ALKBH1 knockdown.
3.4 The demethylase activity of ALKBH1 is required for proliferation of HNSCC cells
ALKBH1 acts as a demethylase of DNA 6mA [20], we next assessed whether the DNA 6mA demethylase activity of ALKBH1 is required for regulating HNSCC cells proliferation. Using purified ALKBH1 protein and synthesized 6mA DNA oligo, we first performed in vitro demethylation assay and validated the demethylation activity of ALKBH1 (Fig. 4A). Through dot blot and mass spectrometry (MS), we found that the global DNA 6mA level was significantly increased in HNSCC cells upon ALKBH1 knockdown (Fig. 4B and C). To block ALKBH1 enzymatic activity to the genome DNA 6mA, we generated the ALKBH1 mutant that carried four point mutations, including R24A, R25A, R28A and R31A, the key residues for the demethylation activity (Fig. 4D) [21, 30]. Using this mutant, we restored ALKBH1 expression by inserting wild type (WT) or mutant cDNA to shALKBH1 targeting the 3’UTR to perform the rescue experiments (Fig. 4E). As expected, restoration of WT but not mutant ALKBH1 could revert the level of DNA 6mA in HNSCC cells (Fig. 4F). We also found that restoration of wild WT ALKBH1, but not the mutant, substantially rescued the defects in cellular growth and clonogenic ability of HNSCC cells caused by ALKBH1 deficiency (Fig. 4G and H). Taken together, these results indicate that the DNA 6mA demethylase activity of ALKBH1 is required to promote HNSCC proliferation.

The demethylase activity of ALKBH1 is required for proliferation of HNSCC cells. (A) DNA dot assay showing 6mA levels of 6mA labeled DNA oligonucleotides after in vitro demethylase reaction with recombinant human ALKBH1 proteins. (B) DNA dot assay showing 6mA levels in patient-derived HNSCC cells after ALKBH1 knockdown. Methylene blue detected DNA loading. (C) Mass spectrometry (MS) showing 6mA levels in patient-derived HNSCC cells after ALKBH1 knockdown. (D) Schematic depiction for human ALKBH1 wild type and mutant with 4 point mutations. (E) Quantification of global 6mA levels in HNSCC cells after transduction with indicated lentiviruses. (F) DNA dot assay showing the global 6mA levels in HNSCC cells after transduction with indicated lentiviruses. (G) Growth curves of HNSCC cells after transduction with indicated lentiviruses. (H) Colony-forming unit assay of HNSCC cells after transduction with indicated lentiviruses.
3.5 ALKBH1 regulates DNA 6mA modification of HNSCC cells
To determine the underlying mechanism of how ALKBH1 regulates the proliferation of HNSCC, we first performed methylated DNA immunoprecipitation following sequencing (MeDIP-seq). We observed 18,822 6mA peaks in HNSCC, and these peaks located in intergenic regions (56.94%), gene bodies (38.4%) and promoters (4.66%) (Fig. 5A). As expected, ALKBH1 knockdown significantly increased DNA 6mA peaks (27,316 vs. 18,822 peaks for shALKBH1 and shControl, respectively). However, ALKBH1 knockdown did not alter the distribution of these peaks (Fig. 5B-D). To identify the potential targets of ALKBH1, we next focused on the peaks located in the promoter regions, and found that the enriched 6mA peaks around in the promoter regions upon ALKBH1 knockdown were assigned to 92 target genes (Fig. 5E). We further performed an RNA-seq assay and profiled gene expression alterations in SCC25 cells upon ALKBH1 knockdown. A total of 648 differentially expressed genes (DEGs) (including 256 downregulated and 392 upregulated) were identified in HNSCC cells after knocking down ALKBH1 (Fig. 5F). Since DNA 6mA is known to repress gene transcription, we integrated the downregulated DEGs with the 92 target genes showing enriched 6mA modification upon ALKBH1 knockdown. Intriguingly, we identified 9 potential overlapped targets, including DDX18, RIOX2, RFC3, MTRR, ARL6, NR2F2, CDCA7L, SCN1A, and NARS2 (Fig. 5G). We further confirmed the downregulation of these 9 genes in HNSCC cells upon ALKBH1 knockdown (Fig. 5H). Among these candidates, DDX18 expression displayed the most pronounced decrease with ALKBH1 loss in SCC9 cells (Fig. 5H). To further determine whether DDX18 is a target of ALKBH1 in HNSCC cells, we performed rescue experiments. As expect, we found that restoration of WT ALBKH1 but not the mutant one reverted the expression level of DDX18 mRNA in SCC9 and SCC25 cells (Fig. 5I), suggesting that ALKBH1 regulates DDX18 expression in HNSCC cells, and this effect depends on the enzymatic activity of ALKBH1. Together, these results indicate that ALKBH1 regulates DNA 6mA modification of HNSCC cells, and DDX18 is one of its targets.

ALKBH1 regulates DNA 6mA modification of HNSCC cells. (A) Genome ontology analysis showing the fraction of 6mA peaks present in distal intergenic, gene-body, or promoter. (B) Heatmap showing sites of 6mA enrichment following ALKBH1 knockdown. The signal is shown over a scaled widow 3 kb upstream and downstream of the gained 6mA peaks. Log2 enrichment was normalized to reads per genome coverage. (C) Profile plot showing sites of 6mA enrichment following ALKBH1 knockdown. The signal is shown over a scaled widow 3 kb upstream and downstream of the gained 6mA peaks. Log2 enrichment was normalized to reads per genome coverage. (D) Genome ontology analysis showing the fraction of 6mA peaks following ALKBH1 knockdown present in distal intergenic, gene-body, or promoter. (E) Profile plot showing average distribution of 6mA MeDIP-seq reads at 92 genes. (F) Volcano plots of RNA-seq assay showing differential expression genes in SCC25 cells after ALKBH1 knockdown. (G) Integrated analysis between downregulated DEGs and the 6mA target genes with enriched 6mA modification upon ALKBH1 knockdown. (H and I) qRT-PCR analysis showing the expression levels of candidates in HNSCC cells transduced with the indicated lentiviruses.
3.6 ALKBH1 enhances DDX18 expression by modulating DNA 6mA modification in the promoter region
We sought to decipher the mechanism by which ALKBH1 regulates DDX18 expression. As expected, IGV showed the increased enrichment of 6mA in the DDX18 promoter in HNSCC cells upon ALKBH1 knockdown (Fig. 6A). We further designed five primer pairs from − 393 to -2630 of DDX18 promoter region (Fig. 6B), and MeDIP-qPCR showed that DNA 6mA modification was mainly enriched in the promoter region from − 393 to -1319 in both SCC9 and SCC25 cells (Fig. 6C and D). We next constructed a luciferase reporter system by inserting this DDX18 promoter region into a pGL3-basic vector plasmid. Using this luciferase reporter assays, we found that ALKBH1 knockdown significantly decreased the luciferase activity of DDX18 promoter reporter, and reintroduction of wild type ALKBH1 but not the mutant reversed this phenotype in both HEK293T and SCC25 cells (Fig. 6E). Together, these results suggest that ALKBH1 promotes DDX18 expression by modulating DNA 6mA modification in its promoter region.

ALKBH1 enhances DDX18 expression by modulating DNA 6mA modification in the promoter region. (A) Genomic snapshots of MeDIP-seq reads at the DDX18 Locus for the indicated cells. Log2 enrichment levels were normalized to reads per genome coverage. (B) Schematic diagram of five primer pairs from − 393 to -2630 of the DDX18 promoter. (C and D) MeDIP-qPCR experiments showing the localization of 6mA at the indicated genes in SCC9 and SCC25 cells with the indicated lentiviruses. (E) Relative luciferase activity of the DDX18 promoter in 293T and SCC25 cells.
3.7 DDX18 mediates the function of ALKBH1 in regulating human HNSCC proliferation
DEAD-box RNA helicase DDX18 implicates in controlling cell cycle progression in zebrafish hematopoietic cells [31], which is consistent with our observation in HNSCC with ALKBH1 knockdown. Thus, we examined whether ectopic expression of DDX18 could rescue the phenotypes of ALKBH1-deficient HNSCC cells. As expected, restoration of DDX18 partially rescued the defects in the proliferation of HNSCC cells caused by ALKBH1 knockdown (Fig. 7A and B). We also observed similar phenomena using patient-derived HNSCC organoid system (Fig. 7C and D). As expected, DDX18 overexpression also reversed the cell cycle defect of HNSCC cells caused by ALKBH1 deletion (Fig. 7E). Taken together, these data suggest that DDX18 mediates the function of ALKBH1 in regulating HNSCC proliferation.

DDX18 mediates the function of ALKBH1 in regulating human HNSCC proliferation. (A) qRT‒PCR analysis showing ALKBH1 and DDX18 expression levels after transduction with the indicated lentiviruses. (B) Growth curves of HNSCC cells with the indicated lentiviruses. (C) Colony-forming unit assay using patient-derived HNSCC organoids from three individual patients after knockdown of ALKBH1. (D) Growth curves of patient-derived HNSCC organoids after ALKBH1 knockdown. (E) Cell cycle distribution of HNSCC patient-derived primary cells after ALKBH1 knockdown.
4 Discussion
Although recent works have reported high expression of different ALKBHs in various types of human cancer [11, 32, 33], the role of ALKBH1 in HNSCC remains unknown. Herein, we demonstrate that the expression level of ALKBH1 is significantly increased in patients with HNSCC, and ALKBH1 is required for HNSCC proliferation which is mediated by DDX18. Interestingly, we find that ALKBH1 regulates DDX18 expression through erasing DNA 6mA modification in its promoter. Overall, this work reveals a new role of ALBKH1 in HNSCC.
Our finding shows that the function of ALKBH1 on HNSCC cells depends on its DNA 6mA demethylase activity. DNA methylation is a well-known epigenetic mark and plays crucial roles in numerous biological processes and human diseases. Beyond 5-Methylcytosine (5mC), the best-characterized type of DNA methylation and predominantly found in higher eukaryotes, 6mA is the most prevalent DNA modification in prokaryote and its presence in eukaryotes is gradually uncovered recently with the development of highly sensitive detection methods. 6mA plays potentially conserved roles in various biological processes, such as immune modulation, transcription, nucleosome positioning, DNA damage control and cell cycle regulation [34,35,36]. Despite controversies regarding the presence of 6mA in multicellular eukaryotes, some potential eukaryote-specific biological roles in stress responses, chromatin regulation and tumorigenesis have been revealed in recent studies [20, 26, 37]. To our knowledge, this is the first time to show that ALKBH1-mediated DNA 6mA modification in HNSCC. It would be of great interest to fully investigate the role of DNA 6mA modification in the development of HNSCC in the future.
Our findings show that ALKBH1 promotes the proliferation of HNSCC cells that is partially mediated by DDX18. Previous studies demonstrate that DDX18 deletion causes cell cycle arrest in G1 phase [31, 38]. In addition, a recent work indicates that DDX18 prevents R-loop-induced DNA damage and genome instability via PARP-1, and deletion of DDX18 results in aberrant endogenous R-loop accumulation which leads to DNA replication defects [39]. Thus, the proliferation defect of HNSCC cells due to ALKBH1 deficiency to certain extent, results from downregulation of DDX18. However, it remains unknown if ALKBH1 and DDX18 involves in DNA damage of HNSCC, which needs to be further investigated in the future. In addition, ectopic expression of DDX18 only partially rescue the phenotypes of ALKBH1-deficient HNSCC cells, suggesting that other targets might mediate the function of ALKBH1. For instance, the enrichment of 6mA level was also enriched in the promoter region of RIOX2 and RFC3, and their expression was also regulated by ALKBH1. These data suggest that RIOX1 and RFC3 might be potential downstream targets, which need to be further studied. Moreover, we also found that ALKBH1 deficiency in HNSCC cells decreases the activation of PI3K/AKT signaling pathway, and upregulates the expression of cell cycle inhibitors CDKN1A and CDKN2D (Data not shown), suggesting that these downstream pathways might also mediate the function of ALKBH1 in HNSCC.
Data Availability
The datasets generated, RNA-seq is being deposited in GEO. This paper does not report original code. Any additional information required to access and analyze the data reported in this paper is available from the lead contact upon request.
References
D.E. Johnson et al., Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 6, 92 (2020)
C.R. Leemans, P.J.F. Snijders, R.H. Brakenhoff, The molecular landscape of head and neck cancer. Nat. Rev. Cancer 18, 269–282 (2018)
S.B. Chinn, J.N. Myers, Oral cavity carcinoma: current management, controversies, and future directions. J. Clin. Oncol. 33, 3269–3276 (2015)
J.D. Cramer, B. Burtness, Q.T. Le, R.L. Ferris, The changing therapeutic landscape of head and neck cancer. Nat. Rev. Clin. Oncol. 16, 669–683 (2019)
C.R. Leemans, B.J. Braakhuis, R.H. Brakenhoff, The molecular biology of head and neck cancer. Nat. Rev. Cancer 11, 9–22 (2011)
M.L. Hedberg et al., Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Invest. 126, 169–180 (2016)
N. Stransky et al., The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160 (2011)
J. Nieminuszczy, E. Grzesiuk, Bacterial DNA repair genes and their eukaryotic homologues: 3. AlkB dioxygenase and Ada methyltransferase in the direct repair of alkylated DNA. Acta Biochim. Pol 54, 459–468 (2007)
R. Ougland, T. Rognes, A. Klungland, E. Larsen, Non-homologous functions of the AlkB homologs. J. Mol. Cell. Biol. 7, 494–504 (2015)
V. Rajecka, T. Skalicky, S. Vanacova, The role of RNA adenosine demethylases in the control of gene expression. Biochim. et Biophys. Acta (BBA)-Gene Regul. Mech. 1862, 343–355 (2019)
A. Tan, Y. Dang, G. Chen, Z. Mo, Overexpression of the fat mass and obesity associated gene (FTO) in breast cancer and its clinical implications. Int. J. Clin. Exp. Pathol 8, 13405 (2015)
B. Singh et al., Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PloS one 11, e0159072 (2016)
I. Yamato et al., PCA-1/ALKBH3 contributes to pancreatic Cancer by supporting apoptotic resistance and AngiogenesisPCA-1/ALKBH3 in pancreatic Cancer. Cancer Res 72, 4829–4839 (2012)
S. Zhou et al., FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting β‐catenin through mRNA demethylation. Mol. Carcinog 57, 590–597 (2018)
T.-C.A. Johannessen et al., The DNA repair protein ALKBH2 mediates temozolomide resistance in human glioblastoma cells. Neuro-oncology 15, 269–278 (2013)
V. Kaklamani et al., The role of the fat mass and obesity associated gene (FTO) in breast cancer risk. BMC Med. Genet 12, 1–10 (2011)
S. Lee et al., TP53 regulates human AlkB homologue 2 expression in glioma resistance to photofrin-mediated photodynamic therapy. Br. J. Cancer 103, 362–369 (2010)
T. Fujii, K. Shimada, S. Anai, K. Fujimoto, N. Konishi, ALKBH 2, a novel a lk B homologue, contributes to human bladder cancer progression by regulating MUC 1 expression. Cancer Sci 104, 321–327 (2013)
J. Wang et al., Leukemogenic chromatin alterations promote AML Leukemia Stem cells via a KDM4C-ALKBH5-AXL Signaling Axis. Cell. Stem Cell. 27, 81–97 e88 (2020)
C.L. Xiao et al., N(6)-Methyladenine DNA modification in the Human Genome. Mol. Cell. 71, 306–318 e307 (2018)
L.F. Tian et al., Structural basis of nucleic acid recognition and 6mA demethylation by human ALKBH1. Cell. Res. 30, 272–275 (2020)
C. Shen, K. Wang, X. Deng, J. Chen, DNA N(6)-methyldeoxyadenosine in mammals and human disease. Trends Genet. 38, 454–467 (2022)
L. Ouyang et al., ALKBH1-demethylated DNA N6-methyladenine modification triggers vascular calcification via osteogenic reprogramming in chronic kidney disease. J Clin Invest 131 (2021)
Y. Liu et al., DNA demethylase ALKBH1 promotes adipogenic differentiation via regulation of HIF-1 signaling. J. Biol. Chem. 298, 101499 (2022)
E. Driehuis et al., Oral mucosal organoids as a potential platform for Personalized Cancer Therapy. Cancer Discov 9, 852–871 (2019)
Q. Xie et al., N(6)-methyladenine DNA modification in Glioblastoma. Cell 175, 1228–1243 e1220 (2018)
C.R. Alarcon et al., HNRNPA2B1 is a mediator of m(6)A-Dependent Nuclear RNA Processing events. Cell 162, 1299–1308 (2015)
T.P. Wu et al., DNA methylation on N(6)-adenine in mammalian embryonic stem cells. Nature 532, 329–333 (2016)
E.L. Greer et al., DNA methylation on N6-Adenine in C. elegans. Cell 161, 868–878 (2015)
M. Zhang et al., Mammalian ALKBH1 serves as an N(6)-mA demethylase of unpairing DNA. Cell. Res. 30, 197–210 (2020)
E.M. Payne et al., Ddx18 is essential for cell-cycle progression in zebrafish hematopoietic cells and is mutated in human AML. Blood 118, 903–915 (2011)
K. Hotta et al., Clinical significance and therapeutic potential of prostate cancer antigen-1/ALKBH3 in human renal cell carcinoma. Oncol. Rep 34, 648–654 (2015)
D. Xu et al., FTO expression is associated with the occurrence of gastric cancer and prognosis. Oncol. Rep 38, 2285–2292 (2017)
X. Li et al., The exploration of N6-deoxyadenosine methylation in mammalian genomes. Protein Cell. 12, 756–768 (2021)
K. Boulias, E.L. Greer, Means, mechanisms and consequences of adenine methylation in DNA. Nat. Rev. Genet. 23, 411–428 (2022)
K.J. Wu, The epigenetic roles of DNA N(6)-Methyladenine (6mA) modification in eukaryotes. Cancer Lett. 494, 40–46 (2020)
C. Ma et al., N6-methyldeoxyadenine is a transgenerational epigenetic signal for mitochondrial stress adaptation. Nat. Cell. Biol. 21, 319–327 (2019)
N. Chen, G. Zhang, J. Fu, Q. Wu, Identification of key modules and hub genes involved in esophageal squamous cell Carcinoma Tumorigenesis using WCGNA. Cancer Control 27, 1073274820978817 (2020)
W.L. Lin et al., DDX18 prevents R-loop-induced DNA damage and genome instability via PARP-1. Cell. Rep. 40, 111089 (2022)
Funding
This work is supported by the Fundamental Research Funds for the Central Universities [2042021kf0225]. This work was also supported by grant from the Hubei Provincial Natural Science Fund for Creative Research Groups [2021CFA003].
Author information
Authors and Affiliations
Contributions
C.G. conceived the study, designed and performed the experiments, and analyzed the data. C.G., Z.L. discussed the results and wrote the manuscript. H.Z. supervised the overall study.
Corresponding authors
Ethics declarations
Ethical approval
All experiments involving human samples were conducted in compliance with all relevant ethical regulations, and were approved by the Medical Ethics Committees of the School of Medicine, Wuhan University.
Competing interests
The authors declare no competing financial interests.
Additional information
Zheming Liu and Haojian Zhang contributed equally to this work.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Guo, C., Liu, Z. & Zhang, H. DNA 6mA demethylase ALKBH1 regulates DDX18 expression to promote proliferation of human head and neck squamous cell carcinoma. Cell Oncol. 46, 1097–1111 (2023). https://doi.org/10.1007/s13402-023-00800-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-023-00800-1