
Abstract
Purpose
Tumor microenvironment (TME) affects the progression of rectal cancer (RC), and the clinical relevance of its immune elements was widely reported. Here we aim to delineate the complete TME landscape, including non-immune features, to improve our understanding of RC heterogeneity and provide a better strategy for precision medicine.
Methods
Single-cell analysis of GSE161277 using Seurat and Cellcall was performed to identify cell-cell interactions. The ssGSEA was employed to quantify the TME elements in TCGA patients, which were further clustered into subtypes by hclust. WGCNA and LASSO were combined to construct a degenerated signature for prognosis, and its performance was validated in two GEO datasets.
Results
We proposed a subtyping strategy based on the abundance of both immune and non-immune components, which divided all RC patients into 4 subtypes (Immune-, Canonical-, Dormant- and Stem-like). Different subtypes exhibited distinct mutation landscapes, biological features, immune characteristics, immunotherapy responses and prognoses. Next, WGCNA and LASSO regression were combined to construct a 10-gene signature based on differentially expressed genes among different subtypes. Subgroups divided by this signature also exhibited different clinical parameters and responses to immune checkpoint blockades. Diverse machine learning algorithms were applied to achieve higher accuracy for survival prediction and a nomogram was further established in combination with M stage and age to provide an accurate and visual prediction of prognosis.
Conclusions
We identified four TME-based RC subtypes with distinct biological and clinical features. Based on those subtypes, we also proposed a degenerated 10-gene signature to predict the prognosis and immunotherapy response.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Rectal cancer (RC) is one of the most common cancers, ranking 8th in incidence and 9th in cancer-related deaths worldwide [1]. Recently, advanced treatment approaches such as neoadjuvant therapy, precise surgery and immunotherapy have been emerging [2], increasing the 5-year recurrence-free survival (RFS) rate of RC to almost 70% [3]. However, owing to the heterogeneity of RC, there are still ample patients who fail to obtain a correct prognosis [4]. The clinical decision-making process is largely dependent on meticulous pathological scrutiny, i.e., the tumor-node-metastasis (TNM) staging system [2]. The TNM classification determines whether a RC patient will be suitable for surgery or benefit from chemotherapy [5]. However, this system only provides information on the anatomical regions of the lesions and the degrees of malignancy. Since it contains little information on the molecular basis of the tumor, the TNM staging system is far from satisfactory to suit various comprehensive RC treatment options.
The tumor microenvironment (TME) represents a complicated heterogeneous mixture of malignant cells and nonmalignant components [6]. It is widely reported that TME components affect cancer cell survival, metabolism, local invasion and metastatic dissemination. Tumor-associated macrophages (TAMs), for example, provide malignant cells with nutrition to shield them against anticancer therapies [7]. Dendritic cells (DCs) initiate anticancer responses as professional antigen-presenting cells [8]. Cancer-associated fibroblasts (CAFs) secrete pro-tumor factors and remodel the extracellular matrix to support cancer stemness [9]. Hence, the presence and abundance of different TME components may serve as prognostic indicators and for the prediction of chemotherapy and immunotherapy responses [10,11,12,13,14].
Recently, various cancer subtyping strategies have been proposed based on immune cell infiltration to provide a precise prediction of prognosis [15,16,17,18,19]. However, non-immune cells in the TME were largely neglected in these studies, even though cross-talk among cancer cells, infiltrated immune cells and other stromal components have already been identified as vital biological processes during tumor development, progression and metastasis. Specific clusters of endothelial cells and fibroblasts have, for instance, been found to promote tissue remodeling and angiogenesis in lung adenocarcinoma [20]. Moreover, CAFs have been shown to promote dysregulated metabolism of breast cancer cells [21]. Thus, we believe that integration of the complete TME landscape will enhance our understanding of cancer heterogeneity and help to improve its treatment.
In this study, we show that the abundance of stromal and epithelial cells and their interactions with immune cells may serve as potential prognostic indicators, underscoring the crucial role of non-immune components in tumor progression. Based on different compositions of the TME, we have divided all RC patients into four subtypes, i.e., the Immune subgroup, the Canonical subgroup, the Stem-like subgroup and the Dormant subgroup. We found that the different RC subtypes not only possessed different mutation profiles and different activated signaling pathways, but also indicated different prognostic tendencies. To construct a simple and effective TME prognostic model, we established a workflow to screen for TME prognostic genes with high efficiency and orthogonality by integrating WGCNA and LASSO. With the help of clinical information and machine learning algorithms, we have largely boosted the prognostic capability of the model and as well as its ability to predict immunotherapy responses, and visualized it in a nomogram.
2 Materials ad methods
2.1 Study design
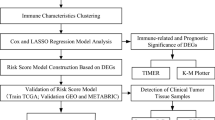
A schematic workflow of this study is depicted in Fig. 1A. First, we evaluated the cellular interactions between immune and non-immune cells on colorectal cancer (CRC) tissues and revealed their correlation with prognosis. Subsequently, we identified four distinctive microenvironment subtypes from all RC patients and revealed their biological features. Next, we generated a concise signature based on differences in microenvironmental features by WGCNA and LASSO regression. Subsequently, we tested this degenerated subtyping system in different datasets and validated the differences in clinical outcomes, mutational features and microenvironmental levels between different subgroups.

Graphic abstract. A. The outline of this study contains three main parts: 1. Identification of four distinct microenvironmental subtypes from all RC patients and their biological features; 2. Generation of a concise signature based on the microenvironmental components by a combination of WGCNA and LASSO regression; 3. Verification of this degenerated signature from a multi-omics perspective in different datasets. B. Two-dimensional tSNE plot identifying 6 distinct cell type in GSE161277. C. Chord diagram showing ligand-receptor interactions between each cell category. D. Bubble plot showing cancer signaling pathways affected by ligand-receptor interplays; the former cell categories at the bottom indicate the ligands and the latter ones indicate the receptors
2.2 Single-cell sequencing data analysis
Single-cell RNA sequence data from the GEO (https://www.ncbi.nlm.nih.gov/geo/) database (i.e., GSE161277) was employed to reveal interactions between each cell category. Four carcinoma tissues (i.e., GSM4904234, GSM4904236, GSM4904239, GSM4904245) with a RNA expression matrix were processed using the Seurat R package. We set the threshold at <25% mitochondrial genes and 200–5000 featured RNAs to exclude low-quality or biased cells. After this, 12,228 cells remained for subsequent analysis. We normalized the data and removed batch effects among different samples via the Seurat R package [22]. PCA (principal component analysis) was performed for dimensionality reduction of the highly variable genes after which the top 15 PCs were selected for graph-based cell clustering. The t-distributed stochastic neighbor embedding (tSNE) R package was employed to show cell distributions in a two-dimensional space [23]. The SingleR package was employed to annotate each cell type by corresponding featured genes [24]. After cell annotation, we employed the Cellcall R package to investigate interactions among receptors, ligands and transcription factors between different cells and revealed the cancer signaling pathways involved in those cell-cell interplays [25].
2.3 Data collection of bulk sequence data
RNA expression profiles and clinical information of 153 RC patients from TCGA (https://portal.gdc.cancer.gov/cart) database were selected as training dataset. Validation datasets were obtained from the GEO database (i.e., GSE161158 and GSE17537), which contains both mRNA expression profiles and clinical data of colorectal cancer patients. Somatic mutation data of the RC patients were downloaded from TCGA, and the mutation rate and the detailed mutational category of the top 30 mutated genes were calculated. Evaluation of immunotherapy responses was performed through the IMvigor210 study, GSE78220, and immunophenoscores (IPS) from TCIA databases [26, 27].
2.4 TME scoring in RC patients
Featured markers of representative immune components were obtained from a classic study (Tables 1 and S1) [28]. Other non-immune stromal cell markers were obtained from the CellMarker website (http://biocc.hrbmu.edu.cn/CellMarker/index.jsp) (Tables 1 and S1) [29]. Based on representative markers, we calculated enrichment levels of different TME components via the GSVA R package, which was deemed a popular algorithm for detecting pathway activities and cell infiltrations [30]. To reveal distinctive TME characteristics with clinical outcomes and biological features, hierarchical clustering was performed to divide patients into 4 subgroups based on different TME scores. We adopted tSNE for dimensional reduction and visualized the distribution of different samples in a two-dimensional image using the tSNE R package. Correlations among different TME cells were evaluated using Pearsons’ correlation coefficients.
2.5 Analysis of different TME subtypes
The Estimate R package was used to calculate the microenvironmental levels of each patient, including immune scores, stromal scores, estimate scores and tumor purity scores [31]. Furthermore, representative markers of both TME-related and DNA repair-associated pathways were obtained from previous literature [26, 32], which were employed to validate the TME homeostasis and genomic stability among different subgroups (Tables S2 and S3). The Survival R package was employed for survival analysis. To identify specific genes in each subgroup, the limma R package was employed for performing differential analysis between subgroup 2 and the other 3 subgroups. |log2fold change| > 0.585 and FDR < 0.05 were used as thresholds to identify differentially expressed genes (DEGs). Cox regression analysis was performed on the DEGs to identify prognostic genes. The GSEA R package was employed to investigate different molecular functions and pathways associated with specific subgroups.
2.6 Prognostic TME-featured gene-based modeling
The WGCNA R package was employed to divide all DEGs into different modules and to calculate correlations between distinct modules with different TME cells and clinical parameters [33]. To reveal connections among all prognostic genes in different modules, we calculated correlations between the prognostic genes via Pearsons’ correlation coefficients. Subsequently, we employed Cytoscape software to depict an interaction network for the genes with a correlation degree above 0.5. The clusterProfiler R package was employed to perform functional enrichment analysis including terms of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) in each module [34]. The top 5 prognostic genes in each module were selected for lasso regression to obtain the final gene signature.
2.7 Evaluation and validation of the degenerated TME signature
To assess the predictive ability of this gene signature on clinical characteristics and TME components, we divided all RC patients into different risk groups by the median risk score. Survival R package was employed to validate differences in the overall survival, disease-specific survival and recurrence-free survival between high- and low-risk groups. We further adopted the ROC curve and the calibration curve for validation of the performance of the degenerated TME signature. To obtain a better performance, different machine learning algorithms, including SVM (support vector machine), RF (random forest) and LR (logistic regression) were used to improve the prediction of the signature. Univariate and multivariate Cox prognosis analyses were performed on this signature with other clinical parameters. The limma R package was employed to evaluate correlations between risk scores with different clinical parameters. The pRRophetic R package was employed to predict chemotherapeutic responses, and the IC50 differences suggested different drug sensitivities between high- and low-risk groups [35]. TIMER (https://cistrome.shinyapps.io/timer/) was employed to calculate the association between the infiltration of diverse immune cells and the levels of DEGS1 and RRAGD. Moreover, the GSE161158 and GSE17537 datasets were used to validate the degenerated TME signature.
2.8 Construction of a nomogram based on the degenerated TME signature
Firstly, we validated the prognostic value of this TME signature in subpopulations stratified by different clinical statuses including age, gender and TNM stage. Next, we employed the rms R package to depict a predictive nomogram consisting of the prognostic signature and other independent prognostic factors we identified [36]. We also depicted the ROC curve and calibration curve for the evaluation of the performance of the nomogram.
2.9 Statistical analysis
Computation and visualization of all data were implemented by R (4.1.1 version) and cystoscope (3.7.0 version) software. The detailed R packages used in each step are listed above, and p < 0.05 results were considered as statistically significant. For multiple comparisons a false discovery rate (FDR) < 0.05 threshold was used to control the rate of the type I error.
3 Results
3.1 Interactions among TME cells
First, the cells were separated into 16 different clusters, after which a heatmap revealed the top 3 featured genes of each cell cluster (Fig. S1A, S1B). We further annotated each type via the SingleR package, through which cells were mainly separated into 6 distinct categories (Fig. 1B). The prediction of receptor-ligand-transcription factor interactions indicated ubiquitous cellular interplays between each cell category (Fig. 1C), especially for epithelial cells of which the receptors exerted cellular communications with the ligands of other cells. Moreover, we found that the interactions between epithelial cells and other cells affected tumor progression (Fig. 1D). For instance, interplays between B-epithelial, NK-epithelial or monocyte-epithelial cells were found to modify the Hippo signaling pathway, the ErbB signaling pathway and cellular senescence, respectively. The T-epithelial interaction also affected platinum drug resistance and the PD1-PDL1-related pathway. The concrete receptor-ligand pairs displayed in Fig. S1C shows the valves that drive cell-to-cell interactions, which may provide potential targets to activate or block cellular communication.
3.2 TME-based molecular subtypes of rectal cancer
Here, we curated the main biomarkers representing different cell types in the TME, including 19 immune-correlated components (e.g. immune cells, immune-related pathways and immune responses), 9 non-immune stromal cells and 7 epithelial cells (Table 1). Detailed lists of these biomarkers are shown in Supplementary Table 1. Using the ssGSEA algorithm, we annotated the abundance of those TME components in each TCGA-READ patient according to the expression of their featured biomarkers. We found that the infiltration levels of myofibroblasts, goblet cells, goblet progenitor cells and Paneth cells were correlated with the overall survival status of the RC patients (Fig. S1D-G), indicating a non-negligible role of non-immune components on tumor progression. Next, the patients were separated into 4 subgroups by hierarchical clustering based on ssGSEA (Fig. S2, Fig. 2A). Subgroup 1 (Immune) exhibited the highest abundance of both immune and non-immune stromal cells, while subgroup 2 (Canonical) showed moderate immune and non-immune stromal cell levels. Both subgroup 3 and subgroup 4 retained the lowest number of immune cells, but subgroup 4 (Stem-like) exhibited a lower non-immune stromal cell abundance and a higher cancer stem cell and goblet cell level compared to subgroup 3 (Dormant).

TME-based molecular subtypes of rectal cancer. A. Heatmap exhibiting the classification of RC patients based on the abundance of immune cells, non-immune stromal cells and epithelial cells by the ssGSEA algorithm. The majority of the patients were categorized to subgroup 2 (Canonical), which showed moderate immune and non-immune stromal cell levels. Patients with higher immune and stromal levels are categorized to Subgroup 1 (Immune). Subgroup 3 (Dormant) and subgroup 4 (Stem-like) retained the lowest immune cells, and subgroup 4 exhibited a lower non-immune stromal cell abundance and a higher cancer stem cell and goblet cell level compared to subgroup 3. B. Heatmap showing the results of Pearson’s correlation analysis on 35 TME components. C. Scatter plot indicating that all RC patients could be unsupervisedly divided into 4 subgroups by two-dimensional tSNE analysis, validating the results of hierarchical clustering. D. Violin plot showing TME scores for RC patients in different subgroups. E. Kaplan–Meier survival plot revealing different survival statuses of different subgroups. F. Mutation landscape of the top 30 mutated genes in different subgroups. The upper plot represents tumor mutational burden (TMB), the left column represents the mutation frequency of each gene and the right column the detailed mutational type. G. Boxplot showing the expression profiles of each HLA protein in different subgroups. H. Boxplot showing the expression profiles of immune checkpoint genes in different subgroups. I. Difference of immunophenoscore (IPS) of anti-PD1, anti-CTLA4, and a combination of both among the four subgroups by TCIA. Statistical analyses on different TME subgroups were performed using the one-way ANOVA algorithm, *p < 0.05, **p < 0.01, ***p < 0.001
In the Pearson’s correlation analysis, Paneth cells were strongly correlated with the infiltration of macrophages and many other immune cells (Fig. 2B), which implies potential capacities of intestine cells to affect immune infiltration. Moreover, myofibroblasts, stem cells, vascular endothelial cells, platelets, mesenchymal cells, fibroblasts and pericytes were positively correlated with each other (Fig. 2B), suggesting that stromal cell infiltration was probably associated with cell stemness.
Then we applied tSNE analysis for dimensional reduction (Fig. 2C) and obtained similar results as by hierarchical clustering analysis (Fig. 2A), suggesting that the subtyping system is mathematically robust. A microenvironmental-related scoring of patients in different subgroups was performed by ESTIMATE. We found that the Immune subgroup exhibited the highest, while the Stem-like subgroup exhibited the lowest ESTIMATE scores (Fig. 2D). Remarkably, the patients of the different TME subtypes showed distinct survival curves. The Immune subgroup had the worst prognosis compared to other groups during the first two years, whereas the Stem-like subgroup exhibited more unfavored outcomes after three years (Fig. 2E).
3.3 Molecular features of different TME subtypes
The mutation data showed that different TME subtypes shared a similar list of most commonly mutated genes, i.e., APC and TP53, and that the most common mutation category was missense mutation (Fig. 2F). Despite the consistency of the top-ranked mutated genes, we found that the detailed mutation profiles varied among the four subgroups (Fig. S3A-D). Remarkably, patients in the Immune group showed the highest frequency of SYNE1 and LRP1B mutations, whereas the Canonical group showed relatively higher FAT4 and RYR1 mutation frequencies compared to other groups. The featured mutated gene in the Dormant group was NRAS, while the Stem-like group was marked by the highest KRAS and PIK3CA mutation rates (Fig. S3E). In addition, we found that patients in the distinct subgroups also exhibited different levels of DNA repair-related pathways (Fig. S4A), further underscoring the heterogeneity of genomic mutation statuses among different TME subgroups.
We also employed other datasets to investigate TME levels of different subgroups. The results well matched the subgroup characteristics. For example, patients in the Immune subgroup exhibited the highest enrichment levels of the TME-related pathways, followed by the Canonical subgroup (Fig. S4B). Although the Dormant and the Stem-like groups exhibited similar extents of immune correlated components (e.g. CD8 T effector and Immune checkpoint), the Stem-like group showed lower epithelial-mesenchymal transition (EMT) and PAN-F-TBRS (Pan-fibroblast TGF β response signature) levels. In addition, we found that the levels of HLA proteins, immune-related stimulators, immune-related inhibitors and microenvironmental molecules varied among the different subgroups (Figs. 2G-H and S5). Results from TCIA revealed that patients in the four subgroups exhibited different immune-response predictions including those of anti-PD1, anti-CTLA4 and a combination of both (Fig. 2I).
3.4 Differentially expressed genes among TME subtypes
We further explored the featured genes of the different subgroups. As the Canonical subgroup included the largest number of patients and exhibited moderate immune and non-immune stromal cell levels, we used this subgroup as a reference. First, we found that 1994 genes were overexpressed and 150 genes were downregulated in the Immune subgroup compared to the Canonical group (Fig. S6A). Further GSEA analysis revealed that patients in the Immune subgroup exhibited higher expression levels in the FC receptor signaling pathway and the antigen processing and presentation pathway compared to the Canonical group (Fig. S6B, C). Second, we found that 9 genes were overexpressed and 472 genes were downregulated in the Dormant group compared to the Canonical group (Fig. S7A). Patients in the Dormant group exhibited comparatively inferior expression levels in the T cell receptor signaling pathway and interferon-gamma mediated signaling pathway (Fig. S7B, C). Third, 108 genes were found to be overexpressed and 1279 genes to be downregulated in the Stem-like group compared to the Canonical group (Fig. S8A). Patients in the Stem-like group exhibited lower expression levels in the endothelial migration and vascular endothelial growth factor receptor signaling pathway compared to the Canonical group (Fig. S8B, C).
All differentially expressed genes (DEGs) between the Canonical group and the other 3 subgroups are shown in Fig. S9. Among these DEGs, only 36 genes were correlated with the prognosis of RC patients (Fig. S9B), suggesting that less than 1/10 TME featured DEGs contributed most to the prognostic value of the TME subtyping system. Hence, different TME subtypes of RC exhibited distinct clinical outcomes, various molecular features and diverse featured gene lists (Fig. S9C), providing the possibility to construct a microenvironmental-related prognostic signature with those DEGs among the different subtypes.
3.5 Construction of a degenerated TME subtyping method by WGCNA-LASSO
To increase the accuracy and simplicity of the signature, we performed WGCNA analysis to divide the DEGs into different modules. Next, we ranked the prognostic genes in Fig. S9B based on their p-values and selected the top 5 prognostic genes in each module by lasso regression. If there were less than 5 prognostic genes in a module, all the prognostic genes in this module were included. The purpose of this step was to enhance the orthogonality of the genes involved in the signature. As shown in Figs. 3A and S10A, the patients were mainly divided into 5 modules. These different modules were correlated with different microenvironmental factors and different clinical characteristics (Figs. 3B and S10B). Subsequently, we performed a Pearson's correlation analysis on the prognostic genes. The genes with a correlation score higher than 0.5 were visualized by Cytoscape software (Fig. 3C). Additional GO and KEGG analyses also revealed that patients in different groups possessed different enrichment statuses of immune cell adhesion, extracellular organization, homeostasis and immune-related signaling pathways (Fig. 3D, E). Based on these results, the genes that we selected exhibited different microenvironmental statuses and a high orthogonality. Lastly, 19 genes were selected for lasso regression, which belonged to 5 distinct modules (MRAP, PLUNC: Yellow module; IL7R, DEGS1, BCL6, DHRS11 and FAM129A: Blue module; KIF13A, ADAMTSL4, TIMP1, PDGFRA and FNBP1: Turquoise module; WNT5A, RRAGD, MYL9, ACTG2 and HCP5: Brown module; EDIL3 and CFL2: Grey module). Next, a 10-gene signature was generated (Fig. 3F), and we determined the risk score of each patient as the sum of each gene multiplied by the corresponding coefficient (Fig. 3G, Table S4).

Degenerated TME subtyping method by WGCNA-LASSO. A. WGCNA obtained co-expression modules of the DEGs in Fig. S9A. Different modules are represented by different colors. Genes in grey did not belong to any of the modules. B. Heatmap showing the results of correlation analyses of different modules. C. Visualization of the co-expression network for prognostic genes in Fig. S9B with high co-expression scores (> 0.5) with any other gene by Pearson’s correlation analysis. The colors of the nodes refer to the classified modules. D. Bubble KEGG plot analysis identifying cancer-related pathways with genes enriched in each module. E. Bubble GO analysis chart identifying molecular functions with genes enriched in each module. F. Partial likelihood deviance revealed by LASSO regression in the 10-fold cross-validation. The optimal values are shown within the two dotted vertical lines. G. Distribution of the coefficient scores of each signature-selected gene. Red indicates a positive and green indicates a negative correlation with the risk score
Based on the classification, patients in the TCGA cohort were divided into high- and low-risk groups by the median risk score (Fig. 4A). The patients in the high-risk group had a lower overall survival rate than those in the low-risk group (Fig. 4B). The accuracy of the signature was validated by the AUCs of 1- and 3-year survival, which were 0.848 and 0.948, respectively (Fig. 4C). The calibration curve revealed that the actual survival status was quite similar to our prediction (Fig. 4D). Besides, we employed different machine learning algorithms to predict the 5-year survival status. To avoid overfitting effects, we randomly selected half of the samples as the training set and used the whole samples as the test group. Notably, the 10-gene panel integrated with all of the algorithms turned out to be robust in predicting the 5-year overall survival status (Fig. 4E). In addition, this signature predicted the DFS (Fig. 4F) and PFS (Fig. 4G) in RC patients. Noticeably, other clinical factors such as age, positive lymph node status and distant metastasis, were also correlated with the prognosis (Fig. 4H). By integrating all these risk factors, we found that the 10-gene signature score serves as an independent factor for the prognosis of RC patients (Fig. 4I).

Testing of the degenerated TME signature in the TCGA cohort. A. The upper chart shows the division of RC patients into high- and low-risk groups by the median cut-off of the risk score. The lower chart shows the survival time and survival status of patients in the TCGA cohort with different risk scores. B. Kaplan-Meier survival plot revealing the overall survival of RC patients in the high- and low-risk subgroups. C. ROC curve of the signature of 1-year and 3-year survival. D. Calibration curve showing that the actual survival time is similar to the predicting ROC curve. E. ROC curves of the 10-gene panel using different machine learning algorithms at 5-year survival. F. Kaplan-Meier survival plot revealing the disease-free survival of RC patients in the high- and low-risk subgroups. G. Kaplan-Meier survival plot revealing the progression-free survival of RC patients in the high- and low-risk subgroups. H. Univariate Cox regression analysis identifying risk factors correlating with overall survival. I. Multivariate Cox regression analysis showing that the 10-gene signature may serve as an independent prognostic factor
3.6 Verification of the degenerated TME signature by GSE17537 and GSE161158
To evaluate the applicability of the signature, two GEO datasets were employed for validation. The patients were divided into high- and low-risk groups by the median risk score. The GSE17537 results showed that patients in the high-risk group have a worse overall survival (OS; Fig. 5A) and disease-free survival (DFS; Fig. 5D) than those in the low-risk group. The accuracy of the signature was validated by the AUC of 1-, 3- and 5-year, which exhibited a good prognostic performance in both OS (Fig. 5B) and DFS (Fig. 5E). The calibration curve further showed that the actual prognostic outcome matched well with our prediction results (Fig. 5C, F). In GSE161158, patients in the high-risk group also exhibited a worse DFS (Fig. 5G) than those in the low-risk group. The 1-, 3-, and 5-year AUC values of this signature were 0.641, 0.680 and 0.657, respectively (Fig. 5H). The calibration curve showed that the actual prognostic outcome matched well with our predictions (Fig. 5I). We found that patients in GSE161158 with increased risk scores were strongly correlated with advanced tumor stages, indicating that this signature correlates with tumor progression (Fig. 5J). We also verified this 10-gene signature as an independent prognostic factor by integrating it with other clinical factors (Fig. 5K, L).

Verification of the degenerated TME signature by GSE17537 and GSE161158 cohorts. A. Kaplan-Meier survival plot revealing overall survival between the high- and low-risk subgroups in the GSE17537 cohort. B. ROC curve of this signature of 1-, 3- and 5-year overall survival. C. Calibration curve revealing that the actual survival time is similar to the predicting overall survival ROC curve. D. Kaplan-Meier survival plot revealing disease-free survival between the high- and low-risk subgroups in the GSE17537 cohort. E. ROC curve of the signature at 1-, 3- and 5-year disease-free survival. F. Calibration curve revealing that the actual survival time is similar to the predicting disease-free survival ROC curve. G. Kaplan-Meier survival plot revealing disease-free survival between the high- and low-risk subgroups in the GSE161158 cohort. H. ROC curve of the signature of 1-, 3- and 5-year disease-free survival. I. Calibration curve revealing that the actual survival time is quite similar to the predicting disease-free survival ROC curve. J. Boxplot showing the risk score distribution of patients in different tumor stages in the GSE161158 cohort. K. Univariate Cox regression analysis identifying the risk factors correlating with overall survival in the GSE161158 cohort. L. Multivariate Cox regression analysis showing that the 10-gene signature may serve as an independent prognostic factor in the GSE161158 cohort. The statistical analysis of the risk score distributions in different tumor stages was tested using the Wilcox algorithm
3.7 Features of the degenerated TME signature and its clinical application
To infer the effectiveness of this 10-gene signature from a biological and clinical perspective, we investigated its correlation with clinical factors and different biological processes. We found that patients with increased risk scores were correlated with advanced tumor stage, growing tumor size and node metastasis (Fig. 6A-D). Subsequently, GSEA was used to reveal the biological features of this signature. The high-risk group was mainly enriched in RNA localization and mRNA transport (Fig. 6E), while the low-risk group was comparatively enriched in metabolic processes, antigen processing and presentation of endogenous antigens (Fig. 6F). Compared with the TME classification, the high-risk patients matched more closely with the Immune group and the Stem-like group, while the low-risk patients matched more closely with the Canonical group and the Dormant group (Fig. 6G). Moreover, the low-risk patients matched more closely with the POLE and GS categories (Fig. 6G).

Correlations between the risk score and different features. A-D. Boxplots showing that patients with different risk scores exhibit different clinical statuses. Patients with advanced stage, large tumor size and positive node metastasis are positively correlated with higher risk scores. E. Enrichment plot showing the biological features of which the high-risk group is associated with. F. Enrichment plot showing the biological functions of which the low-risk group is associated with. G. Circle plot showing sample distributions between the risk groups, TME subtypes and TCGA classifications. H. Discrepancy of IPS for anti-PD1 immunotherapy between the two subgroups. I. Kaplan-Meier survival plot showing the overall survival status between high- and low-risk groups in the IMvigor210 dataset. J. Box plot showing the distributions of risk scores between CR/PR (complete response/partial response) and SD/PD (stable disease/ progressive disease) patients in IMvigor210. K. Kaplan-Meier survival plot showing the overall survival status between high- and low-risk groups in the GSE78220 dataset. L. Box plot showing the distributions of risk scores between R (response) and NR (non- response) patients in GSE78220. M-P. Box plots showing the IC50 values of different chemotherapeutic drugs between high- and low-risk patients. Q. The mutation landscape of patients in the high-risk group. R. Mutation landscape of patients in the low-risk group. The statistical analyses of Fig. 6A-D, H, J, L-P were all performed using the Wilcox test, *p < 0.05
We also explored whether this signature could be used for other treatment decision-making scenarios. First, the prediction of TCIA suggested a better effect of anti-PD1 immunotherapy on the low-risk group (Fig. 6H). Furthermore, two immunotherapy datasets, IMvigor210 and GSE78220, were employed for the evaluation of this 10-gene panel in immunotherapy response prediction. We found that the patients with a higher risk score not only showed a worse prognosis in the two datasets (Fig. 6I, K), but also showed worse responses to anti-PDL1 (Fig. 6J) and anti-PD1 (Fig. 6L) therapy. Hence, we suggest this 10-gene panel as an effective prediction tool for immune checkpoint-based immunotherapy, especially for PD1/PDL1 blockades. Moreover, we found that the effectiveness of chemotherapeutic drugs (e.g. Imatinib and Sorafenib) could also be predicted by the signature. Patients in the low-risk group exhibited lower IC50 values for these chemotherapeutic drugs (Fig. 6M-P).
Through the analysis of mutation profiles, we found that the most frequent mutated gene list was largely shared by both subgroups (Fig. 6Q, R). However, the high-risk group (Fig. 6Q) showed a comparatively lower overall mutation rate compared to the low-risk group (Fig. 6R), which could be a partial explanation for the different therapeutic responses noted.
Among the 10 genes in the signature, we found that DEGS1 and RRAGD were upregulated in the high-risk group (Fig. 7A, B). Upregulation of these two genes was correlated with higher risk degrees (Fig. 7C, D). Moreover, we observed strong correlations between DEGS1/RRAGD overexpression and tumor stage (Fig. 7E, F) and tumor size (Fig. 7G, H). Additionally, downregulation of RRAGD was observed in stage I tumors (Fig. 7F). Concordantly, we found that RRAGD expression in T4 was significantly higher than in T1–3 (Fig. 7H). Subsequent TIMER analysis revealed that DEGS1 was correlated with the infiltration of CD8+ T cells, macrophages, neutrophils and dendritic cells (Fig. 7I). RRAGD expression was found to be correlated with the infiltration of CD4+ T cells, macrophages, neutrophils and dendritic cells (Fig. 7J).

DEGS1 and RRAGD are correlated with clinical parameters and immune infiltration. A. Upregulation of DEGS1 in the high-risk group. B. Upregulation of RRAGD in the high-risk group. C. Spearmans’ correlation plot showing that the increasing expression level of DEGS1 is positively correlated with the risk score. D. Spearmans’ correlation plot showing that the increasing expression level of RRAGD is positively correlated with the risk score. E. Correlation of DEGS1 expression with tumor stage. F. Correlation of RRAGD expression with tumor stage. G. Correlation of DEGS1 expression with tumor size. H. Correlation of RRAGD expression with tumor size. I. TIMER platform revealing Spearmans’ correlations between DEGS1 and the infiltration of different immune cells. J. TIMER platform revealing Spearmans’ correlations between RRAGD and the infiltration of different immune cells. The statistical analyses of gene expression levels in different risk groups and risk score distributions in different clinical parameters were tested using the Wilcox test
We also performed stratification survival analyses. In most subpopulations, the high-risk group exhibited worse survival rates compared to the low-risk group (Fig. 8A-L). In addition, a nomogram was constructed combining this signature with age and M stage to predict the survival of RC patients (Fig. 8M). The ROC curve showed the AUC for 1 year, 3 year and 5 year survival rates were 0.875, 0.957 and 1.000, respectively (Fig. 8N). The actual survival of the patients was deduced from the calibration curve, which was similar to our prediction by the pre-established nomogram (Fig. 8O).

Clinical application of the 10-gene signature in combination with other clinical parameters to predict the prognosis of RC patients. A-L. Stratified analyses showing that patients in the high-risk group are correlated with poor overall survival. M. Nomogram curve showing that the 10-gene signature combines age and M stage to predict the survival status. N. ROC curve of the established nomogram. O. Calibration curve showing that the actually observed 1-, 3- and 5-year overall survival status is close to our prediction
4 Discussion
Previous studies on TME-based stratification of CRC mainly focused on immune cells, while the putative importance of non-immune cells was neglected. Chen et al., for instance, identified three TME subtypes based on tumor-infiltrating lymphocytes (TILs) [37]. Zhao et al. used T cell, B cell and Macrophage (TBM) scores to predict the metastasis of CRC, thereby establishing a prognostic Immune Scoring System [38]. Chong et al. divided CRCs into three clusters using m6A regulators and explored the immune cell subsets in each cluster [39]. Besides, other studies have investigated correlations between the TME and other biological processes, including pyroptosis [40, 41] and chemotherapy response [42]. Here, we identified evident interplays between epithelial cells and immune cells. Those interactions were shown to affect classical cancer signaling pathways, highlighting the crucial role of non-immune components in the TME. Furthermore, we identified the abundance of myofibroblasts, goblet cells, goblet progenitor cells and Paneth cells as prognosis-related factors. Hence, it would be more suitable to define the TME considering the integration of representative immune, stromal and epithelial components. Based on overall TME features (immune-correlated components, non-immune stromal cells and epithelial cells), all RC patients could be separated into Immune-, Canonical -, Dormant- and Stem-like subgroups. Compared to other stratification studies, our subtyping results not only enabled the identification of differences in clinical features, especially prognosis, but also provided a biological interpretability of those differences. Consequently, the TME features in distinct RC subgroups further emphasized a crucial role of non-immune stromal cells.
It should also be noted that CRC is a broad definition encompassing both colon cancer (CC) and RC. Different anatomical features of cancer may bring different risk factors and different biological processes during carcinogenesis. For example, there is a wide difference in not only clinical features (e.g. prognosis, lesion morphology, stage distribution), but also biological characteristics (e.g. mutation profile, CpG island methylator phenotype, microsatellite instability) between right- and left-sided CC [43, 44]. Thus, it would not be appropriate to simply adapt the CRC TME subtyping system to RC stratification. However, few studies probed into the stratification of RC. Here we separated all RC patients into four distinct subgroups based on the estimated abundance of immune cells, non-immune stromal cells and epithelial cells. The patients in the different subgroups not only exhibited different TME features, but also distinct prognostic statuses. The Immune subgroup had the worst prognosis compared to other groups in the first two years, whereas the Stem-like subgroup exhibited more unfavored outcomes after three years. Considering that cancer stem cells (CSCs) may stay static for a long time while their stemness allows them to accumulate carcinogenic and mutagenic inducers including inflammation and oxidative stress, CSCs are regarded as precursors of tumor recurrence and metastases [45], which well fits the trends of the survival curve of the Stem-like subgroup.
The mutation landscape may reflect the genetic carcinogenic path to some extent. Despite commonly mutated genes such as APC, other high-frequency mutated genes showed different mutation rates in different subgroups. LRP1B and SYNE1 were the most representative mutated genes in the Immune subgroup. Previous studies have already reported the occurrence of somatic LRP1B and SYNE1 mutations during the progression of various cancers [46,47,48]. LRP1B mutations have been correlated with TMB and could serve as an independent prognostic factor in hepatocellular carcinoma (HCC) [46] and glioblastoma [47]. Similarly, SYNE1 mutations have been correlated with a higher TMB level and a worse prognosis [48]. Those findings are in good accordance with our conclusion that the Immune subgroup has the worst prognosis compared to other groups in the first two years. KRAS and PIK3CA were the most representative mutated genes in the Stem-like subgroup, which showed disappointing outcomes after three years. It has already been reported that PIK3CA mutations are commonly accompanied by KRAS mutations [49], which is in line with our results. Li et al revealed that KRAS mutations were correlated with lymphovascular tumor embolism and perineural infiltration [50]. Moreover, KRAS mutations in exons 3/4 and PIK3CA mutations were simultaneously found to be correlated with a worse prognosis in CRC patients [49], which could partially explain the dramatic decline in the survival curve in the Stem-like subgroup after three years.
As predicted by TCIA, the four RC subtypes exhibited different responses to immunotherapies including anti-PD1, anti-CTLA4 and a combination of both. The ESTIMATE scores and HLA levels of the four RC subtypes were also different from each other. Corresponding to the difference in estimated immune cell abundance, those distinct immunological features provided a molecular basis for a different sensitivity to immunotherapy. Noticeably, Chen et al. proposed to classify cancers into immune-desert, immune-excluded and immune-inflamed phenotypes. They also provided meticulous biological mechanisms for the interpretation of different immune responses of different immune phenotypes [51]. Hereafter, this classification frame was widely referred to and recreated in numerous studies. For instance, Chen et al. separated gastric cancer patients into 3 subgroups via an unsupervised consensus algorithm [52]. These subgroups exhibited dissimilar immune features and the immune-inflamed subgroup was correlated with a strong immune activation and a better prognosis compared to the other groups. Moreover, the immune-inflamed subgroup showed the highest sensitivity to anti-PD1 immunotherapy and chemotherapy [53]. Here, we also found that the patients with the highest level of immune cell infiltration (the Immune subtype) exhibited the worst survival, which seems to be contradictory to previous findings. Thus, the Immune subtype we identified could not be simply mapped to the immune-inflamed cancers. The highest level of non-immune stromal cell infiltration in the Immune subtype would provide good support for cancer cell proliferation and metastasis [54], providing a partial explanation for its unfavorable prognosis.
Even though the four subtypes we identified from all RC patients could facilitate our understanding of the heterogeneity of RC, it is not suitable to reproduce this complicated subtyping in clinical scenarios. Hence, we employed integrated bioinformatics tools to develop a degenerated prognostic model. WCGNA is a powerful algorithm to explore potential gene-gene correlations in an expression matrix. We obtained five modules of DEGs via WGCNA and selected orthogonal prognostic genes for modeling without subjectivity and biases. Next, we applied LASSO regression to screen the top-ranked genes identified by WCGNA in each module and established a 10-gene prognostic model. Then, we applied several machine learning algorithms to integrate those genes to achieve a higher accuracy for survival prediction and a nomogram was adopted for further including clinical variables and visualization. The final degenerated prognostic model we established could well distinguish patients with different prognoses in both the testing dataset and the validation datasets. Moreover, it could predict different responses to immune checkpoint blockade immunotherapy and the application of chemotherapeutic drugs.
The DEGS1, MRAP, PLUNC, RRAGD, EDIL3, IL7R, DHRS11, HCP5, WNT5A and KIF13A were selected to construct a 10-gene signature. Many of these genes have already been proven to participate in carcinogenesis and the TME. As a major dihydroceramide desaturase in human neuroblastoma cells, DEGS1 was identified as being crucial for cell proliferation via cell cycle checkpoint evasion [55]. ABTL0812, an anti-cancer molecule, has been proven to suppress DEGS1 activity to promote cytotoxic autophagy in cancer cells [56]. Moreover, inhibition of DEGS1 has been found to activate endoplasmic reticulum stress and autophagy, which is essential in maintaining immunophenotypic and functional stem cells [57]. RRAGD is a monomeric guanine nucleotide-binding protein that enhances nutritious metabolism and tumor proliferation via activating the mTOR pathway [58]. RRAGD has also been reported to be overexpressed and to correlated with an unfavorable outcome in HCC patients [59]. Here, we identified DEGS1 and RRAGD as being overexpressed in the high-risk RC group, and to be correlated with advanced tumor stage and tumor size, further highlighting the clinical value of evaluating their expression in cancer. We also found that DEGS1 was correlated with the infiltration of different immune cells, especially neutrophils, providing a clue for the potential immunomodulatory role of DEGS1.
In conclusion, we divided all RC patients into four subtypes based on different compositions of TME components and afforded the biological interpretability for those differences. Moreover, the differences in clinical characteristics and prognosis among different RC subtypes enabled us to establish a robust and concise model by including only 10 DEGs, which provided predictions for both prognosis and immunotherapy response. We believe that both the non-predefined four subtypes we established by unsupervised clustering and the corresponding degenerated 10-gene prognostic model may facilitate our understanding of RC heterogeneity, help the clinical stratification of RC patients and assist the clinical decision-making process.
Data availability
All sequencing data were public available datasets (TCGA https://portal.gdc.cancer.gov/cart and GEO https://www.ncbi.nlm.nih.gov/geo/). All other data supporting the conclusions of this article are presented within the article and its supplementary files.
References
H. Sung, J. Ferlay, R.L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal, F. Bray, Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. (2021). https://doi.org/10.3322/caac.21660
D.S. Keller, M. Berho, R.O. Perez, S.D. Wexner, M. Chand, The multidisciplinary management of rectal cancer. Nat. Rev. Gastroenterol. Hepatol. (2020). https://doi.org/10.1038/s41575-020-0275-y
W.J. Tan, H.J. Tan, S.R. Dorajoo, F.J. Foo, C.L. Tang, M.H. Chew, Rectal Cancer surveillance-recurrence patterns and survival outcomes from a cohort followed up beyond 10 years. J. Gastrointest. Cancer (2018). https://doi.org/10.1007/s12029-017-9984-z
R. Braun, L. Anthuber, D. Hirsch, D. Wangsa, J. Lack, N.E. McNeil, K. Heselmeyer-Haddad, I. Torres, D. Wangsa, M.A. Brown, A. Tubbs, N. Auslander, E.M. Gertz, P.R. Brauer, M.C. Cam, D.L. Sackett, J.K. Habermann, A. Nussenzweig, E. Ruppin, et al., Single-cell-derived primary rectal carcinoma cell lines reflect Intratumor heterogeneity associated with treatment response. Clin. Cancer Res. (2020). https://doi.org/10.1158/1078-0432.CCR-19-1984
Z. Lakkis, G. Manceau, V. Bridoux, A. Brouquet, S. Kirzin, L. Maggiori, C. de Chaisemartin, J.H. Lefevre, Y. Panis, Management of rectal cancer: The 2016 French guidelines. Color. Dis. (2017). https://doi.org/10.1111/codi.13550
N.M. Anderson, M.C. Simon, The tumor microenvironment. Curr. Biol. (2020). https://doi.org/10.1016/j.cub.2020.06.081
I. Vitale, G. Manic, L.M. Coussens, G. Kroemer, L. Galluzzi, Macrophages and metabolism in the tumor microenvironment. Cell Metab. (2019). https://doi.org/10.1016/j.cmet.2019.06.001
M. Saxena, N. Bhardwaj, Re-emergence of dendritic cell vaccines for Cancer treatment. Trends Cancer (2018). https://doi.org/10.1016/j.trecan.2017.12.007
J.J. Loh, S. Ma, The role of Cancer-associated fibroblast as a dynamic player in mediating Cancer Stemness in the tumor microenvironment. Front. Cell Dev. Biol. (2021). https://doi.org/10.3389/fcell.2021.727640
S. Sui, X. An, C. Xu, Z. Li, Y. Hua, G. Huang, S. Sui, Q. Long, Y. Sui, Y. Xiong, M. Ntim, W. Guo, M. Chen, W. Deng, X. Xiao, M. Li, An immune cell infiltration-based immune score model predicts prognosis and chemotherapy effects in breast cancer. Theranostics (2020). https://doi.org/10.7150/thno.49451
W. Jiang, D. Zhu, C. Wang, Y. Zhu, An immune relevant signature for predicting prognoses and immunotherapeutic responses in patients with muscle-invasive bladder cancer (MIBC). Cancer Med. (2020). https://doi.org/10.1002/cam4.2942
R. Zhou, J. Zhang, D. Zeng, H. Sun, X. Rong, M. Shi, J. Bin, Y. Liao, W. Liao, Immune cell infiltration as a biomarker for the diagnosis and prognosis of stage I-III colon cancer. Cancer Immunol. Immunother. (2019). https://doi.org/10.1007/s00262-018-2289-7
P. Ge, W. Wang, L. Li, G. Zhang, Z. Gao, Z. Tang, X. Dang, Y. Wu, Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed. Pharmacother. (2019). https://doi.org/10.1016/j.biopha.2019.109228
M. Binnewies, E.W. Roberts, K. Kersten, V. Chan, D.F. Fearon, M. Merad, L.M. Coussens, D.I. Gabrilovich, S. Ostrand-Rosenberg, C.C. Hedrick, R.H. Vonderheide, M.J. Pittet, R.K. Jain, W. Zou, T.K. Howcroft, E.C. Woodhouse, R.A. Weinberg, M.F. Krummel, Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. (2018). https://doi.org/10.1038/s41591-018-0014-x
Z. Wu, K. Zhu, Q. Liu, Y. Liu, L. Chen, J. Cui, H. Guo, N. Zhou, Y. Zhu, Y. Li, B. Shi, Profiles of immune infiltration in bladder Cancer and its clinical significance: An integrative genomic analysis. Int. J. Med. Sci. (2020). https://doi.org/10.7150/ijms.42151
J. Liu, Z. Tan, J. He, T. Jin, Y. Han, L. Hu, J. Song, S. Huang, Identification of three molecular subtypes based on immune infiltration in ovarian cancer and its prognostic value. Biosci. Rep. (2020). https://doi.org/10.1042/BSR20201431
P. Hu, Y. Gao, Y. Huang, Y. Zhao, H. Yan, J. Zhang, L. Zhao, Gene expression-based immune cell infiltration analyses of prostate Cancer and their associations with survival outcome. DNA Cell Biol. (2020). https://doi.org/10.1089/dna.2020.5371
S. Yang, T. Liu, Y. Cheng, Y. Bai, G. Liang, Immune cell infiltration as a biomarker for the diagnosis and prognosis of digestive system cancer. Cancer Sci. (2019). https://doi.org/10.1111/cas.14216
X. Liu, S. Wu, Y. Yang, M. Zhao, G. Zhu, Z. Hou, The prognostic landscape of tumor-infiltrating immune cell and immunomodulators in lung cancer. Biomed. Pharmacother. (2017). https://doi.org/10.1016/j.biopha.2017.08.003
N. Kim, H.K. Kim, K. Lee, Y. Hong, J.H. Cho, J.W. Choi, J.I. Lee, Y.L. Suh, B.M. Ku, H.H. Eum, S. Choi, Y.L. Choi, J.G. Joung, W.Y. Park, H.A. Jung, J.M. Sun, S.H. Lee, J.S. Ahn, K. Park, et al., Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. (2020). https://doi.org/10.1038/s41467-020-16164-1
L.M. Becker, J.T. O'Connell, A.P. Vo, M.P. Cain, D. Tampe, L. Bizarro, H. Sugimoto, A.K. McGow, J.M. Asara, S. Lovisa, K.M. McAndrews, R. Zielinski, P.L. Lorenzi, M. Zeisberg, S. Raza, V.S. LeBleu, R. Kalluri, Epigenetic reprogramming of Cancer-associated fibroblasts deregulates glucose metabolism and facilitates progression of breast Cancer. Cell Rep. (2020). https://doi.org/10.1016/j.celrep.2020.107701
A. Butler, P. Hoffman, P. Smibert, E. Papalexi, R. Satija, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. (2018). https://doi.org/10.1038/nbt.4096
M.C. Cieslak, A.M. Castelfranco, V. Roncalli, P.H. Lenz, D.K. Hartline, T-distributed stochastic neighbor embedding (t-SNE): A tool for eco-physiological transcriptomic analysis. Mar. Genomics (2020). https://doi.org/10.1016/j.margen.2019.100723
Q. Huang, Y. Liu, Y. Du, L.X. Garmire, Evaluation of cell type annotation R packages on single-cell RNA-seq data. Genomics Proteomics Bioinformatics (2021). https://doi.org/10.1016/j.gpb.2020.07.004
Y. Zhang, T. Liu, X. Hu, M. Wang, J. Wang, B. Zou, P. Tan, T. Cui, Y. Dou, L. Ning, Y. Huang, S. Rao, D. Wang, X. Zhao, CellCall: Integrating paired ligand-receptor and transcription factor activities for cell-cell communication. Nucleic Acids Res. (2021). https://doi.org/10.1093/nar/gkab638
S. Mariathasan, S.J. Turley, D. Nickles, A. Castiglioni, K. Yuen, Y. Wang, E.I. Kadel, H. Koeppen, J.L. Astarita, R. Cubas, S. Jhunjhunwala, R. Banchereau, Y. Yang, Y. Guan, C. Chalouni, J. Ziai, Y. Senbabaoglu, S. Santoro, D. Sheinson, et al., TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature (2018). https://doi.org/10.1038/nature25501
P. Charoentong, F. Finotello, M. Angelova, C. Mayer, M. Efremova, D. Rieder, H. Hackl, Z. Trajanoski, Pan-cancer Immunogenomic analyses reveal genotype-Immunophenotype relationships and predictors of response to checkpoint blockade. Cell Rep. (2017). https://doi.org/10.1016/j.celrep.2016.12.019
G. Bindea, B. Mlecnik, M. Tosolini, A. Kirilovsky, M. Waldner, A.C. Obenauf, H. Angell, T. Fredriksen, L. Lafontaine, A. Berger, P. Bruneval, W.H. Fridman, C. Becker, F. Pages, M.R. Speicher, Z. Trajanoski, J. Galon, Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity (2013). https://doi.org/10.1016/j.immuni.2013.10.003
X. Zhang, Y. Lan, J. Xu, F. Quan, E. Zhao, C. Deng, T. Luo, L. Xu, G. Liao, M. Yan, Y. Ping, F. Li, A. Shi, J. Bai, T. Zhao, X. Li, Y. Xiao, CellMarker: A manually curated resource of cell markers in human and mouse. Nucleic Acids Res. (2019). https://doi.org/10.1093/nar/gky900
S. Hanzelmann, R. Castelo, J. Guinney, GSVA: Gene set variation analysis for microarray and RNA-seq data. Bmc Bioinformatics (2013). https://doi.org/10.1186/1471-2105-14-7
J. Wu, L. Li, H. Zhang, Y. Zhao, H. Zhang, S. Wu, B. Xu, A risk model developed based on tumor microenvironment predicts overall survival and associates with tumor immunity of patients with lung adenocarcinoma. Oncogene (2021). https://doi.org/10.1038/s41388-021-01853-y
R. Cao, B. Ma, G. Wang, Y. Xiong, Y. Tian, L. Yuan, Characterization of hypoxia response patterns identified prognosis and immunotherapy response in bladder cancer. Mol. Ther. Oncolytics (2021). https://doi.org/10.1016/j.omto.2021.06.011
P. Langfelder, S. Horvath, WGCNA: An R package for weighted correlation network analysis. Bmc Bioinformatics (2008). https://doi.org/10.1186/1471-2105-9-559
T. Wu, E. Hu, S. Xu, M. Chen, P. Guo, Z. Dai, T. Feng, L. Zhou, W. Tang, L. Zhan, X. Fu, S. Liu, X. Bo, G. Yu, clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation (Camb.) (2021). https://doi.org/10.1016/j.xinn.2021.100141
P. Geeleher, N. Cox, R.S. Huang, pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One (2014). https://doi.org/10.1371/journal.pone.0107468
D. Sun, Y. Zhu, H. Zhao, T. Bian, T. Li, K. Liu, L. Feng, H. Li, H. Hou, Loss of ARID1A expression promotes lung adenocarcinoma metastasis and predicts a poor prognosis. Cell Oncol. (Dordr.) (2021). https://doi.org/10.1007/s13402-021-00616-x
Y. Chen, J. Zhao, Identification of an immune gene signature based on tumor microenvironment characteristics in Colon adenocarcinoma. Cell Transplant. (2021). https://doi.org/10.1177/09636897211001314
Y. Zhao, C. Chen, X. Xu, X. Ge, K. Ding, S. Zheng, J. Wang, L. Sun, An efficient prognostic immune scoring system for colorectal Cancer patients with peritoneal metastasis. Oncoimmunology (2021). https://doi.org/10.1080/2162402X.2021.1901464
W. Chong, L. Shang, J. Liu, Z. Fang, F. Du, H. Wu, Y. Liu, Z. Wang, Y. Chen, S. Jia, L. Chen, L. Li, H. Chen, M(6)a regulator-based methylation modification patterns characterized by distinct tumor microenvironment immune profiles in colon cancer. Theranostics (2021). https://doi.org/10.7150/thno.52717
J. Rao, W. Li, C. Chen, Pyroptosis-mediated molecular subtypes and tumor microenvironment infiltration characterization in Colon Cancer. Front. Cell Dev. Biol. (2021). https://doi.org/10.3389/fcell.2021.766503
W. Song, J. Ren, R. Xiang, C. Kong, T. Fu, Identification of pyroptosis-related subtypes, the development of a prognosis model, and characterization of tumor microenvironment infiltration in colorectal cancer. Oncoimmunology (2021). https://doi.org/10.1080/2162402X.2021.1987636
X. Zhu, X. Tian, L. Ji, X. Zhang, Y. Cao, C. Shen, Y. Hu, J. Wong, J.Y. Fang, J. Hong, H. Chen, A tumor microenvironment-specific gene expression signature predicts chemotherapy resistance in colorectal cancer patients. NPJ Precis. Oncol. (2021). https://doi.org/10.1038/s41698-021-00142-x
C.A. Doubeni, D.A. Corley, V.P. Quinn, C.D. Jensen, A.G. Zauber, M. Goodman, J.R. Johnson, S.J. Mehta, T.A. Becerra, W.K. Zhao, J. Schottinger, V.P. Doria-Rose, T.R. Levin, N.S. Weiss, R.H. Fletcher, Effectiveness of screening colonoscopy in reducing the risk of death from right and left colon cancer: A large community-based study. Gut (2018). https://doi.org/10.1136/gutjnl-2016-312712
R. Yaeger, W.K. Chatila, M.D. Lipsyc, J.F. Hechtman, A. Cercek, F. Sanchez-Vega, G. Jayakumaran, S. Middha, A. Zehir, M. Donoghue, D. You, A. Viale, N. Kemeny, N.H. Segal, Z.K. Stadler, A.M. Varghese, R. Kundra, J. Gao, A. Syed, et al., Clinical sequencing defines the genomic landscape of metastatic colorectal Cancer. Cancer Cell (2018). https://doi.org/10.1016/j.ccell.2017.12.004
S. Talukdar, P. Bhoopathi, L. Emdad, S. Das, D. Sarkar, P.B. Fisher, Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. (2019). https://doi.org/10.1016/bs.acr.2018.12.002
F. Liu, W. Hou, J. Liang, L. Zhu, C. Luo, LRP1B mutation: A novel independent prognostic factor and a predictive tumor mutation burden in hepatocellular carcinoma. J. Cancer (2021). https://doi.org/10.7150/jca.53124
E. Tabouret, M. Labussiere, A. Alentorn, Y. Schmitt, Y. Marie, M. Sanson, LRP1B deletion is associated with poor outcome for glioblastoma patients. J. Neurol. Sci. (2015). https://doi.org/10.1016/j.jns.2015.09.345
P. Li, J. Xiao, B. Zhou, J. Wei, J. Luo, W. Chen, SYNE1 mutation may enhance the response to immune checkpoint blockade therapy in clear cell renal cell carcinoma patients. Aging (Albany NY) (2020). https://doi.org/10.18632/aging.103781
Q. Luo, D. Chen, X. Fan, X. Fu, T. Ma, D. Chen, KRAS and PIK3CA bi-mutations predict a poor prognosis in colorectal cancer patients: A single-site report. Transl. Oncol. (2020). https://doi.org/10.1016/j.tranon.2020.100874
S. Jang, M. Hong, M.K. Shin, B.C. Kim, H.S. Shin, E. Yu, S.M. Hong, J. Kim, S.M. Chun, T.I. Kim, K.C. Choi, Y.W. Ko, J.W. Kim, KRAS and PIK3CA mutations in colorectal adenocarcinomas correlate with aggressive histological features and behavior. Hum. Pathol. (2017). https://doi.org/10.1016/j.humpath.2017.01.010
D.S. Chen, I. Mellman, Elements of cancer immunity and the cancer-immune set point. Nature (2017). https://doi.org/10.1038/nature21349
Y. Chen, Z. Sun, W. Chen, C. Liu, R. Chai, J. Ding, W. Liu, X. Feng, J. Zhou, X. Shen, S. Huang, Z. Xu, The immune subtypes and landscape of gastric Cancer and to predict based on the whole-slide images using deep learning. Front. Immunol. (2021). https://doi.org/10.3389/fimmu.2021.685992
C. Tang, J. Ma, X. Liu, Z. Liu, Identification of four immune subtypes in bladder Cancer based on immune gene sets. Front. Oncol. (2020). https://doi.org/10.3389/fonc.2020.544610
M. Sasaki, N. Miyoshi, S. Fujino, K. Saso, T. Ogino, H. Takahashi, M. Uemura, H. Yamamoto, C. Matsuda, M. Yasui, M. Ohue, T. Mizushima, Y. Doki, H. Eguchi, The meiosis-specific cohesin component stromal antigen 3 promotes cell migration and chemotherapeutic resistance in colorectal cancer. Cancer Lett. (2021). https://doi.org/10.1016/j.canlet.2020.10.006
J.M. Kraveka, L. Li, Z.M. Szulc, J. Bielawski, B. Ogretmen, Y.A. Hannun, L.M. Obeid, A. Bielawska, Involvement of dihydroceramide desaturase in cell cycle progression in human neuroblastoma cells. J. Biol. Chem. (2007). https://doi.org/10.1074/jbc.M700647200
P. Munoz-Guardiola, J. Casas, E. Megias-Roda, S. Sole, H. Perez-Montoyo, M. Yeste-Velasco, T. Erazo, N. Dieguez-Martinez, S. Espinosa-Gil, C. Munoz-Pinedo, G. Yoldi, J.L. Abad, M.F. Segura, T. Moran, M. Romeo, J. Bosch-Barrera, A. Oaknin, J. Alfon, C. Domenech, et al., The anti-cancer drug ABTL0812 induces ER stress-mediated cytotoxic autophagy by increasing dihydroceramide levels in cancer cells. Autophagy (2021). https://doi.org/10.1080/15548627.2020.1761651
S.Z. Xie, L. Garcia-Prat, V. Voisin, R. Ferrari, O.I. Gan, E. Wagenblast, K.B. Kaufmann, A. Zeng, S.I. Takayanagi, I. Patel, E.K. Lee, J. Jargstorf, G. Holmes, G. Romm, K. Pan, M. Shoong, A. Vedi, C. Luberto, M.D. Minden, et al., Sphingolipid modulation activates Proteostasis programs to govern human hematopoietic stem cell self-renewal. Cell Stem Cell (2019). https://doi.org/10.1016/j.stem.2019.09.008
C. Di Malta, D. Siciliano, A. Calcagni, J. Monfregola, S. Punzi, N. Pastore, A.N. Eastes, O. Davis, R. De Cegli, A. Zampelli, L.G. Di Giovannantonio, E. Nusco, N. Platt, A. Guida, M.H. Ogmundsdottir, L. Lanfrancone, R.M. Perera, R. Zoncu, P.G. Pelicci, et al., Transcriptional activation of RagD GTPase controls mTORC1 and promotes cancer growth. Science (2017). https://doi.org/10.1126/science.aag2553
L. Ding, X. Liang, Ras related GTP binding D promotes aerobic glycolysis of hepatocellular carcinoma. Ann. Hepatol. (2021). https://doi.org/10.1016/j.aohep.2021.100307
Acknowledgements
We acknowledge the owners of TCGA, GEO and all other databases for providing valuable platforms and for making all those meaningful data available. All the contributions to those public datasets are deeply appreciated.
Funding
This work was supported by grants from the Beijing Nova Program of Science and Technology (Z191100001119128), the National Natural Science Foundation of China (82073390, 81702314), the Beijing Municipal Science and Technology Project (Z191100006619081), the Beijing Municipal Administration of Hospitals’ Youth Programme (QML20180108), the Funding Program for Excellent Talents of Beijing (2017000021469G212) and the Digestive Medical Coordinated Development Center of Beijing Municipal Administration of Hospitals (XXZ02, XXZ01). The study sponsors had no role in the design of the study and the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
Study concept and design: FB and LM. Data acquisition and data cleaning: YZ and FB. Data analysis and interpretation: FB, YSZ, YC and LS. Drafting of the manuscript: FB, XY, YZ and YSZ. Supervision: LM and SZ. Proofreading and revision: LM, SZ, LS, and YC. Critical revision of the manuscript for important intellectual content: All authors.
Corresponding authors
Ethics declarations
Ethics approval and inform consent
This study was conducted based on the open-source data from the TCGA and GEO databases. The patients involved in those databases have already provided ethical approval. All users were permitted to download relevant data for research and to publish relevant articles. Therefore, there are no ethical issues and other conflicts of interest regarding this article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bu, F., Zhao, Y., Zhao, Y. et al. Distinct tumor microenvironment landscapes of rectal cancer for prognosis and prediction of immunotherapy response. Cell Oncol. 45, 1363–1381 (2022). https://doi.org/10.1007/s13402-022-00725-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-022-00725-1