
Abstract
Background
Increasing evidence indicates that obesity is associated with tumor development and progression. Leptin is an adipocyte-related hormone with a key role in energy metabolism and whose circulating levels are elevated in obesity. The effect of leptin on cancer progression and metastasis and its underlying mechanisms are still unclear. Leptin can impact various steps in tumor metastasis, including epithelial-mesenchymal transition, cell adhesion to the extracellular matrix (ECM), and proteolysis of ECM components. To do so, leptin binds to its receptor (OB-Rb) to activate signaling pathways and downstream effectors that participate in tumor cell invasion as well as distant metastasis.
Conclusions
In this review, we describe metastasis steps in detail and characterize metastasis-related molecules activated by leptin, which may help to develop a roadmap that guides future work. In addition, we conclude that a profound understanding of the fundamental molecular processes that contribute to leptin-induced metastasis may pave the way for the development of new prognostic molecules and appropriate approaches to the treatment of obesity-related cancers.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Obesity is considered one of the most important health challenges in recent decades [1]. According to the world health organization (WHO), obesity prevalence has nearly tripled between 1975 and 2016 [2]. Obesity is a well-established risk factor for many disorders, such as type 2 diabetes and cardiovascular disease [3, 4]. In addition, epidemiologic studies have suggested a relationship between obesity and different cancer types [1, 5].
Cancer is one of the most common causes of death globally, with approximately 8 million deaths each year [6]. Several studies have indicated associations between obesity and different cancer types [7, 8], including hematological malignancies [9, 10], colon cancer [11], gastrointestinal malignancies [12], bladder and renal carcinoma [13], endometrial cancer [14, 15], ovarian cancer [16], hepatocellular carcinoma [17], and gastric, gallbladder [1], pancreatic [18, 19], prostate [20, 21] and breast cancer [22,23,24,25,26]. Several mechanisms have been proposed to explain the link between obesity and cancer progression, including increased lipid profiles in cancer, pro-tumorigenic signaling lipids, inflammation, insulin resistance and alterations in adipokine regulation [27]. Leptin is an important adipokine secreted by adipocytes and plays a key role in energy homeostasis regulation [4, 28]. Previous studies have shown mitogenic and anti-apoptotic effects of leptin in various cancer types. Leptin also influences cell migration and invasion, which are two important steps in tumor progression and metastasis [29,30,31,32].
Metastasis is the most important feature of malignant tumors and is the leading cause of cancer-related death [33]. This process includes several unique biological steps in which tumor cells separate from the primary site, migrate along primary tissue and invade into the blood or lymphatic circulation system until they reach distant target tissues [33, 34]. Molecular interactions between tumor cells, the extracellular matrix (ECM), and stromal cells are important in the metastasis process. Epithelial-mesenchymal transition (EMT) is a crucial step in the metastasis process and is characterized by conversion of epithelial cells into mesenchymal cells [35]. During EMT, epithelial cells obtain mesenchymal properties, including motility and invasiveness [36]. In this metastasis step, gene expression profiles switch from E-cadherin to N-cadherin, and cells manifest a migratory morphology [37]. Cell migration starts in response to an external signal associated with changes in cytoskeletal machinery that results in “leading front” extension. Thereafter, the leading front of tumor cells adheres to ECM components by means of adhesion molecules, including integrins and cadherins [38, 39]. Actin-myosin contraction is essential to generate a traction force, which promotes slow sliding of the cell body forward [40]. Actin-myosin assembly is regulated by various modulatory molecules, such as the Rho-GTPase family, Rho-associated kinase (ROCK), Myosin light-chain kinase (MLCK), Myosin light-chain phosphatase (MLCP), mammalian homolog of Drosophila diaphanous (mDia1), LIM kinases and the actin-depolymerizing factor (ADF)/cofilin family [41].
Another step in cell invasion is ECM degradation, mediated by proteolytic enzymes that are secreted by anchored tumor cells. This process provides a locally modified region in the matrix for invasive cells to migrate through the ECM [42, 43]. Matrix metalloproteinase (MMPs) [42] and the plasminogen activator (PA) system [44] are two types of hydrolyzing enzymes that participate in ECM degradation. Activation of different signaling pathways, such as Janus Kinase-Signal Transducer and Activator of Transcription-3 (JAK-STAT3) [45], Phosphatidylinositol 3-Kinase and AKT (PI3K/AKT) [46], and Mitogen-activated Protein Kinase (MAPK) [47] pathways, by cytokines and growth factors is responsible for regulating the expression of proteins contributing to the metastasis process. JAKs are tyrosine kinases that are activated and phosphorylated in response to cytokine receptor activation. Activated JAKs induce transphosphorylation and dimerization of STATs, which is crucial for their entrance into the nucleus [48]. PI3K is a key effector downstream of receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs) that generates phospholipids, activates several downstream effectors (such as AKT and mTOR), and produces intracellular messages in response to different growth factors and cytokines [49]. In addition, the MAPK family consists of three major groups: extracellular signal-regulated kinase (ERK), Jun N-terminal kinase (JNK) and p38 isoforms. The MAPKs are phosphorylated by kinases upon ligand-mediated activation of receptor tyrosine kinases [47].
Past studies have demonstrated that leptin stimulates cell migration and invasion by promoting the expression of proteins that participate in different steps in the metastasis process. In this review, we describe a number of molecular mechanisms that contribute to different metastasis steps and their roles in leptin-induced cancer progression. We focus on the different roles of EMT-associated markers, cell adhesion to ECM molecules, and particularly protease enzymes in leptin-induced metastasis. We also discuss the role of adiponectin, another adipokine, which has an effect opposite that of leptin on cancer progression. Finally, we review all the leptin antagonist molecules that can be considered as promising therapeutic agents in the treatment of obesity-related cancers. Inquiries for this review were carried out in bibliographic databases, including Google Scholar, PubMed, and Science Direct. We screened all studies regarding leptin and cancer metastasis published up to 2018. Studies that investigated the effects of leptin on cancer cell proliferation, cell growth, cell cycling or cell apoptosis or studies that demonstrated leptin effects on noncancerous cells were excluded.
2 Leptin and leptin receptors
Leptin, a 16-kDa protein encoded by the ob (obese) gene, comprises 167 amino acids and is predominantly produced by adipocytes [50]. While white adipose tissue is the main site of leptin secretion, other tissues can also secrete leptin, such as brown interscapular fat, placenta, ovaries, endometrium, stomach, hypothalamus, pituitary and cancer cells [51, 52]. The primary role of leptin is regulation of energy homeostasis by controlling energy intake and appetite through its effects on the hypothalamus nucleus [50]. Furthermore, leptin has important roles in the endocrine and immune systems and influences other processes, such as glucose homeostasis, bone formation, tissue repair and inflammation [53]. Leptin may directly act on various types of cancer cells and induce cancer initiation and progression [7].
The blood level of leptin is influenced by adipose tissue mass [54], and they have a positive correlation [55]. Serum leptin level is typically maintained at a concentration of less than 10 ng/ml in healthy subjects but may increase up to 50 ng/ml in obese individuals [56]. Serum leptin level is also affected by several physical, physiological, chemical, neurological, and genetic conditions. Alteration in body mass, energy balance, fasting/overfeeding, composition of diet, nutritional status, cigarette smoking, exercise and serum levels of molecules such as insulin, glucocorticoids, estrogen and testosterone isoproterenol, β-adrenergic receptor agonists, tumor necrosis factor-α (TNF-α), and interleukin (IL)-1 are among the main factors affecting the serum leptin level [50, 55, 57].
Several in vitro studies have demonstrated leptin-induced cell invasion and migration in different cancer cells, such as hepatocellular carcinoma [58, 59], lung cancer [60], breast cancer [31, 61,62,63,64,65], prostate cancer [66], colorectal cancer [67], melanoma [68], ovarian cancer [69] and renal carcinoma [70] cells. Recently, Huang et al. demonstrated that cell migration and invasion were increased through acetyl-CoA acetyltransferase 2 (ACAT2) upregulation after leptin stimulation in breast cancer. They found that the PI3K/AKT/Sterol regulatory element-binding protein 2 (SREBP-2) signaling pathway is involved in leptin-induced ACAT2 upregulation and the invasive effect of leptin [71]. In another study, it was shown that leptin enhances IL-18 upregulation and secretion in tumor-associated macrophages (TAMs), leading to increased migration and invasion of breast cancer cells. Pretreatment with a nuclear factor kappa-light-chain-enhancer of activated B cells (NF-Κβ) inhibitor significantly inhibited the stimulatory effects of leptin, signifying a role of NF-κB in leptin-induced IL-18 expression and cell invasion [72]. In contrast with these findings, Meerson et al. reported that leptin and insulin-induced microRNA (miR)-4443 overexpression blocked nuclear receptor coactivator 1 (NCOA1) and TNF receptor-associated factor-4 (TRAF4) expression and attenuated cell invasion of human colon cancer cells [73].
Leptin exerts its effects through leptin receptors (ObRs) expressed in the brain as well as in peripheral tissues. ObR, a member of the class I cytokine receptor family, has at least six isoforms: a long form (OB-Rb), four short forms (OB-Ra, OB-Rc, OB-Rd, and OB-Re) and a soluble form (OB-Re). They are produced via alternative mRNA splicing of the diabetes (db) gene [74, 75]. OB-Rb contains a long intracellular domain that is essential for the full signaling capability responsible for the biological effects of leptin [76]. Leptin binding to OB-Rb induces activation of several signaling pathways, such as JAK/STAT3, PI3K/AKT, and MAPK/ERK. These signaling pathways participate in stimulatory effects of leptin on the hallmarks of cancer development, including cell proliferation, apoptosis, cell migration and invasion, angiogenesis and vascular stimulation [58, 77,78,79,80,81,82,83].
The polymorphism of leptin and OB-Rb genes has been evaluated in several cancers [84,85,86,87,88,89,90,91,92,93,94,95,96,97]. A number of in vitro studies has shown the involvement of OB-Rb in cell invasion and metastasis in various cancer cell lines [98]. Moreover, in vivo studies demonstrated a positive correlation between OB-Rb overexpression and cancer progression [84, 99,100,101,102,103,104]. How obesity is correlated with the survival of patients suffering from metastatic cancer compared with nonobese patients is not fully understood. Recently, Kato et al. observed that overweight cancer patients had higher serum leptin levels than healthy nonobese patients. In addition, they found a positive correlation between serum leptin levels and ascites levels, and they observed higher noticeable ascites levels among overweight and obese patients compared with healthy nonobese cancer patients. Their results indicated that the leptin/OB-Rb pathway, and maybe other inflammatory cytokines, particularly in obese women, could participate in the survival of floating cancer cells in ascites or the abdominal cavity following debulking surgery, helping their migration to secondary sites and recurrence and progression of cancer [105]. They demonstrated a higher OB-Rb expression level in ascites and metastatic tumors than in benign subjects and reported that OB-Rb has a key role in crucial metastasis steps, controlling processes such as migration and cell invasion [105]. In another study on ovarian cancer, overexpression of OB-Rb was found to be correlated with an unfavorable outcome in Middle Eastern patients [106]. Furthermore, Fan et al. found that OB-Rb is overexpressed in pancreatic cancer tissues and that its activation by leptin promotes cell invasion in pancreatic cancer [107]. Similarly, in renal cancer, it was reported that OB-Rb overexpression is associated with venous invasion, histological type and lymph node metastasis [108]. Moreover, Ishikawa et al. analyzed 207 gastric cancer tissues (100 early and 107 advanced carcinomas) and detected lymph node metastasis in 49.5% of subjects with high leptin expression and in 50.5% of OB-Rb-positive subjects [109]. In colorectal cancer, it was noticed that the expression of OB-R is significantly higher in metastatic colorectal tissues than in local colorectal cancer tissues [110]. In a cohort study of 173 subjects, a strong correlation was found between OB-R expression and nodal metastasis and advanced stage in medullary thyroid carcinoma (MTC) [111]. Furthermore, it was shown that ObR is markedly overexpressed in metastatic breast cancer tissues compared with normal tissues [112, 113]. Additionally, overexpression of OB-Rb was detected in 62% of non-small-cell lung cancer tissues versus 31% of noncancer lung tissues [114].
3 EMT-associated markers
EMT is defined by loss of epithelial properties with concomitant gain of a mesenchymal-like phenotype and participates in processes such as cancer cell invasion and metastasis [35]. During EMT, epithelial cells lose their cell polarity and cell-cell adhesion ability and gain migratory and invasive characteristics [36].
Among cell adhesion molecules, the cadherin superfamily consists of more than 80 members. E-cadherin (CDH1), a major member of the cadherin family, is extensively found in epithelial cells and plays a crucial role in adherens junctions and the stability of cell-cell connections [115, 116]. Epithelial cells link to their neighboring cells through the extracellular domain of two E-cadherins. Moreover, the cytoplasmic face of adherens junctions links to the actin cytoskeleton through binding of E-cadherin to β-catenin, α-catenin, and p120-catenin [117, 118]. In EMT, E-cadherin gene expression is modulated by certain transcriptional factors, such as SNAI1, SNAI2, and ZEB2 or Twist, leading to destabilization of adherens junctions [35]. In addition, upregulation of mesenchymal markers, such as vimentin and N-cadherin, results in cytoskeleton remodeling. N-cadherin (CDH2) is another member of the cadherin superfamily and is involved in cell motility and migration [119]. The E- to N-cadherin switch is often seen in EMT and is recognized as a cancer progression hallmark [37]. In transformed epithelial cells, cell-cell adhesion is maintained via N-cadherin-mediated cell-cell contacts, which results in directional persistence of migration [120].
Altogether, these changes eventually lead to cell detachment from epithelial cell clusters and enhance cell motility [121]. Several previous studies have evaluated the role of leptin and its receptor in breast cancer EMT, a key step in cell invasion and metastasis. A recent study indicated that leptin treatment changes CDH1 and SNAI2 expression via the transforming growth factor beta 1 (TGFβ1) signaling pathway and consequently promotes EMT in breast cancer [122]. In another study, Wei et al. reported that leptin enhances vimentin and fibronectin expression, attenuates E-cadherin expression and promotes EMT in breast cancer cells via pyruvate kinase M2 (PKM2) upregulation through the PI3K/AKT signaling pathway [123]. Moreover, Wang et al. reported that IL-8 contributes to leptin-induced EMT in breast cancer cells through PI3K/AKT activation. They observed that the use of a blocking antibody against IL-8 and AKT inhibitor significantly blocked leptin-induced vimentin and fibronectin expression [124]. In addition, Yan et al. reported that cell treatment with leptin reduced E-cadherin expression as well as EMT in breast cancer. They also found that β-catenin activation through the Akt/GSK3 and MTA1/Wnt1 pathways is required for leptin-induced EMT [125].
Considering other cancer types, Trevellin et al. demonstrated that incubation of OE33 cells with conditioned medium (CM) collected from cultured biopsies of adipose tissue altered the expression of leptin, ObR and EMT markers. They concluded that leptin, as a crucial player, contributes to peritumoral adipose tissue-induced cell invasion in esophageal adenocarcinoma [126]. In ovarian cancer, Kato et al. demonstrated that leptin prompts the expression of EMT markers, such as N-cadherin, vimentin, SNAIL, and ZEB2, and contributes to stemness maintenance [105]. A study on the effect of leptin on EMT in lung cancer showed vimentin overexpression and E-Cadherin downregulation in leptin-treated A549 cells. In addition, they found that blocking of TGFβ with small interfering RNA (siRNA) significantly reduces EMT as well as vimentin expression, suggesting key roles of TGFβ in leptin-induced EMT in lung cancer [60]. Moreover, two other studies using animal models revealed that EMT marker levels in sera from obese mice are altered and that EMT is promoted in LNCaP and PacMetUT1 prostate cancer cells [66] and B16BL6 melanoma cancer cells [127].
Previous studies have demonstrated the role of Notch components in EMT and the acquisition of tumor aggressive phenotypes [128,129,130]. It is also reported that leptin promotes the expression of Notch components via IL-1 signaling in breast [131, 132] and pancreatic [133, 134] cancer cells. Geo et al. demonstrated novel crosstalk between Notch, IL-1 and leptin (NILCO) that promotes cell proliferation, migration and tumor angiogenesis in breast cancer cells [135, 136]. In an in vivo study, Gonzalez-Perez et al. also showed the important role of NILCO in breast cancer progression. They reported that leptin, IL-1, and Notch molecules are differentially expressed in breast cancer, suggesting potential prognostic biomarker value in breast cancer [137]. In endometrial cancer, Daley-Brown et al. reported key roles for NILCO in leptin-induced cell invasion [138]. Accordingly, these results demonstrated that NILCO can contribute to leptin-related cancer progression.
The role of miRNAs in EMT and cancer progression has been reported by several investigators [139]. It was found that some miR200 family members inhibit EMT, self-renewal, and differentiation of cancer stem cells (CSCs) [140]. Previous studies have demonstrated a role of miR200 members in suppression of these processes by interacting with Notch signaling and ZEB1/2 ZEB1 in pancreatic and breast cancer cells [141,142,143]. Menthol et al. preliminarily analyzed pancreatic cancer biopsies using TCGA Databank and reported higher miR21 expression compared with miR200, suggestive of a positive association between pancreatic cancer progression and miR21 upregulation. They also analyzed data from Pathway Studio Platform and determined a potential relationship between leptin signaling, CSC markers, histone deacetylases (HDACs), miR21 (oncogenic), and miR200 (tumor suppressor) resulting in pancreatic cancer progression. They also showed that leptin can promote the expression of CSC (ALDH1 and CD44) markers as well as miR21 expression. On the other hand, miR200 members may attenuate the expression of OB-Rb and CSC markers, while miR21 enhances their expression. In addition, further analysis by Menthol et al. showed that leptin could indirectly impact HDAC expression via miRNA or CSC markers. These findings suggest that dysregulation of HDACs, miR21, miR200, leptin signaling and CSC markers plays a crucial role in pancreatic cancer progression [144]. In colon cancer, an inhibitory role of miR-4443 in leptin-induced cell invasion was reported by Meerson et al. They demonstrated that miR-4443 blocks cell invasion through downregulation of TRAF4 and NCOA1 [73].
4 Cell-to-ECM adhesion molecules
The ECM serves as a scaffold for the generation of traction force, which is necessary for migratory cell movement. To this end, adhesion molecules on the surface of tumor cells bind to ECM components, including fibronectin, collagen, and laminin [39]. Adhesion molecules, also known as CAMs, are highly specific transmembrane glycoproteins involved in cell-cell or cell-ECM interactions [145]. Several families of adhesion molecules have been identified, such as integrins, the immunoglobulin family, selectins, cadherins, and lymphocyte homing receptors. Many CAM members play essential roles in pathological processes, including tumor metastasis. One of the most important adhesion molecules participating in cell-ECM interactions are integrins. These transmembrane proteins comprise two alpha and beta subunits linked to each other via noncovalent bonds and penetrate into the cell membrane [146]. The NH2-terminal ends of the alpha and beta subunits contribute to the constitution of ECM-cell bridges [147]. In addition, the cytoplasmic ends of integrins are directly linked with signaling proteins, such as focal adhesion kinase (FAK) and other adapter proteins [148]. FAK, a cytoplasmic tyrosine kinase, is activated via autophosphorylation at Tyr-397 when ECM components bind to integrins. These proteins prepare a scaffold for interaction between ECM proteins and integrins [149]. The roles of integrins and FAK activation are well recognized in tumor formation and metastasis.
Another adhesion molecule involved in cell-ECM interactions is intercellular adhesion molecule-1 (ICAM-1), a member of the immunoglobulin superfamily. This transmembrane glycoprotein can bind to macrophage-1 antigen (Mac-1) and lymphocyte function-associated antigen-1 (LFA-1) to facilitate cancer cell invasion [150, 151]. An association between ICAM-1 upregulation and more aggressive lesions has been reported in several tumor types, such as gastric [152] and lung [153] cancer. Moreover, Chen et al. reported that ICAM-1 overexpression participates in prostate cancer cell invasion, which can be attenuated by suppression of ICAM-1 expression [154].
The role of many integrin family members, including β3, αvβ5, and αvβ3, as well as that of FAK, in leptin-induced cell invasion has been investigated in several studies (see Table 1). How leptin affects each of these integrins has been explained by several investigators. Spina et al. showed that in MDA-MB-231 breast cancer cells, elevation of intracellular cyclic adenosine monophosphate (cAMP) markedly blocks leptin-mediated ERK1/2 and STAT3 phosphorylation in breast cancer [155]. In another study, they demonstrated that the activation of both protein kinas A (PKA) and protein exchange directly activated by cAMP (EPAC) in response to cAMP elevation inhibited leptin-induced cell migration via β3 integrin and FAK downregulation [156]. Previously, it was revealed that activation of FAK via a cluster of integrins is potentiated by autophosphorylation of FAK at Tyr397 and attachment of src family kinases to this binding site [157, 158]. Another mechanism underlying leptin-directed integrin activation was demonstrated by Yang et al. They found that leptin promotes cell migration through αvβ3 integrin upregulation. Furthermore, they showed that blocking of leptin receptor, insulin receptor substrate (IRS)-1, phosphatidylinositol 3-kinase (PI3K), Akt and NF-kB pathways inhibited leptin-mediated cell migration as well as upregulation of αvβ3 integrin, indicating key roles of these pathways in leptin-induced effects in chondrosarcoma cells [159]. The role of OBR1/IRS-1/PI3K/AKT/NF-κB signaling pathways in leptin-induced αvβ3 integrin expression and cell migration has also been evaluated in prostate cancer cells [160].
Upregulation of CAM-1 by leptin was demonstrated by Wong et al. for the first time. They observed that leptin has a key role in the migration of eosinophils and allergic inflammation induced by increased ICAM-1 expression [161]. In gastric cancer, Dong et al. reported a strong positive correlation between leptin and ICAM-1 overexpression in metastatic gastric cancer tissues. It was also reported that knockdown of ICAM-1 with siRNA significantly decreased leptin-induced cell migration of AGS and MKN-45 gastric cancer cell lines [162]. Another study conducted by Suzukawa et al. revealed that ICAM-1 expression was increased in human primary airway epithelial cells and the human airway epithelial cell line, BEAS-2B, after treatment with leptin. The leptin-mediated ICAM-1 expression and cell migration were attenuated by BAY11 7082, a specific inhibitor of NF-κB, or by dexamethasone [163].
The connection between adhesion molecules and the actin-myosin cytoskeleton is essential for the formation of focal adhesion complexes and cell body contraction, which are important in directional motility during migration [39]. In focal adhesion complexes, integrins bind to actin through vinculin and talin, membrane-cytoskeletal proteins [148]. Vinculin is recruited to nascent adhesions and has crucial roles in focal adhesion formation. It binds directly to actin filaments and the talin rod domain and stabilizes interactions between actin and talin. This essential stabilization function of vinculin is necessary for binding of integrins to the actin cytoskeleton [164]. Previous studies have demonstrated that loss of vinculin enhances cell invasion and metastasis [165, 166]. Polymerization of actin and its interaction with myosin generates a contractile force in the cell body, which is necessary for directional migration. The tension of the actin-myosin cytoskeleton induces body cell shortening, promotes internal tension generation and subsequently stimulates slow sliding of the cell rear edge forward [167,168,169].
Actin cytoskeletal reorganization is regulated by various signaling proteins, such as Rho family GTPase proteins [41]. This family belongs to the Ras superfamily and contains several members, including RhoA, Rac and CDC42 proteins [170]. Rho family proteins serve as a binary molecular switch cycling between an inactive, GDP-bound form, and an active, GTP-bound form [171]. The effects of RhoA on actin polymerization and actin-myosin contractility appear to be mediated by several downstream effectors [41]. RhoA induces actin polymerization through activation of mDia or inhibition of cofilin through the ROCK-LIM kinases (LIMK) pathway [172, 173]. On the other hand, it has been shown that RhoA-ROCK activity leads to actin-myosin contractility by blocking myosin phosphatase and by activation of myosin light chain (MLC) [41]. The involvement of RhoA in cancer cell invasion and metastasis has been demonstrated in previous studies [174].
To demonstrate the mechanisms of leptin-induced cell migration and tumor metastasis, a number of investigators reported that leptin affects the RhoA/ROCK signaling pathway and its downstream targets to promote cell invasion in different cancer types. In a recent study on ovarian cancer, we reported that leptin promotes RhoA and ROCK activation as well as cell invasion in OVCAR3 and SKOV3 cell lines, which was significantly inhibited by knockdown of OB-Rb using siRNA. We also found no effect of leptin on RhoA/ROCK activation in the CaoV-3 cell line, which does not substantially express OB-Rb protein. Moreover, pretreatment with RhoA and ROCK inhibitors markedly attenuated the invasive ability of cells, suggesting a role of RhoA/ROCK in leptin-induced cell invasion in ovarian cancer [175]. In another study, through F-actin and vinculin staining and immunocytochemistry, Kato et al. detected the formation of new focal adhesion complexes in an ovarian cell line incubated with leptin. The authors of this study also reported that leptin induces both active and total forms of RhoA in a time-dependent manner. They also described a significant increase in activation of myosin phosphatase target subunit 1 (MYPT1), a downstream target of RhoA involved in cytoskeletal reorganization, in HEY and SKOV3 cells after incubation with leptin [105]. Furthermore, Dong et al. reported an increase of RhoA activation and ROCK phosphorylation in AGS and MKN-45 gastric cancer cell lines after leptin stimulation. In addition, they observed that leptin-induced cell migration is markedly inhibited in the presence of either C3 (Rho inhibitor) or Y-27632 (ROCK inhibitor) in both AGS and MKN-45 cells, demonstrating the crucial role of the RhoA/ROCK pathway in gastric cancer cell invasion [162]. Considering colon cancer, Jaffe et al. evaluated the signaling pathways involved in leptin-mediated cell motility. Their results demonstrated that leptin activates Rac1 and Cdc42 but not RhoA via PI3K and controls RhoA activation through the Src kinase rather than JAK2 pathway. Moreover, they concluded that leptin-mediated cell invasiveness and locomotion are essentially dependent on Rac1 and Cdc42 but not RhoA activation. RhoA activation only occurs in the absence of Rac1 and Cdc42 or a high level of leptin [176]. In addition to cancer cell invasion, the roles of leptin-induced Rho activation and its downstream targets in physiological control of actin cytoskeleton dynamics in normal tissues have been investigated.
In an animal model study on Lepr+/+ and Lepr−/− mice, Xie et al. cultured and purified the Sca-1+ progenitor cells from the vessel wall and subsequently investigated the effect of leptin on cell migration participating in neointimal lesions in vessel injury. They found that leptin is upregulated after vessel injury and that leptin-promoted cell migration in Sca-1+ progenitor cells through Rac1/Cdc42 activation results in an increase in neointimal formation [177]. In a study on pancreatic β-cells, it was reported that Rac-mediated actin remodeling and myosin 2 are involved in kATP channel trafficking to the plasma. It was also shown that leptin-induced AMP-dependent Protein Kinase (AMPK) activation increases Rac activation and the phosphorylation of myosin regulatory light chain (MRLC), which promotes cytoskeletal remodeling. Leptin-mediated actin remodeling is attenuated when cells are pretreated with Rac and myosin ATPase inhibitors or are transfected with Rac siRNA [178]. Leptin-induced RhoA/ROCK activation is also involved in cytoskeleton reorganization in nucleus pulposus cells. Thus, Li et al. demonstrated that leptin-mediated actin remodeling and stress fiber formation are associated with RhoA-LIMK1 activation and cofilin phosphorylation. They observed that blocking of RhoA/ROCK with pharmacological inhibitors significantly abrogated the impacts of leptin on actin assembly and stress fiber formation as well as phosphorylation of LIMK1 and cofilin. Their findings suggest that activation of RhoA/ROCK/LIMK/cofilin-2 pathways by leptin is involved in cytoskeleton remodeling in nucleus pulpous cells [179].
5 ECM component proteolysis
Proteolytic cleavage of ECM proteins is necessary for passage of metastatic cancerous cells through the basement membrane [39]. For this purpose, cells expand specialized protrusions known as invadopodia with a capacity for ECM degradation. These structures have been observed in aggressive tumors and play a key role in migration, invasion, and metastasis of cancer cells [180]. They are rich in actin filaments, actin regulatory proteins, adhesion molecules and matrix-degrading proteases [180, 181]. The degradation of ECM components by invadopodia is mediated by two types of proteases, MMPs and components of PA.
MMPs constitute a zinc-dependent endopeptidase family containing 24 members that are structurally related and have an ability to degrade ECM components [182]. Typically, the MMP structure consists of four regions: a signal peptide region, a calcium-dependent catalytic region, a linker region, and a hemopexin-like domain. These proteases are usually secreted as a pro-peptide and are then activated via proteolytic cleavage in the NH2 terminus of the latent form [183]. They are classified according to substrate specificity, structure, and subcellular localization into five main groups, namely, gelatinases, stromelysins, matrilysins, collagenases, and membrane-type (MT)-MMPs [184,185,186]. There is growing evidence that MMPs contribute to several physiological and pathological processes, including wound healing, reproduction, fetus development, tissue degradation, inflammation, cell invasion and tumor metastasis in different organs [187]. Several studies have reported that leptin induces cell invasion and migration through MMP overexpression in various cancer types [188,189,190,191,192,193,194,195,196] (see Table 1 for a summary of the relevant literature).
MMP-2 and 9 (known as gelatinase A and B) are two members of the gelatinase MMP family, characterized by a high capacity to degrade gelatin, a product of collagen degradation. MMP-2 and MMP-9 have additional fibronectin repeats in their catalytic domain compared with other MMPs [187]. They can be distinguished by differences such as molecular weight (MMP2: 72 kDa; MMP9: 95 kDa), enzyme efficiency for gelatin degradation (higher for MMP-2), type of substrate, type of inhibitor (TIMP-2 and TIMP-1) and tissue distribution [185, 197]. MMP2 is produced by keratinocytes, endothelial cells, chondrocytes, osteoblasts, and monocytes, whereas MMP9 is predominantly expressed in normal alveolar macrophages, polymorphonuclear leukocytes, osteoclasts, and trophoblasts [197].
MMP-2 and MMP-9, relative to other MMPs, have been the particular focus of research in relation to their roles in tumor progression and metastasis. According to these studies, elevated levels of both MMP-2 and 9 have been observed in different types of malignancies [183, 198]. They promote cancer cell invasion and metastasis through degradation of type IV collagen, laminin-5 and other ECM components, such as proteoglycans, osteonectin, and entactin [199]. Various studies have demonstrated that leptin enhances the expression and activation of MMP-2 and MMP-9 in different types of tissues. In a recent study, Santos et al. found that leptin promotes the expression levels of both MMP-2 and MMP-9, leading to enhanced migration of oral squamous carcinoma cells. They also reported that gallic acid, a polyphenolic anticancer agent, modulates leptin-mediated cell migration through downregulation of MMP-2 and MMP-9 mRNA expression in oral squamous cell carcinoma cells [200].
In gallbladder cancer (GBC), leptin treatment may cause a significant increase in MMP-9 expression and activation as well as cell invasion in GBC cells. These responses to leptin can be inhibited by short hairpin RNA (shRNA) targeting OB-Rb, which demonstrates the significant role of OB-Rb in leptin-induced MMP-9 activation and suggests that the leptin/OB-Rb axis may be a therapeutic target for GBC treatment [201]. In another study, Ahn et al. evaluated the role of leptin in cell migration and invasion as well as MMP-2 expression in human endometriotic cells. The authors reported a potential increase in cell migration and invasion in response to leptin treatment. Blocking of MMP-2 using siRNA and/or gelatinase inhibitors markedly reduced leptin-induced migration and invasion. They concluded that upregulation of MMP-2 mediated by leptin plays a crucial role in cell migration and invasion in endometriotic cancer [202].
Another MMP type involved in cell migration and invasion are collagenases, which are characterized by a broad substrate specificity. Similarly, enzymatic studies have demonstrated a difference between MMP-1, 8, and 13 in affinity for various types of collagen [203, 204]. In addition, while MMP-1 and MMP-13 are generated by different cell types and secreted immediately after synthesis, MMP-8 is predominantly produced by neutrophils and stored until required [205]. MMP-13 has been shown to play a role in different cancer types. Elevated levels of MMP-13 have been associated with cell invasion and metastasis in papillary thyroid carcinoma [206] and breast cancer [207]. MMP-13 has also been studied as a prognostic indicator in colon [208] and ovarian cancer [209]. In one study, Yeh et al. evaluated the role of MMP-2, MMP-9, and MMP-13 in leptin-induced cell invasion of glioma cells. They observed that MMP-13 but not MMP-2 and MMP-9 expression was enhanced in C6 glioma cells after leptin treatment. Leptin-enhanced migration and invasion were attenuated by a neutralizing antibody or siRNA against MMP-13, demonstrating that MMP-13 is involved in these effects of leptin in glioma cells. Moreover, a significant decrease was observed in leptin-promoted MMP-13 expression in response to p38 MAP kinase and NF-kB inhibitors, suggesting that leptin enhances MMP-13 expression via p38 and NF-kB pathways [210]. In another study, the contribution of MMP-13 in leptin-mediated cell invasion was investigated in human pancreatic cancer by Fan et al. They revealed that OB-Rb overexpression is associated with increased MMP-13 expression in human pancreatic cancer tissues. Furthermore, it was shown that leptin treatment markedly promoted cell invasion and migration as well as MMP-13 expression in both PANC-1 and AsPC-1 pancreatic cell lines. In view of that, the investigators reported that leptin stimulates cell invasion and migration of pancreatic cancer cells through MMP-13 overexpression, raising the possibility of the leptin/MMP-13 axis as a therapeutic target for pancreatic cancer therapy [107].
In another study, Iliopoulos et al. inspected the effect of leptin epigenetic regulation on the expression of MMP-3, 9, and 13 in osteoarthritic chondrocytes. Increased expression of leptin was positively associated with MMP-13 activity in chondrocytes. Inhibition of leptin expression via siRNA significantly attenuated MMP-13 expression as well as cartilage destruction, indicating the importance of leptin in osteoarthritis therapy [211]. Moreover, several studies have shown that leptin induces MMP-1, MMP-8 and MMP-3 production in some diseases; however, their role in leptin-mediated cancer progression remains unclear [212,213,214].
MMP-7, the smallest member of the MMP family, has been reported as a key player in metastasis and cancer progression [215]. MMP-7 is also proposed to have a crucial role in leptin-mediated cancer cell invasion. A recent study revealed that leptin enhances MMP-7 expression as well as ovarian cell migration and invasion. Pretreatment with ERK1/2 or JNK1/2 inhibitors significantly attenuated leptin-mediated effects, suggesting that these signaling pathways are involved in leptin impacts on ovarian cancer progression. In addition, inhibition of MMP-7 gene expression using siRNA markedly reduced leptin-induced MMP-9 activation, indicating that MMP-7 has an important role in leptin-mediated MMP-9 activation [216]. In another study, Lin et al. reported that leptin enhances MMP-7 expression and the invasive potential of human colon cancer cells. MMP-7 gene silencing markedly blocked the leptin-promoted invasiveness of human colon cancer. Furthermore, they observed a significant decrease in the stimulatory effects of leptin on colon cancer progression after pretreatment with PI3K/AKT and MAPK/ERK inhibitors. They concluded that leptin-induced MMP-7 upregulation plays a crucial role in colon cancer progression [217].
In addition, previous studies have described a role of membrane type 1-matrix metalloproteinase (MT1-MMP, also known as MMP14) in leptin-mediated cell migration and invasion. This MMP family comprises surface-anchored proteinases shown to be upregulated in aggressive tumors and to have a key role in cell invasion [218, 219]. In another study, the surface expression of MT1-MMP in gastric cancer cells was detected using a cell surface biotinylation assay and flow cytometry in the presence of leptin. They observed that using siRNA against kinesin family member 1B (KIF1B), a microtubule-dependent motor involved in gastric cancer, abolishes the influence of leptin on MT1-MMP expression. Moreover, overexpression of leptin, MT1-MMP, and KIF1B were revealed in gastric cancer tissues by immunohistochemistry. Taken together, these findings suggest that leptin-induced cell invasion in gastric cancer is dependent on MT1-MMP and KIF1B overexpression [220]. Wang et al. characterized the role of MT1-MMP in the stimulatory effect of leptin on human extravillous trophoblast (EVT) invasion. Increased expression of MT1-MMP was observed in invasive cells in response to leptin stimulation, and blocking of MT1-MMP significantly inhibited the invasive potential of EVT cells. They concluded that MT1-MMP overexpression is required for leptin-enhanced invasion in EVTs [221].
Similar to MMPs, upA also participates in tumor metastasis and cancer progression [222]. The serine protease upA is released as a zymogen (pro-upA) and activated by proteases such as plasmin and trypsin following binding to its plasma membrane receptor, uPAR. Then, active uPA catalyzes plasminogen, a zymogen, converting it to plasmin, an active serine protease, which is able to proteolytically degrade the ECM and basement membrane, facilitating cell invasion and migration [222, 223]. Plasminogen activator inhibitor type-1 (PAI-1) is an inhibitor of uPA and has a key role in processes such as signal transduction, cell adherence, and cell migration [224]. In a recent study, it was demonstrated that leptin promotes ovarian cancer cell invasion and migration through upA overexpression. A time- and concentration-dependent increase in upA expression was observed in ovarian cancer cell lines after incubation with leptin. Cell transfection with OB-Rb siRNA and pretreatment with specific PI3K/AKT, JAK/STAT, and NF-kB inhibitors markedly decreased leptin-mediated upA expression, illustrating that these pathways contribute to leptin-dependent upA expression [175]. In another study, Singh et al. reported that different concentrations of leptin significantly increase PAI expression but do not affect tissue-type plasminogen activator (tPA) expression or tPA activity. Blocking of the ERK1/2 pathway by specific inhibitors attenuated the elevation in PAI-1 induced by leptin. They concluded that leptin enhances PAI expression through the ERK1/2 pathway but has no regulatory effect on tPA [225].
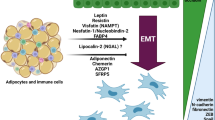
6 Adiponectin reverses the impact of leptin on cancer cell invasion
Unlike leptin, the circulating level of adiponectin, another adipocyte-derived hormone involved in obesity-related cancers, is inversely correlated with the amount of white adipose tissue [226, 227]. Numerous studies have conveyed a negative relationship between circulating adiponectin levels and cancer risk. Accordingly, decreased levels of adiponectin are observed in colorectal cancer [228], endometrial cancer [229], esophageal cancer, prostate cancer [230], and breast cancer [231]. Several in vitro studies have reported that adiponectin negatively influences leptin-induced cell invasion in some cancer types [232]. In esophageal carcinoma cells, incubation with adiponectin significantly reduced the impact of leptin on cell migration and invasion as well as MMP-2 and MMP-9 activation. Additionally, a decreased effect of leptin on JAK and STAT activation was observed in response to adiponectin treatment. These effects of adiponectin were attenuated by a protein tyrosine phosphatase 1B (PTP1B) inhibitor, suggesting that adiponectin blocks the leptin-induced effect on cancer progression in a PTP1B-sensitive manner by increasing the expression and activation of PTP1B [233]. PTP1B is an important regulator of different signaling pathways that contribute to diabetes, obesity, and cancer [234]. Several pieces of evidence have demonstrated that PTP1B negatively regulates signaling pathways mediated by leptin [235]. In another study, Taliaferro-Smith et al. reported the key roles of PTP1B upregulation and activation in adiponectin-mediated blocking of the stimulatory effects of leptin in breast cancer. In animal experiments, they observed an inhibitory effect of adenovirus-mediated adiponectin on leptin-promoted tumorigenesis in nude mice [236]. The inhibitory effect of adiponectin on leptin activity was also demonstrated in hepatocellular carcinoma by Sharma et al. They reported that adiponectin significantly inhibited leptin-mediated invasion through upregulation of suppressor of cytokine signaling (SOCS3) in HepG2 and Huh7 cell lines. In addition, their results showed that adiponectin markedly decreases leptin-mediated tumor burden in nude mice [237]. Moreover, Wu et al. found that the AMPK signaling pathway is involved in adiponectin suppression of leptin-induced aggressive endometrial cancer cell phenotypes. They reported downregulation of JAK/STAT3 in leptin-treated cells after incubation with adiponectin [238].
7 Leptin/OB-Rb as therapeutic targets in leptin-mediated cancers
Modulation of leptin, OB-Rb, or their downstream signaling pathway may be a conceivable drug discovery target for treating obesity-associated diseases, especially obesity-related cancers. In this line, investigations have been directed toward designing and developing leptin antagonists that may block ObR and thereby control or delay leptin-related cancers.
Leptin muteins are a class of potent leptin antagonists that are produced by a series of point mutations in human leptin. Among the different types of leptin muteins, R128Q [239], D40N and S127D influence the biological effects of leptin without changing its receptor binding [240]. S120A/T121A is another leptin mutein with powerful antagonist capacity that is able to bind the CRH2 domain of ObR and inhibit the JAK/STAT pathway [241]. The effect of this mutein on glutamate-mediated apoptosis through ERK phosphorylation was investigated by Park et al. They reported that leptin may block this effect of glutamate but leptin S120A/T121A mutein was unable to reverse this leptin effect [242]. Subsequently, L39A/D40A/F41A (LDF or Lan1) and L39A/D40A/F41A/I42A (LDFI or Lan2), triple and quadruple muteins respectively, were assembled using molecular modeling accomplished with site-directed mutations. These muteins blocked the biological activity of leptin through interaction with the IgD domain of ObR [243, 244]. In prostate cancer, LDF markedly suppressed the inductive effect of leptin on JAK2, ERK1/2, and Akt/PKB phosphorylation as well as activation of apoptotic proteins [245]. In addition, LDFI suppressed leptin-induced cell proliferation in MCF-7 and SKBR-3 breast cancer cells [246]. Fiedor et al. evaluated the influences of two leptin receptor antagonists on the carcinogenic effects of leptin in ovarian cancer and human folliculoma. They reported that leptin-related carcinogenesis was significantly abrogated by both LDFI and a superactive human leptin antagonist (SHLA) [247, 248]. SHLA is a leptin mutein with more binding affinity for the receptor and more antagonistic activity than other muteins and blocks leptin activity through inhibition of the JAK2 pathway. Finally, preparation of PEGylated forms of these leptin muteins has improved their bioavailability in the body, increased their antagonist activity (but decreased their receptor affinity), and raised them as potential therapeutic agents in leptin-related diseases [249, 250]. In gastro-esophageal adenocarcinoma, it was indicated that SHLA alone or in combination with cisplatin markedly decreased the tumorigenic effect of leptin [251]. Furthermore, Fiedor et al. recently reported that SHLA and Lan2 reverse the negative influences of leptin on the expression of HDACs in ovarian cancer cell lines [252].
Another class of leptin antagonists, peptide antagonists, have primarily been synthesized by Grasso et al. and correspond to the sequences of native leptin (167 amino acids) with/without some modifications [253]. Leptin peptide antagonist-1 (LPA-1, leptin demine 70–95) [254] and leptin peptide antagonist-2 (LPA-2, leptin demine 3–34) [255] exhibit high-affinity binding to ObR and block its signaling pathway in the presence of leptin [256]. In addition, Aca1, Allo-Aca, and D-Ser are three modified peptides (121–129) that show pure antagonist properties. Among these, Allo-Aca exhibits the highest serum half-life and antagonist efficacy and has been shown to be a potent antagonist to inhibit the biological activity of leptin in breast cancer [257, 258]. In one study, Beccari et al. generated a novel analog of Allo-aca and evaluated its anti-proliferative activity in breast and colon cancer cell lines. They found that this Allo-aca analog inhibited leptin-mediated cell proliferation and its related signaling pathway in both cancer types [259]. The effectiveness of nanoparticle-linked leptin antagonists in combination with chemotherapeutics on breast cancer cell viability was investigated. The inhibitory effect of a nanoparticle-linked antagonist on leptin signaling was demonstrated in MDA-MB-231 cells [260]. In another study, a synthetic OB3 peptide blocked leptin-mediated cancer progression by suppressing STAT3 and ERα activation [261, 262].
Some small chemical molecules, such as 2-aminopurine (2-AP) [258], benzyl isothiocyanate (BITC) [263] and pyridine derivatives [264], have also been designed and synthesized as leptin antagonists. These molecules directly interact with leptin-related signaling pathways, leading to inhibition of leptin signaling-induced processes. Kim et al. revealed the potential effect of BITC against influences of leptin in MDA-MB-231 and MCF-7 human breast cancer cells and suggested that this could be a therapeutic strategy for breast carcinoma in obese patients [263].
Monoclonal antibodies (mAbs) have also been reported as potent leptin antagonists that block leptin effects in a dose-dependent manner. For example, decreased leptin activity was observed in breast cancer cells following 9F8 mAb treatment, suggesting this as a drug candidate for leptin-related cancers [265]. Recently, other leptin antagonists have also been obtained using a random nanobody-based approach, consisting of monomeric antibody-derived therapeutic proteins targeted against the extracellular portion of ObR [266].
8 Conclusions and perspectives
To date, ample studies have demonstrated that obesity is associated with an increase in leptin levels. According to the abovementioned studies, leptin and OB-Rb play key roles in obesity-mediated cancers. The leptin/OB-Rb axis induces cancer progression through various biological molecules that participate in EMT, cell-ECM interactions and ECM proteolysis following activation of related signaling pathways, such as the JAK/STAT, MAPK and PI3K/AKT pathways. Therefore, it is clear that leptin and OB-Rb can be considered important targets for the prevention and treatment of obesity-mediated diseases, especially cancer. Several studies have focused on leptin muteins, peptide molecules, small chemical molecules, mAb and nanobody-based approaches to antagonize leptin/OB-Rb functions as conceivable therapeutic strategies for obesity-mediated cancer. These molecules might be promising therapeutic agents based on their remarkable antagonist activity in many diseases in which leptin signaling is involved, particularly obesity-related cancer. Overall, bearing in mind that the activity of all these leptin antagonist molecules has been determined in cell culture and animal experiments, further preclinical and clinical experiments are needed. Figure 1 depicts an integrated perspective based on the findings presented in this review.

Leptin may affect tumor cells through binding to their OB-Rb receptor and subsequent activation of its associated signaling pathways, which in turn may lead to activation of downstream effectors involved in cell migration and invasion. Strategies such as manipulation of leptin/OB-Rb antagonists, as well as integrative approaches, have been proposed as effective options for the treatment of leptin-mediated cancers
References
I. Vucenik, J.P. Stains, Obesity and cancer risk: evidence, mechanisms, and recommendations. Ann. N. Y. Acad. Sci. 1271, 37–43 (2012)
E.-J. Choi, H.-R. Kim, J.-H. Kie, B.-I. Moon and J.-Y. Seoh, Attenuation of obesity and related metabolic disorders by the individual or combination treatment with IL-2/anti-IL-2 complex and hyperbaric oxygen. bioRxiv. 351841 (2018)
M.I. Goran, G.D. Ball, M.L. Cruz, Obesity and risk of type 2 diabetes and cardiovascular disease in children and adolescents. J. Clin. Endocrinol. Metab. 88, 1417–1427 (2003)
A. Khodabakhshi, M. Ghayour-Mobarhan, H. Rooki, R. Vakili, S. Hashemy, S. Mirhafez, M. Shakeri, R. Kashanifar, R. Pourbafarani, H. Mirzaei, Comparative measurement of ghrelin, leptin, adiponectin, EGF and IGF-1 in breast milk of mothers with overweight/obese and normal-weight infants. Eur. J. Clin. Nutr. 69, 614–618 (2015)
L. Mazzarella, Why does obesity promote cancer? Ecancermedicalscience 9, 554 (2015)
J. Ferlay, I. Soerjomataram, R. Dikshit, S. Eser, C. Mathers, M. Rebelo, D.M. Parkin, D. Forman, F. Bray, Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–386 (2015)
G.A. Colditz, L.L. Peterson, Obesity and cancer: evidence, impact, and future directions. Clin. Chem. Clinchem. 64, 154–162 (2018)
X. Fang, J. Wei, X. He, J. Lian, D. Han, P. An, T. Zhou, S. Liu, F. Wang, J. Min, Q uantitative association between body mass index and the risk of cancer: A global meta-analysis of prospective cohort studies. Int. J. Cancer 143, 1595–1603 (2018)
M.A. Lichtman, Obesity and the risk for a hematological malignancy: leukemia, lymphoma, or myeloma. The Oncologist 15, 1083–1101 (2010)
S. Ladhani, B. Empringham, K.-W. Wang, C. Portwine, L. Banfield, R.J. de Souza, L. Thabane, M.C. Samaan, Overweight and obesity management strategies in survivors of paediatric acute lymphoblastic leukaemia: a systematic review protocol. BMJ Open 8, e022530 (2018)
A. Tarasiuk, P. Mosińska, J. Fichna, The mechanisms linking obesity to colon cancer: an overview. Obes. Res. Clin. Pract. 12, 251–259 (2018)
S.G. Krishna, H. Hussan, Z. Cruz-Monserrate, L.F. Conteh, K. Mumtaz, D.L. Conwell, A review of the impact of obesity on common gastrointestinal malignancies. Integr. Cancer Sci. Ther. 4 (2017). https://doi.org/10.15761/ICST.1000223
P. Gild, B. Ehdaie, L.A. Kluth, Effect of obesity on bladder cancer and renal cell carcinoma incidence and survival. Curr. Opin. Urol. 27, 409–414 (2017)
D. Sharma, N. Saxena, P. Vertino, F. Anania, Leptin promotes the proliferative response and invasiveness in human endometrial cancer cells by activating multiple signal-transduction pathways. Endocr. Relat. Cancer 13, 629–640 (2006)
S. Kitson, J. Duffy, N. Ryan, M. MacKintosh, E. Crosbie, Interventions for weight reduction in obesity to improve survival in women with endometrial cancer. Cochrane Database Syst. Rev. (2018). https://doi.org/10.1002/14651858.CD012513.pub2
I. Huang-Doran, S. Franks, Genetic rodent models of obesity-associated ovarian dysfunction and subfertility: insights into polycystic ovary syndrome. Front. Endocrinol. 7, 53 (2016). https://doi.org/10.3389/fendo.2016.00053
L. Gan, Z. Liu, C. Sun, Obesity linking to hepatocellular carcinoma: A global view. Biochim. Biophys. Acta Rev. Cancer 1869, 97–102 (2018)
D. Li, J.S. Morris, J. Liu, M.M. Hassan, R.S. Day, M.L. Bondy, J.L. Abbruzzese, Body mass index and risk, age of onset, and survival in patients with pancreatic cancer. JAMA. 301, 2553–2562 (2009)
M. Xu, X. Jung, O.J. Hines, G. Eibl, Y. Chen, Obesity and pancreatic cancer: overview of epidemiology and potential prevention by weight loss. Pancreas 47, 158–162 (2018)
R.J. MacInnis, D.R. English, Body size and composition and prostate cancer risk: systematic review and meta-regression analysis. Cancer Causes Control. 17, 989–1003 (2006)
M. Bandini, G. Gandaglia, A. Briganti, Obesity and prostate cancer. Curr. Opin. Urol. 27, 415–421 (2017)
E.H.B.C.C. Group, Body mass index, serum sex hormones, and breast cancer risk in postmenopausal women. J. Natl. Cancer Inst. 95, 1218–1226 (2003)
Y. Geng, J. Wang, R. Wang, K. Wang, Y. Xu, G. Song, C. Wu, Y. Yin, Leptin and HER-2 are associated with gastric cancer progression and prognosis of patients. Biomed. Pharmacother. 66, 419–424 (2012)
S. Mahbouli, A. Der Vartanian, S. Ortega, S. Rougé, M.-P. Vasson, A. Rossary, Leptin induces ROS via NOX5 in healthy and neoplastic mammary epithelial cells. Oncol. Rep. 38, 3254–3264 (2017)
H.G. Kim, S.W. Jin, Y.A. Kim, T. Khanal, G.H. Lee, S.J. Kim, S. Dal Rhee, Y.C. Chung, Y.J. Hwang, T.C. Jeong, Leptin induces CREB-dependent aromatase activation through COX-2 expression in breast cancer cells. Food Chem. Toxicol. 106, 232–241 (2017)
M. Hosney, S. Sabet, M. El-Shinawi, K.M. Gaafar, M.M. Mohamed, Leptin is overexpressed in the tumor microenvironment of obese patients with estrogen receptor positive breast cancer. Exp. Ther. Med. 13, 2235–2246 (2017)
S.M. Louie, L.S. Roberts, D.K. Nomura, Mechanisms linking obesity and cancer. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1831, 1499–1508 (2013)
C. Bjorbæk, B.B. Kahn, Leptin signaling in the central nervous system and the periphery. Recent Prog. Horm. Res. 59, 305–332 (2004)
D. Dutta, S. Ghosh, K. Pandit, P. Mukhopadhyay, S. Chowdhury, Leptin and cancer: Pathogenesis and modulation. Ind. J. Endocrin. Metabolism 16, S596 (2012)
M.N. VanSaun, Molecular pathways: adiponectin and leptin signaling in cancer. Clin. Cancer Res. 19, 1926–1932 (2013)
N.K. Saxena, L. Taliaferro-Smith, B.B. Knight, D. Merlin, F.A. Anania, R.M. O'Regan, D. Sharma, Bidirectional crosstalk between leptin and insulin-like growth factor-I signaling promotes invasion and migration of breast cancer cells via transactivation of epidermal growth factor receptor. Cancer Res. 68, 9712–9722 (2008)
V.K. Clements, T. Long, R. Long, C. Figley, D. Smith, S. Ostrand-Rosenberg, Frontline Science: High fat diet and leptin promote tumor progression by inducing myeloid-derived suppressor cells. J. Leukoc. Biol. 103, 395–407 (2018)
T.N. Seyfried, L.C. Huysentruyt, On the origin of cancer metastasis. Crit. Rev. Oncog. 18, 43 (2013)
A.W. Lambert, D.R. Pattabiraman, R.A. Weinberg, Emerging biological principles of metastasis. Cell 168, 670–691 (2017)
A. Voulgari, A. Pintzas, Epithelial–mesenchymal transition in cancer metastasis: mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta Rev. Cancer 1796, 75–90 (2009)
J.H. Tsai and J. Yang, Epithelial–mesenchymal plasticity in carcinoma metastasis. Genes Dev. 27, 2192–2206 (2013)
M.J. Wheelock, Y. Shintani, M. Maeda, Y. Fukumoto, K.R. Johnson, Cadherin switching. J. Cell Sci. 121, 727–735 (2008)
D.A. Lauffenburger, A.F. Horwitz, Cell migration: a physically integrated molecular process. Cell 84, 359–369 (1996)
G. Bozzuto, P. Ruggieri, A. Molinari, Molecular aspects of tumor cell migration and invasion. Ann. Ist. Super. Sanita. 46, 66–80 (2010)
T.D. Pollard, G.G. Borisy, Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 (2003)
K. O'Connor, M. Chen, Dynamic functions of RhoA in tumor cell migration and invasion. Small GTPases 4, 141–147 (2013)
B. Davidson, R. Reich, B. Risberg, J. Nesland, The biological role and regulation of matrix metalloproteinases (MMP) in cancer. Arkh. Patol. 64, 47–53 (2002)
A.M. Weaver, Invadopodia: specialized cell structures for cancer invasion. Clin. Exp. 23, 97–105 (2006)
M. Duffy, (Portland Press Limited, 2002),
Y. Teng, J.L. Ross, J.K. Cowell, The involvement of JAK-STAT3 in cell motility, invasion, and metastasis. Jak-Stat. 3, e28086 (2014)
B. Li, W.W. Xu, A.K.Y. Lam, Y. Wang, H.-F. Hu, X.Y. Guan, Y.R. Qin, N. Saremi, S.W. Tsao, Q.-Y. He, Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget 8, 38755 (2017)
A.S. Dhillon, S. Hagan, O. Rath, W. Kolch, MAP kinase signalling pathways in cancer. Oncogene 26, 3279–3290 (2007)
F. Seif, M. Khoshmirsafa, H. Aazami, M. Mohsenzadegan, G. Sedighi, M. Bahar, The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Comm. Signal. 15, 23 (2017)
P. Liu, H. Cheng, T.M. Roberts, J.J. Zhao, Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 8, 627–644 (2009)
K.L. Houseknecht, C.A. Baile, R.L. Matteri, M.E. Spurlock, The biology of leptin: a review. J. Anim. Sci. 76, 1405–1420 (1998)
C.A. Siegrist-Kaiser, V. Pauli, C.E. Juge-Aubry, O. Boss, A. Pernin, W.W. Chin, I. Cusin, F. Rohner-Jeanrenaud, A.G. Burger, J. Zapf, Direct effects of leptin on brown and white adipose tissue. J. Clin. Invest. 100, 2858–2864 (1997)
S. Blüher, C.S. Mantzoros, Leptin in humans: lessons from translational research. Am. J. Clin. Nutrition 89, 991S–997S (2009)
S. Margetic, C. Gazzola, G. Pegg, R. Hill, Leptin: a review of its peripheral actions and interactions. Int. J. Obes. Relat. Metab. Disord. 26, 1407–1433 (2002)
C.S. Mantzoros, The role of leptin in human obesity and disease: a review of current evidence. Ann. Intern. Med. 130, 671–680 (1999)
H. Shimizu, Y. Shimomura, R. Hayashi, K. Ohtani, N. Sato, T. Futawatari, M. Mori, Serum leptin concentration is associated with total body fat mass, but not abdominal fat distribution. Int. J. Obes. Relat. Metab. Disord. 21, 536–541 (1997)
R.V. Considine, M.K. Sinha, M.L. Heiman, A. Kriauciunas, T.W. Stephens, M.R. Nyce, J.P. Ohannesian, C.C. Marco, L.J. McKee, T.L. Bauer, Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 334, 292–295 (1996)
J. Licinio, C. Mantzoros, A.B. Negrão, G. Cizza, M.-L. Wong, P.B. Bongiorno, G.P. Chrousos, B. Karp, C. Allen, J.S. Flier, Human leptin levels are pulsatile and inversely related to pituitary–ardenal function. Nat. Med. 3, 575–579 (1997)
N.K. Saxena, D. Sharma, X. Ding, S. Lin, F. Marra, D. Merlin, F.A. Anania, Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 67, 2497–2507 (2007)
Y. Ding, Y. Cao, B. Wang, L. Wang, Y. Zhang, D. Zhang, X. Chen, M. Li, C. Wang, APPL1-mediating leptin signaling contributes to proliferation and migration of cancer cells. PLoS ONE 11, e0166172 (2016)
H. Feng, Q. Liu, N. Zhang, L. Zheng, M. Sang, J. Feng, J. Zhang, X. Wu, B. Shan, Leptin promotes metastasis by inducing an epithelial-mesenchymal transition in A549 lung cancer cells. Oncol. Res. 21, 165–171 (2014)
A.L. Strong, J.F. Ohlstein, B.A. Biagas, L.V. Rhodes, D.T. Pei, H.A. Tucker, C. Llamas, A.C. Bowles, M.F. Dutreil, S. Zhang, Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 17, 112 (2015)
S.D.H. Ahmed, F. Idrees, M. Ahsan, A. Khanam, N. Sultan, N. Akhter, Association of serum leptin with serum estradiol in relation to breast carcinogenesis: a comparative case-control study between pre-and postmenopausal women. Turkish J. Med. Sciences 48, 305–310 (2018)
L.W. Bowers, E.L. Rossi, S.B. McDonell, S.S. Doerstling, S.A. Khatib, C.G. Lineberger, J.E. Albright, X. Tang, S.D. Hursting, Leptin signaling mediates obesity-associated CSC enrichment and EMT in preclinical TNBC models. Mol. Cancer Res. 16, 869–879 (2018)
P.S. Thiagarajan, Q. Zheng, M. Bhagrath, E.E. Mulkearns-Hubert, M.G. Myers, J.D. Lathia, O. Reizes, STAT3 activation by leptin receptor is essential for TNBC stem cell maintenance. Endocr. Relat. Cancer 24, 415–426 (2017)
H. Cao, Y. Huang, L. Wang, H. Wang, X. Pang, K. Li, W. Dang, H. Tang, L. Wei, M. Su, Leptin promotes migration and invasion of breast cancer cells by stimulating IL-8 production in M2 macrophages. Oncotarget 7, 65441 (2016)
R. Price, D. Cavazos, R. De Angel, S. Hursting, Obesity-related systemic factors promote an invasive phenotype in prostate cancer cells. Prostate Cancer Prostatic Dis. 15, 135–143 (2012)
K.-W. Yoon, S.-Y. Park, J.-Y. Kim, S.-M. Lee, C.H. Park, S.-B. Cho, W.-S. Lee, Y.-E. Joo, J.H. Lee, H.S. Kim, Leptin-induced adhesion and invasion in colorectal cancer cell lines. Oncol. Rep. 31, 2493–2498 (2014)
J. Oba, W. Wei, J.E. Gershenwald, M.M. Johnson, C.M. Wyatt, J.A. Ellerhorst, E.A. Grimm, Elevated serum leptin levels are associated with an increased risk of sentinel lymph node metastasis in cutaneous melanoma. Medicine (Baltimore) 95, e3073 (2016)
X. Wei, Y. Liu, C. Gong, T. Ji, X. Zhou, T. Zhang, D. Wan, S. Xu, P. Jin, X. Yang, Targeting leptin as a therapeutic strategy against ovarian cancer peritoneal metastasis. Anti-Cancer Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Anti-Cancer Agents). 17, 1093–1101 (2017)
F. Campo-Verde-Arbocco, J.D. López-Laur, L.R. Romeo, N. Giorlando, F.A. Bruna, D.E. Contador, G. López-Fontana, F.E. Santiano, C.V. Sasso, L.E. Zyla, Human renal adipose tissue induces the invasion and progression of renal cell carcinoma. Oncotarget 8, 94223 (2017)
Y. Huang, Q. Jin, M. Su, F. Ji, N. Wang, C. Zhong, Y. Jiang, Y. Liu, Z. Zhang, J. Yang, Leptin promotes the migration and invasion of breast cancer cells by upregulating ACAT2. Cell. Oncol. 40, 537–547 (2017)
K. Li, L. Wei, Y. Huang, Y. Wu, M. Su, X. Pang, N. Wang, F. Ji, C. Zhong, T. Chen, Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 48, 2479–2487 (2016)
A. Meerson, H. Yehuda, Leptin and insulin up-regulate miR-4443 to suppress NCOA1 and TRAF4, and decrease the invasiveness of human colon cancer cells. BMC Cancer. 16, 882 (2016)
I. Tourkantonis, M. Kiagia, E. Peponi, S. Tsagouli, K.N. Syrigos, The role of leptin in cancer pathogenesis. J. Cancer Ther. 4, 640 (2013)
G. Newman, R.R. Gonzalez-Perez, Leptin–cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 382, 570–582 (2014)
L.A. Tartaglia, The leptin receptor. J. Biol. Chem. 272, 6093–6096 (1997)
G. Frühbeck, Intracellular signalling pathways activated by leptin. Biochem. J. 393, 7–20 (2006)
D. Shida, J. Kitayama, K. Mori, T. Watanabe, H. Nagawa, Transactivation of epidermal growth factor receptor is involved in leptin-induced activation of janus-activated kinase 2 and extracellular signal–regulated kinase 1/2 in human gastric cancer cells. Cancer Res. 65, 9159–9163 (2005)
J.W. Park, C.R. Han, L. Zhao, M. Willingham, S.-y. Cheng, Inhibition of STAT3 activity delays obesity-induced thyroid carcinogenesis in a mouse model. Endocr. Relat. Cancer. ERC-15-0417 (2015)
I. Haque, A. Ghosh, S. Acup, S. Banerjee, K. Dhar, A. Ray, S. Sarkar, S. Kambhampati, S.K. Banerjee, Leptin-induced ER-α-positive breast cancer cell viability and migration is mediated by suppressing CCN5-signaling via activating JAK/AKT/STAT-pathway. BMC Cancer 18, 99 (2018)
J. Kumar, H. Fang, D.R. McCulloch, T. Crowley, A.C. Ward, Leptin receptor signaling via Janus kinase 2/Signal transducer and activator of transcription 3 impacts on ovarian cancer cell phenotypes. Oncotarget 8, 93530 (2017)
H. Zahid, K. Subbaramaiah, N.M. Iyengar, X.K. Zhou, I.-C. Chen, P. Bhardwaj, A. Gucalp, M. Morrow, C.A. Hudis, A.J. Dannenberg, Leptin regulation of the p53-HIF1α/PKM2-aromatase axis in breast adipose stromal cells: a novel mechanism for the obesity–breast cancer link. Int. J. Obes. 42, 711 (2018)
C. Giordano, F. Chemi, S. Panza, I. Barone, D. Bonofiglio, M. Lanzino, A. Cordella, A. Campana, A. Hashim, P. Rizza, Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 7, 1262 (2016)
S. Zhang, J. Jiang, Z. Chen, Y. Wang, W. Tang, C. Liu, L. Liu, Y. Chen, investigation of LEP and LEPR polymorphisms with the risk of hepatocellular carcinoma: a case–control study in eastern chinese han population. OncoTargets and Therapy 11, 2083 (2018)
C.-R. Liu, Q. Li, C. Hou, H. Li, P. Shuai, M. Zhao, X.-R. Zhong, Z.-P. Xu, J.-Y. Li, Changes in body mass index, leptin, and leptin receptor polymorphisms and breast cancer risk. DNA Cell Biol. 37, 182–188 (2018)
H. Qiu, X. Lin, W. Tang, C. Liu, Y. Chen, H. Ding, M. Kang, S. Chen, Investigation of TCF7L2, LEP and LEPR polymorphisms with esophageal squamous cell carcinomas. Oncotarget 8, 109107 (2017)
J. Bieńkiewicz, H. Romanowicz, A. Malinowski, B. Smolarz, Association of single nucleotide polymorphism-2548 G/A (rs12112075) of leptin gene with endometrial cancer and uterine leiomyomas. Eur. J. Obstet. Gynecol. Reprod. Biol. 218, 113–118 (2017)
H. Luan, H. Zhang, Y. Li, P. Wang, L. Cao, H. Ma, Q. Cui, G. Tian, Association of two obesity-related gene polymorphisms LEPG2548A rs7799039 and LEPRQ223R rs1137101 with the risk of breast cancer. Oncotarget 8, 59333 (2017)
X. Yuan, Z. Xu, C. Liu, L. Yan, P. Tao, P. Xiong, Q. Li, M. Zhou, H. Li, M. Zhao, Study of the association between polymorphism of persistent obesity, human leptin gene/leptin receptor gene and molecular subtypes of breast cancer. Zhonghua yu fang yi xue za zhi (Chinese Journal of Preventive Medicine) 51, 533–538 (2017)
T. Amer, R. El-Baz, A.-R. Mokhtar, S. El-Shaer, R. Elshazli and A. Settin, Genetic polymorphisms of IL-23R (rs7517847) and LEP (rs7799039) among Egyptian patients with hepatocellular carcinoma. Arch. Physiol. Biochem. 123, 279–285 (2017)
M.A.-B. El-Hussiny, M.A. Atwa, W.E. Rashad, D.A. Shaheen, N.M. Elkady, Leptin receptor Q223R polymorphism in Egyptian female patients with breast cancer. Contemp. Oncol. 21, 42 (2017)
A. Méndez-Hernández, M.P. Gallegos-Arreola, H. Moreno-Macías, J.E. Fematt, R. Pérez-Morales, LEP rs7799039, LEPR rs1137101, and ADIPOQ rs2241766 and 1501299 polymorphisms are associated with obesity and chemotherapy response in Mexican women with breast cancer. Clin. Breast Cancer 17, 453–462 (2017)
C. Rodrigo, K.H. Tennekoon, E.H. Karunanayake, K. De Silva, I. Amarasinghe, A. Wijayasiri, Circulating leptin, soluble leptin receptor, free leptin index, visfatin and selected leptin and leptin receptor gene polymorphisms in sporadic breast cancer. Endocr. J. 64, 393–401 (2017)
D.R. Farias, A.B. Franco-Sena, F. Rebelo, G.F. Salles, C.J. Struchiner, M.C. Martins, G. Kac, Polymorphisms of leptin (G2548A) and leptin receptor (Q223R and K109R) genes and blood pressure during pregnancy and the postpartum period: A Cohort. Am. J. Hypertens. 30, 130–140 (2017)
A. Babic, Y. Bao, Z.R. Qian, C. Yuan, E.L. Giovannucci, H. Aschard, P. Kraft, L.T. Amundadottir, R.Z. Stolzenberg-Solomon, V. Morales-Oyarvide, Pancreatic cancer risks associated with prediagnostic plasma levels of leptin and leptin receptor genetic polymorphisms. Cancer Res. 1699, 7160–7167 (2016)
T. Mahmoudi, H. Farahani, H. Nobakht, R. Dabiri, M.R. Zali, Genetic variations in leptin and leptin receptor and susceptibility to colorectal cancer and obesity. Iran J Cancer Prev. 9, e7013 (2016)
P. Rodrigues, L. Maia, M. Santos, G. Peterle, L. Alves, J. Takamori, R. Souza, W. Barbosa, A. Mercante, F. Nunes, Leptin receptor expression and Gln223Arg polymorphism as prognostic markers in oral and oropharyngeal cancer. Genet. Mol. Res. 14, 14979–14988 (2015)
A.M. Mendonsa, M.C. Chalfant, L.D. Gorden, M.N. VanSaun, Modulation of the leptin receptor mediates tumor growth and migration of pancreatic cancer cells. PLoS ONE 10, e0126686 (2015)
M. Szyszka, M. Tyczewska, P. Milecka, K. Jopek, P. Celichowski, L.K. Malendowicz, M. Rucinski, Effects of leptin on leptin receptor isoform expression and proliferative activity in human normal prostate and prostate cancer cell lines. Oncol. Rep. 39, 182–192 (2018)
R. Sultana, A.C. Kataki, B.B. Borthakur, T.K. Basumatary, S. Bose, Imbalance in leptin-adiponectin levels and leptin receptor expression as chief contributors to triple negative breast cancer progression in Northeast India. Gene. 621, 51–58 (2017)
Y. Pan, F. Zhou, C. He, L. Hui, T. Huang, Y. Wei, Leptin-LepRb expressed in gastric cancer patients and related to cancer-related depression. Biomed. Res. Int. 2017, 1–7 (2017)
Y.-C. Lee, W.-J. Wu, H.-H. Lin, W.-M. Li, C.-N. Huang, W.-C. Hsu, L.-L. Chang, C.-C. Li, H.-C. Yeh, C.-F. Li, Prognostic value of leptin receptor overexpression in upper tract urothelial carcinomas in Taiwan. Clin. Genitourin. Cancer 15, e653–e659 (2017)
H. Feng, P. Guo, J. Wang, Q. Liu, J. Xu, H. Yang, J. Zhang, Association of the expression of leptin and leptin receptor with bone metastasis in pulmonary adenocarcinoma. Zhonghua zhong liu za zhi (Chinese Journal of Oncology) 38, 840–844(2016)
N. Saetang, T. Boonpipattanapong, A. Palanusont, W. Maneechay, S. Sangkhathat, Alteration of leptin and adiponectin in multistep colorectal tumorigenesis. Asian Pac. J. Cancer Prev. 17, 2119–2123 (2016)
S. Kato, L. Abarzua-Catalan, C. Trigo, A. Delpiano, C. Sanhueza, K. García, C. Ibañez, K. Hormazábal, D. Diaz, J. Brañes, Leptin stimulates migration and invasion and maintains cancer stem-like properties in ovarian cancer cells: an explanation for poor outcomes in obese women. Oncotarget 6, 21100 (2015)
S. Uddin, R. Bu, M. Ahmed, J. Abubaker, F. Al-Dayel, P. Bavi, K.S. Al-Kuraya, Overexpression of leptin receptor predicts an unfavorable outcome in Middle Eastern ovarian cancer. Mol. Cancer. 8, 74 (2009)
Y. Fan, Y. Gan, Y. Shen, X. Cai, Y. Song, F. Zhao, M. Yao, J. Gu, H. Tu, Leptin signaling enhances cell invasion and promotes the metastasis of human pancreatic cancer via increasing MMP-13 production. Oncotarget 6, 16120 (2015)
A. Horiguchi, M. Sumitomo, J. Asakuma, T. Asano, R. Zheng, T. Asano, D.M. Nanus, M. Hayakawa, Increased serum leptin levels and over expression of leptin receptors are associated with the invasion and progression of renal cell carcinoma. J. Urol. 176, 1631–1635 (2006)
M. Ishikawa, J. Kitayama, H. Nagawa, Expression pattern of leptin and leptin receptor (OB-R) in human gastric cancer. World J. Gastroenterol. 12, 5517 (2006)
N. Erkasap, M. Ozkurt, S. Erkasap, F. Yasar, K. Uzuner, E. Ihtiyar, S. Uslu, M. Kara, O. Bolluk, Leptin receptor (Ob-R) mRNA expression and serum leptin concentration in patients with colorectal and metastatic colorectal cancer. Braz. J. Med. Biol. Res. 46, 306–310 (2013)
Y.L. Fan, X.Q. Li, Expression of leptin and its receptor in thyroid carcinoma: distinctive prognostic significance in different subtypes. Clin. Endocrinol. 83, 261–267 (2015)
C. Garofalo, M. Koda, S. Cascio, M. Sulkowska, L. Kanczuga-Koda, J. Golaszewska, A. Russo, S. Sulkowski, E. Surmacz, Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin. Cancer Res. 12, 1447–1453 (2006)
M. Ishikawa, J. Kitayama, H. Nagawa, Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 10, 4325–4331 (2004)
Y.-J. Xu, Y.-F. Shao, X. Zhao, Y.-T. Geng, K. Wang, Y.-M. Yin, Expression and clinical significance of leptin, the functional receptor of leptin (OB-Rb) and HER-2 in non-small-cell lung cancer: a retrospective analysis. J. Cancer Res. Clin. Oncol. 137, 1841 (2011)
U.H. Frixen, J. Behrens, M. Sachs, G. Eberle, B. Voss, A. Warda, D. Löchner, W. Birchmeier, E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J. Cell Biol. 113, 173–185 (1991)
K. Strumane, G. Berx, F. Van Roy, in Cell adhesion, (Springer, 2004), p. 69–103
F. Van Roy, G. Berx, The cell-cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 65, 3756–3788 (2008)
B.M. Gumbiner, Regulation of cadherin-mediated adhesion in morphogenesis. Nat. Rev. Mol. Cell Biol. 6, 622–634 (2005)
W. Shih, S. Yamada, N-cadherin as a key regulator of collective cell migration in a 3D environment. Cell Adh. Migr. 6, 513–517 (2012)
W. Shih, S. Yamada, N-cadherin-mediated cell-cell adhesion promotes cell migration in a three-dimensional matrix. J. Cell Sci. 125, 3661–3670 (2012)
S. Lamouille, J. Xu, R. Derynck, Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196 (2014)
A.K. Mishra, C.R. Parish, M.-L. Wong, J. Licinio, A.C. Blackburn, Leptin signals via TGFB1 to promote metastatic potential and stemness in breast cancer. PLoS ONE 12, e0178454 (2017)
L. Wei, K. Li, X. Pang, B. Guo, M. Su, Y. Huang, N. Wang, F. Ji, C. Zhong, J. Yang, Leptin promotes epithelial-mesenchymal transition of breast cancer via the upregulation of pyruvate kinase M2. J. Exp. Clin. Cancer Res. 35, 166 (2016)
L. Wang, C. Tang, H. Cao, K. Li, X. Pang, L. Zhong, W. Dang, H. Tang, Y. Huang, L. Wei, Activation of IL-8 via PI3K/Akt-dependent pathway is involved in leptin-mediated epithelial-mesenchymal transition in human breast cancer cells. Cancer Biol. Ther. 16, 1220–1230 (2015)
D. Yan, D. Avtanski, N.K. Saxena, D. Sharma, Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires β-catenin activation via Akt/GSK3-and MTA1/Wnt1 protein-dependent pathways. J. Biol. Chem. 287, 8598–8612 (2012)
E. Trevellin, M. Scarpa, A. Carraro, F. Lunardi, A. Kotsafti, A. Porzionato, L. Saadeh, M. Cagol, R. Alfieri, U. Tedeschi, Esophageal adenocarcinoma and obesity: peritumoral adipose tissue plays a role in lymph node invasion. Oncotarget 6, 11203 (2015)
K. Kushiro, N.P. Núñez, Ob/ob serum promotes a mesenchymal cell phenotype in B16BL6 melanoma cells. Clin. Exp. Metastasis 28, 877–886 (2011)
Z. Wang, Y. Li, D. Kong, F. H Sarkar, The role of Notch signaling pathway in epithelial-mesenchymal transition (EMT) during development and tumor aggressiveness. Curr. Drug Targets 11, 745–751 (2010)
Y. Li, J. Ma, X. Qian, Q. Wu, J. Xia, L. Miele, F. H Sarkar, Z. Wang, Regulation of EMT by Notch signaling pathway in tumor progression. Curr. Cancer Drug Targets 13, 957–962 (2013)
A. Harbuzariu, R.R. Gonzalez-Perez, Leptin-Notch axis impairs 5-fluorouracil effects on pancreatic cancer. Oncotarget 9, 18239 (2018)
M. Battle, C. Gillespie, A. Quarshie, V. Lanier, T. Harmon, K. Wilson, M. Torroella-Kouri, R.R. Gonzalez-Perez, Obesity induced a leptin-Notch signaling axis in breast cancer. Int. J. Cancer. 134, 1605–1616 (2014)
C.C. Lipsey, A. Harbuzariu, D. Daley-Brown, R.R. Gonzalez-Perez, Oncogenic role of leptin and Notch interleukin-1 leptin crosstalk outcome in cancer. World J. Methodology 6, 43–55 (2016)
A. Harbuzariu, G.M. Oprea-Ilies, R.R. Gonzalez-Perez, The role of notch signaling and leptin-notch crosstalk in pancreatic cancer. Medicines (Basel, Switzerland). 5, E68 (2018)
A. Harbuzariu, A. Rampoldi, D.S. Daley-Brown, P. Candelaria, T.L. Harmon, C.C. Lipsey, D.J. Beech, A. Quarshie, G.O. Ilies, R.R. Gonzalez-Perez, Leptin-Notch signaling axis is involved in pancreatic cancer progression. Oncotarget 8, 7740 (2017)
S. Guo, R.R. Gonzalez-Perez, Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS ONE 6, e21467 (2011)
L.S. Colbert, K. Wilson, S. Kim, Y. Liu, G. Oprea-Ilies, C. Gillespie, T. Dickson, G. Newman, R.R. Gonzalez-Perez, NILCO biomarkers in breast cancer from Chinese patients. BMC Cancer 14, 249 (2014)
C. Gillespie, A. Quarshie, M. Penichet, R. Gonzalez-Perez, Potential role of leptin signaling in DMBA-induced mammary tumors by non-responsive C57BL/6J mice fed a high-fat diet. J Carcinogene Mutagene 3, 20–23 (2012)
D. Daley-Brown, G. Oprea-Iles, K.T. Vann, V. Lanier, R. Lee, P.V. Candelaria, A. Quarshie, R. Pattillo, R.R. Gonzalez-Perez, Type II endometrial Cancer overexpresses NILCO: A preliminary evaluation. Dis. Markers 2017, 1–14 (2017)
A. Zaravinos, The regulatory role of microRNAs in EMT and cancer. J. Oncol. 2015, 1–13 (2015)
J. Yu, K. Ohuchida, K. Mizumoto, N. Sato, T. Kayashima, H. Fujita, K. Nakata, M. Tanaka, MicroRNA, hsa-miR-200c, is an independent prognostic factor in pancreatic cancer and its upregulation inhibits pancreatic cancer invasion but increases cell proliferation. Mol. Cancer 9, 169 (2010)
S. Brabletz, K. Bajdak, S. Meidhof, U. Burk, G. Niedermann, E. Firat, U. Wellner, A. Dimmler, G. Faller, J. Schubert, The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 30, 770–782 (2011)
U. Burk, J. Schubert, U. Wellner, O. Schmalhofer, E. Vincan, S. Spaderna, T. Brabletz, A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 9, 582–589 (2008)
C.C. Chang, M.J. Wu, J.Y. Yang, I. Carmarillo, C.-J. Chang, Leptin-STAT3-G9a signaling promotes obesity-mediated breast cancer progression. Cancer Res. 75, 2375–2386 (2015)
C.I.T. Mantho, A. Harbuzariu, R.R. Gonzalez-Perez, Histone deacetylases, microRNA and leptin crosstalk in pancreatic cancer. World J. Clin. Oncol. 8, 178 (2017)
J. Joseph-Silverstein, R.L. Silverstein, Cell adhesion molecules: an overview. Cancer Invest. 16, 176–182 (1998)
B. Leitinger, E. Hohenester, Mammalian collagen receptors. Matrix Biol. 26, 146–155 (2007)
P. Caswell, J. Norman, Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol. 18, 257–263 (2008)
E. Zamir, B. Geiger, Components of cell-matrix adhesions. J. Cell Sci. 114, 3577–3579 (2001)
S.K. Mitra, D.A. Hanson, D.D. Schlaepfer, Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 (2005)
M.S. Diamond, D.E. Staunton, A.R. De Fougerolles, S.A. Stacker, J. Garcia-Aguilar, M.L. Hibbs, T.A. Springer, ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 111, 3129–3139 (1990)
N. Makrilia, A. Kollias, L. Manolopoulos, K. Syrigos, Cell adhesion molecules: role and clinical significance in cancer. Cancer Invest. 27, 1023–1037 (2009)
Y. Maruo, A. Gochi, A. Kaihara, H. Shimamura, T. Yamada, N. Tanaka, K. Orita, ICAM-1 expression and the soluble ICAM-1 level for evaluating the metastatic potential of gastric cancer. Int. J. Cancer. 100, 486–490 (2002)
A. Grothey, P. Heistermann, S. Philippou, R. Voigtmann, Serum levels of soluble intercellular adhesion molecule-1 (ICAM-1, CD54) in patients with non-small-cell lung cancer: correlation with histological expression of ICAM-1 and tumour stage. Br. J. Cancer 77, 801–807 (1998)
P.-C. Chen, T.-H. Lin, H.-C. Cheng, C.-H. Tang, CCN3 increases cell motility and ICAM-1 expression in prostate cancer cells. Carcinogenesis 33, 937–945 (2012)
A. Spina, F. Di Maiolo, A. Esposito, L. Sapio, E. Chiosi, L. Sorvillo, S. Naviglio, cAMP elevation down-regulates β 3 integrin and focal adhesion kinase and inhibits leptin-induced migration of MDA-MB-231 breast cancer cells. BioResearch 1, 324–332 (2012)
S. Naviglio, D. Di Gesto, F. Illiano, E. Chiosi, A. Giordano, G. Illiano, A. Spina, Leptin potentiates antiproliferative action of cAMP elevation via protein kinase A down-regulation in breast cancer cells. J. Cell. Physiol. 225, 801–809 (2010)
J. Ratke, F. Entschladen, B. Niggemann, K.S. Zänker, K. Lang, Leptin stimulates the migration of colon carcinoma cells by multiple signaling pathways. Endocr. Relat. Cancer. 17, 179–189 (2010)
N.-M. Heida, M. Leifheit-Nestler, M.R. Schroeter, J.-P. Müller, I.-F. Cheng, S. Henkel, A. Limbourg, F.P. Limbourg, F. Alves, J.P. Quigley, Leptin enhances the potency of circulating angiogenic cells via src kinase and integrin αvβ5: implications for angiogenesis in human obesity. Arterioscler. Thromb. Vasc. Biol. 30, 200 (2010)
S.-N. Yang, H.-T. Chen, H.-K. Tsou, C.-Y. Huang, W.-H. Yang, C.-M. Su, Y.-C. Fong, W.-P. Tseng, C.-H. Tang, Leptin enhances cell migration in human chondrosarcoma cells through OBRl leptin receptor. Carcinogenesis 30, 566–574 (2009)
C.Y. Huang, H.S. Yu, T.Y. Lai, Y.L. Yeh, C.C. Su, H.H. Hsu, F.J. Tsai, C.H. Tsai, H.C. Wu, C.H. Tang, Leptin increases motility and integrin up-regulation in human prostate cancer cells. J. Cell. Physiol. 226, 1274–1282 (2011)
C.K. Wong, P.F.Y. Cheung, C.W. Lam, Leptin-mediated cytokine release and migration of eosinophils: Implications for immunopathophysiology of allergic inflammation. Eur. J. Immunol. 37, 2337–2348 (2007)
Z. Dong, S. Fu, X. Xu, Y. Yang, L. Du, W. Li, S. Kan, Z. Li, X. Zhang, L. Wang, Leptin-mediated regulation of ICAM-1 is Rho/ROCK dependent and enhances gastric cancer cell migration. Br. J. Cancer 110, 1801–1810 (2014)
M. Suzukawa, R. Koketsu, S. Baba, S. Igarashi, H. Nagase, M. Yamaguchi, N. Matsutani, M. Kawamura, S. Shoji, A. Hebisawa, Leptin enhances ICAM-1 expression, induces migration and cytokine synthesis, and prolongs survival of human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 309, L801–L811 (2015)
M. Yilmaz, G. Christofori, EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 28, 15–33 (2009)
W.H. Goldmann, V. Auernheimer, I. Thievessen, B. Fabry, Vinculin, cell mechanics and tumour cell invasion. Cell Biol. Int. 37, 397–405 (2013)
T. Li, H. Guo, Y. Song, X. Zhao, Y. Shi, Y. Lu, S. Hu, Y. Nie, D. Fan, K. Wu, Loss of vinculin and membrane-bound β-catenin promotes metastasis and predicts poor prognosis in colorectal cancer. Mol. Cancer. 13, 263 (2014)
P. Friedl, K. Wolf, Tumour-cell invasion and migration: diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374 (2003)
S.P. Palecek, J.C. Loftus, M.H. Ginsberg, D.A. Lauffenburger, A.F. Horwitz, Integrin-ligand binding properties govern cell migration speed through cell-substratum adhesiveness. Nature 385, 537 (1997)
C. Ballestrem, B. Hinz, B.A. Imhof, B. Wehrle-Haller, Marching at the front and dragging behind. J. Cell Biol. 155, 1319–1332 (2001)
C.D. Nobes, A. Hall, Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 81, 53–62 (1995)
K. Wennerberg, C.J. Der, Rho-family GTPases: it's not only Rac and Rho (and I like it). J. Cell Sci. 117, 1301–1312 (2004)
S. Narumiya, M. Tanji, T. Ishizaki, Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 28, 65–76 (2009)
A.L. Bishop, H. Alan, Rho GTPases and their effector proteins. Biochem. J. 348, 241–255 (2000)
A.J. Ridley, Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 (2015)
A. Ghasemi, S.I. Hashemy, M. Aghaei, M. Panjehpour, RhoA/ROCK pathway mediates leptin-induced uPA expression to promote cell invasion in ovarian cancer cells. Cell. Signal. 32, 104–114 (2017)
T. Jaffe, B. Schwartz, Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int. J. Cancer. 123, 2543–2556 (2008)
Y. Xie, C.M. Potter, A. Le Bras, W.N. Nowak, W. Gu, S.I. Bhaloo, Z. Zhang, Y. Hu, L. Zhang, Q. Xu, Leptin induces Sca-1+ progenitor cell migration enhancing neointimal lesions in vessel-injury mouse models. Arterioscler. Thromb. Vasc. Biol. 37, 2114–2127 (2017)
Y.-E. Han, A. Lim, S.-H. Park, S. Chang, S.-H. Lee, W.-K. Ho, Rac-mediated actin remodeling and myosin II are involved in KATP channel trafficking in pancreatic β-cells. Exp. Mol. Med. 47, e190 (2015)
Z. Li, J. Liang, W.K.K. Wu, X. Yu, J. Yu, X. Weng, J. Shen, Leptin activates RhoA/ROCK pathway to induce cytoskeleton remodeling in nucleus pulposus cells. Int. J. Mol. Sci. 15, 1176–1188 (2014)
S.S. Stylli, A.H. Kaye, P. Lock, Invadopodia: at the cutting edge of tumour invasion. J. Clin. Neurosci. 15, 725–737 (2008)
W.T. Chen, Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J. Exp. Zool. A Ecol. Genet. Physiol. 251, 167–185 (1989)
R.E. Vandenbroucke, C. Libert, Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 13, 904–927 (2014)
A. Alaseem, K. Alhazzani, P. Dondapati, S. Alobid, A. Bishayee, A. Rathinavelu, in Semin. Cancer Biol., (Elsevier, 2017),
Y. Gong, U.D. Chippada-Venkata, W.K. Oh, Roles of matrix metalloproteinases and their natural inhibitors in prostate cancer progression. Cancer 6, 1298–1327 (2014)
L. Al-Alem, T.E. Curry, Ovarian cancer: involvement of the matrix metalloproteinases. Reproduction 150, R55–R64 (2015)
J. Cathcart, A. Pulkoski-Gross , J. Cao, Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2, 26–34 (2015)
S. Löffek, O. Schilling, C.-W. Franzke, (Eur Respiratory Soc, 2011),
E.M. Sobrinho Santos, T.A. Guimaraes, H.O. Santos, L.M.B. Cangussu, S.F. de Jesus, C.A.d.C. Fraga, C.M. Cardoso, S.H.S. Santos, A.M.B. de Paula, R.S. Gomez, Leptin acts on neoplastic behavior and expression levels of genes related to hypoxia, angiogenesis, and invasiveness in oral squamous cell carcinoma. Tumor Biol. 39, 1010428317699130 (2017)
M. Hoffmann, E. Fiedor, A. Ptak, 17β-Estradiol reverses leptin-inducing ovarian cancer cell migration by the PI3K/Akt signaling pathway. Reproductive Sciences 23, 1600–1608 (2016)
Y.S. Jo, G.S.R. Lee, S.Y. Nam, S.J. Kim, Progesterone inhibits leptin-induced invasiveness of BeWo cells. Int. J.Med. Sci. 12, 773–779 (2015)
G. Han, W. Zhao, L. Wang, Z. Yue, R. Zhao, Y. Li, X. Zhou, X. Hu, J. Liu, Leptin enhances the invasive ability of glioma stem-like cells depending on leptin receptor expression. Brain Res. 1543, 1–8 (2014)
L. Wang, H. Cao, X. Pang, K. Li, W. Dang, H. Tang, T. Chen, The effect of leptin and its mechanisms on the migration and invasion of human breast cancer MCF-7 cells. Xi bao yu fen zi mian yi xue za zhi (Chinese Journal of Cellular and Molecular Immunology) 29, 1272–1276 (2013)
Y. Cha, Y. Kang, A. Moon, HER2 induces expression of leptin in human breast epithelial cells. BMB Reports 45, 719–723 (2012)
M.-C. Lin, F.-Y. Wang, Y.-H. Kuo, F.-Y. Tang, Cancer chemopreventive effects of lycopene: suppression of MMP-7 expression and cell invasion in human colon cancer cells. J. Agric. Food Chem. 59, 11304–11318 (2011)
N. Stefanou, V. Papanikolaou, Y. Furukawa, Y. Nakamura, A. Tsezou, Leptin as a critical regulator of hepatocellular carcinoma development through modulation of human telomerase reverse transcriptase. BMC Cancer 10, 442 (2010)
V. McMurtry, A.-M. Simeone, R. Nieves-Alicea, A.M. Tari, Leptin utilizes Jun N-terminal kinases to stimulate the invasion of MCF-7 breast cancer cells. Clin. Exp. 26, 197–204 (2009)
N. Johansson, M. Ahonen, V.-M. Kähäri, Matrix metalloproteinases in tumor invasion. Cell. Mol. Life Sci. 57, 5–15 (2000)
B. Bauvois, New facets of matrix metalloproteinases MMP-2 and MMP-9 as cell surface transducers: outside-in signaling and relationship to tumor progression. Biochim. Biophys. Acta Rev. Cancer 1825, 29–36 (2012)
M. Björklund, E. Koivunen, Gelatinase-mediated migration and invasion of cancer cells. Biochim. Biophys. Acta Rev. Cancer 1755, 37–69 (2005)
E.M.S. Santos, R.G. da Rocha, H.O. Santos, T.A. Guimarães, C.A. de Carvalho Fraga, L.H. da Silveira, P.R. Batista, P.S.L. de Oliveira, G.A. Melo, S.H. Santos, Gallic acid modulates phenotypic behavior and gene expression in oral squamous cell carcinoma cells by interfering with leptin pathway. Pathol. Res. Pract. 214, 30–37 (2018)
H. Zou, Y. Liu, D. Wei, T. Wang, K. Wang, S. Huang, L. Liu, Y. Li, J. Ge, X. Li, Leptin promotes proliferation and metastasis of human gallbladder cancer through OB-Rb leptin receptor. Int. J. Oncol. 49, 197–206 (2016)
J.-H. Ahn, Y.S. Choi, J.-H. Choi, Leptin promotes human endometriotic cell migration and invasion by up-regulating MMP-2 through the JAK2/STAT3 signaling pathway. MHR: Basic Sci. Reprod. Med. 21, 792–802 (2015)
N. Borkakoti, Structural studies of matrix metalloproteinases. J. Mol. Med. 78, 261–268 (2000)
H. Nagase, J.F. Woessner, Matrix metalloproteinases. J. Biol. Chem. 274, 21491–21494 (1999)
T.E. Curry Jr, K.G. Osteen, The matrix metalloproteinase system: changes, regulation, and impact throughout the ovarian and uterine reproductive cycle. Endocr. Rev. 24, 428–465 (2003)
J. Wang, X. Li, X. Gao, S. An, H. Liu, J. Liang, K. Zhang, Z. Liu, J. Wang, Z. Chen, Expression of MMP-13 is associated with invasion and metastasis of papillary thyroid carcinoma. Eur. Rev. Med. Pharmacol. Sci. 17, 427–435 (2013)
B. Zhang, X. Cao, Y. Liu, W. Cao, F. Zhang, S. Zhang, H. Li, L. Ning, L. Fu, Y. Niu, Tumor-derived matrix metalloproteinase-13 (MMP-13) correlates with poor prognosis of invasive breast cancer. BMC Cancer 8, 83 (2008)
B. Yang, J. Gao, Z. Rao, Q. Shen, Clinicopathological significance and prognostic value of MMP-13 expression in colorectal cancer. Scand. J. Clin. Lab. Invest. 72, 501–505 (2012)
B. Hantke, N. Harbeck, B. Schmalfeldt, I. Claes, O. Hiller, M.-O. Luther, A. Welk, W. Kuhn, M. Schmitt, H. Tschesche, Clinical relevance of matrix metalloproteinase-13 determined with a new highly specific and sensitive ELISA in ascitic fluid of advanced ovarian carcinoma patients. Biol. Chem. 384, 1247–1251 (2003)
W.L. Yeh, D.Y. Lu, M.J. Lee, W.M. Fu, Leptin induces migration and invasion of glioma cells through MMP-13 production. Glia 57, 454–464 (2009)
D. Iliopoulos, K.N. Malizos, A. Tsezou, Epigenetic regulation of leptin affects MMP-13 expression in osteoarthritic chondrocytes: possible molecular target for osteoarthritis therapeutic intervention. Ann. Rheum. Dis. 66, 1616–1621 (2007)
A. Koskinen, K. Vuolteenaho, R. Nieminen, T. Moilanen, E. Moilanen, Leptin enhances MMP-l, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin. Exp. Rheumatol. 29, 57 (2011)
R.C. Williams, A.J. Skelton, S.M. Todryk, A.D. Rowan, P.M. Preshaw, J.J. Taylor, Leptin and pro-inflammatory stimuli synergistically upregulate MMP-1 and MMP-3 secretion in human gingival fibroblasts. PLoS ONE 11, e0148024 (2016)
L. Liberale, A. Bonaventura, F. Carbone, M. Bertolotto, P. Contini, N. Scopinaro, G.B. Camerini, F.S. Papadia, R. Cordera, G.G. Camici, Early reduction of matrix metalloproteinase-8 serum levels is associated with leptin drop and predicts diabetes remission after bariatric surgery. Int. J. Cardiol. 245, 257–262 (2017)
F.q. Wang, J. So, S. Reierstad, D.A. Fishman, Matrilysin (MMP-7) promotes invasion of ovarian cancer cells by activation of progelatinase. Int. J. Cancer 114, 19–31 (2005)
A. Ghasemi, S.I. Hashemy, M. Aghaei, M. Panjehpour, Leptin induces matrix metalloproteinase 7 expression to promote ovarian cancer cell invasion by activating ERK and JNK pathways. J. Cell. Biochem. 119, 2333–2344 (2018)
M.-C. Lin, S.-Y. Tsai, F.-Y. Wang, F.-H. Liu, J.-N. Syu, F.-Y. Tang, Leptin induces cell invasion and the upregulation of matrilysin in human colon cancer cells. Biomedicine 3, 174–180 (2013)
I. Yana, M. Seiki, MT-MMPs play pivotal roles in cancer dissemination. Clin. Exp. Metastasis 19, 209–215 (2002)
Y. Itoh, Membrane-type matrix metalloproteinases: their functions and regulations. Matrix Biol. 44, 207–223 (2015)
Z. Dong, X. Xu, L. Du, Y. Yang, H. Cheng, X. Zhang, Z. Li, L. Wang, J. Li, H. Liu, Leptin-mediated regulation of MT1-MMP localization is KIF1B dependent and enhances gastric cancer cell invasion. Carcinogenesis 34, 974–983 (2013)
H. Wang, H. Cheng, Q. Shao, Z. Dong, Q. Xie, L. Zhao, Q. Wang, B. Kong, X. Qu, Leptin-promoted human extravillous trophoblast invasion is MMP14 dependent and requires the cross talk between Notch1 and PI3K/Akt signaling. Biol. Reprod. 90, 71–10 (2014)
K. Dass, A. Ahmad, A.S. Azmi, S.H. Sarkar, F.H. Sarkar, Evolving role of uPA/uPAR system in human cancers. Cancer Treat. Rev. 34, 122–136 (2008)
C.E. de Bock, Y. Wang, Clinical significance of urokinase-type plasminogen activator receptor (uPAR) expression in cancer. Med. Res. Rev. 24, 13–39 (2004)
P.A. van Dam, A. Coelho, C. Rolfo, Is there a role for urokinase-type plasminogen activator inhibitors as maintenance therapy in patients with ovarian cancer? Eur. J. Surg. Oncol. 43, 252–257 (2017)
P. Singh, T.E. Peterson, K.R. Barber, F.S. Kuniyoshi, A. Jensen, M. Hoffmann, A.S. Shamsuzzaman, V.K. Somers, Leptin upregulates the expression of plasminogen activator inhibitor-1 in human vascular endothelial cells. Biochem. Biophys. Res. Commun. 392, 47–52. (2010)
S. Landskroner-Eiger, B. Qian, E.S. Muise, A.R. Nawrocki, J.P. Berger, E.J. Fine, W. Koba, Y. Deng, J.W. Pollard, P.E. Scherer, Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin. Cancer Res. 15, 3265–3276 (2009)
F. Messaggio, A.M. Mendonsa, J. Castellanos, N.S. Nagathihalli, L. Gorden, N.B. Merchant, M.N. VanSaun, Adiponectin receptor agonists inhibit leptin induced pSTAT3 and in vivo pancreatic tumor growth. Oncotarget 8, 85378 (2017)
W. An, Y. Bai, S.-X. Deng, J. Gao, Q.-W. Ben, Q.-C. Cai, H.-G. Zhang, Z.-S. Li, Adiponectin levels in patients with colorectal cancer and adenoma: a meta-analysis. Eur. J. Cancer Prev. 21, 126–133 (2012)
E. Petridou, C. Mantzoros, N. Dessypris, P. Koukoulomatis, C. Addy, Z. Voulgaris, G. Chrousos, D. Trichopoulos, Plasma adiponectin concentrations in relation to endometrial cancer: a case-control study in Greece. J. Clin. Endocrinol. Metab. 88, 993–997 (2003)
P.C. Konturek, G. Burnat, T. Rau, E.G. Hahn, S. Konturek, Effect of adiponectin and ghrelin on apoptosis of Barrett adenocarcinoma cell line. Dig. Dis. Sci. 53, 597–605 (2008)
S.S. Tworoger, A.H. Eliassen, T. Kelesidis, G.A. Colditz, W.C. Willett, C.S. Mantzoros, S.E. Hankinson, Plasma adiponectin concentrations and risk of incident breast cancer. J. Clin. Endocrinol. Metab. 92, 1510–1516 (2007)
J.A. Handy, N.K. Saxena, P. Fu, S. Lin, J.E. Mells, N.A. Gupta, F.A. Anania, Adiponectin activation of AMPK disrupts leptin-mediated hepatic fibrosis via suppressors of cytokine signaling (SOCS-3). J. Cell. Biochem. 110, 1195–1207 (2010)
I.L. Beales, C. Garcia-Morales, O.O. Ogunwobi, G. Mutungi, Adiponectin inhibits leptin-induced oncogenic signalling in oesophageal cancer cells by activation of PTP1B. Mol. Cell. Endocrinol. 382, 150–158 (2014)
L. Lessard, M. Stuible, M.L. Tremblay, The two faces of PTP1B in cancer. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1804, 613–619 (2010)
Z.-Y. Zhang, G.T. Dodd, T. Tiganis, Protein tyrosine phosphatases in hypothalamic insulin and leptin signaling. Trends Pharmacol. Sci. 36, 661–674 (2015)
L. Taliaferro-Smith, A. Nagalingam, B.B. Knight, E. Oberlick, N.K. Saxena, D. Sharma, Integral role of PTP1B in adiponectin-mediated inhibition of oncogenic actions of leptin in breast carcinogenesis. Neoplasia 15, 23IN10–38IN11 (2013)
D. Sharma, J. Wang, P.P. Fu, S. Sharma, A. Nagalingam, J. Mells, J. Handy, A.J. Page, C. Cohen, F.A. Anania, Adiponectin antagonizes the oncogenic actions of leptin in hepatocellular carcinogenesis. Hepatology 52, 1713–1722 (2010)
X. Wu, Q. Yan, Z. Zhang, G. Du, X. Wan, Acrp30 inhibits leptin-induced metastasis by downregulating the JAK/STAT3 pathway via AMPK activation in aggressive SPEC-2 endometrial cancer cells. Oncol. Rep. 27, 1488–1496 (2012)
S.A. Verploegen, G. Plaetinck, R. Devos, J. Van der Heyden, Y. Guisez, A human leptin mutant induces weight gain in normal mice. FEBS Lett 405, 237–240 (1997)
L. Brunner, S. Whitebread, I. Leconte, A. Stricker-Krongrad, F. Cumin, M. Chiesi, N. Levens, A peptide leptin antagonist reduces food intake in rodents. Int. J. Obes. Relat. Metab. Disord. 23, 463–469 (1999)
F. Peelman, K. Van Beneden, L. Zabeau, H. Iserentant, P. Ulrichts, D. Defeau, A. Verhee, D. Catteeuw, D. Elewaut, J. Tavernier, Mapping of the leptin binding sites and design of a leptin antagonist. J. Biol. Chem. 279, 41038–41046 (2004)
H. Park, S.-H. Ahn, Y. Jung, J.C. Yoon, Y.-H. Choi, Leptin suppresses glutamate-induced apoptosis through regulation of ERK1/2 signaling pathways in rat primary astrocytes. Cell. Physiol. Biochem. 44, 2117–2128 (2017)
G. Salomon, L. Niv-Spector, E.E. Gussakovsky, A. Gertler, Large-scale preparation of biologically active mouse and rat leptins and their L39A/D40A/F41A muteins which act as potent antagonists. Protein Expr. Purif. 47, 128–136 (2006)
L. Niv-Spector, D. Gonen-Berger, I. Gourdou, E. Biener, E.E. Gussakovsky, Y. Benomar, K.V. Ramanujan, M. Taouis, B. Herman, I. Callebaut, Identification of the hydrophobic strand in the A–B loop of leptin as major binding site III: implications for large-scale preparation of potent recombinant human and ovine leptin antagonists. Biochem. J. 391, 221–230 (2005)
S. Samuel-Mendelsohn, M. Inbar, E. Weiss-Messer, L. Niv-Spector, A. Gertler, R.J. Barkey, Leptin signaling and apoptotic effects in human prostate cancer cell lines. The Prostate 71, 929–945 (2011)
S. Catalano, A. Leggio, I. Barone, R. De Marco, L. Gelsomino, A. Campana, R. Malivindi, S. Panza, C. Giordano, A. Liguori, A novel leptin antagonist peptide inhibits breast cancer growth in vitro and in vivo. J. Cell. Mol. Med. 19, 1122–1132 (2015)
E. Fiedor, E.Ł. Gregoraszczuk, The molecular mechanism of action of superactive human leptin antagonist (SHLA) and quadruple leptin mutein Lan-2 on human ovarian epithelial cell lines. Cancer Chemother. Pharmacol. 78, 611–622 (2016)
E. Fiedor, E. Gregoraszczuk, Superactive human leptin antagonist (SHLA), triple Lan1 and quadruple Lan2 leptin mutein as a promising treatment for human folliculoma. Cancer Chemother. Pharmacol. 80, 815–827 (2017)
L. Niv-Spector, M. Shpilman, Y. Boisclair, A. Gertler, Large-scale preparation and characterization of non-pegylated and pegylated superactive ovine leptin antagonist. Protein Expr. Purif. 81, 186–192 (2012)
M. Shpilman, L. Niv-Spector, M. Katz, C. Varol, G. Solomon, M. Ayalon-Soffer, E. Boder, Z. Halpern, E. Elinav, A. Gertler, Development and characterization of high affinity leptins and leptin antagonists. J. Biol. Chem. 286, 4429–4442 (2011)
G. Bain, E. Collie-Duguid, G.I. Murray, F. Gilbert, A. Denison, F. McKiddie, T. Ahearn, I. Fleming, J. Leeds, P. Phull, Tumour expression of leptin is associated with chemotherapy resistance and therapy-independent prognosis in gastro-oesophageal adenocarcinomas. Br. J. Cancer. 110, 1525 (2014)
E. Fiedor, K. Zajda, E.L. Gregoraszczuk, leptin receptor antagonists' action on HDAC expression eliminating the negative effects of leptin in ovarian cancer. Cancer Genomics Proteomics 15, 329–336 (2018)
P. Grasso, M.C. Leinung, S.P. Ingher, D.W. Lee, In vivo effects of leptin-related synthetic peptides on body weight and food intake in female ob/ob mice: localization of leptin activity to domains between amino acid residues 106–140. Endocrinology. 138, 1413–1418 (1997)
R.R. Gonzalez, P.C. Leavis, A peptide derived from the human leptin molecule is a potent inhibitor of the leptin receptor function in rabbit endometrial cells. Endocrine 21, 185–195 (2003)
M. Ramos, B. Rueda, P. Leavis, R. Gonzalez, Leptin serves as an upstream activator of an obligatory signaling cascade in the embryo-implantation process. Endocrinology 146, 694–701 (2005)
A. Leggio, S. Catalano, R. De Marco, I. Barone, S. Andò, A. Liguori, Therapeutic potential of leptin receptor modulators. Eur. J. Med. Chem. 78, 97–105 (2014)
L. Otvos, E. Surmacz, Targeting the leptin receptor: a potential new mode of treatment for breast cancer. Expert Rev. Anticancer Ther. 11, 1147–1150 (2011)
L. Otvos, I. Kovalszky, M. Riolfi, R. Ferla, J. Olah, A. Sztodola, K. Nama, A. Molino, Q. Piubello, J.D. Wade, Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur. J. Cancer 47, 1578–1584 (2011)
S. Beccari, I. Kovalszky, J.D. Wade, L. Otvos Jr, E. Surmacz, Designer peptide antagonist of the leptin receptor with peripheral antineoplastic activity. Peptides 44, 127–134 (2013)
T. Harmon, A. Harbuzariu, V. Lanier, C.C. Lipsey, W. Kirlin, L. Yang, R.R. Gonzalez-Perez, Nanoparticle-linked antagonist for leptin signaling inhibition in breast cancer. World J. Clin. Oncol. 8, 54 (2017)
Y.-T. Chin, L.-M. Wang, M.-T. Hsieh, Y.-J. Shih, A.W. Nana, C.A. Changou, Y.-C.S. Yang, H.-C. Chiu, E. Fu, P.J. Davis, Leptin OB3 peptide suppresses leptin-induced signaling and progression in ovarian cancer cells. J. Biomed. Sci. 24, 51 (2017)
Y.-C.S. Yang, Y.-T. Chin, M.-T. Hsieh, H.-Y. Lai, C.-C. Ke, D.R. Crawford, O.K. Lee, E. Fu, S.A. Mousa, P. Grasso, Novel leptin OB3 peptide-induced signaling and progression in thyroid cancers: comparison with leptin. Oncotarget 7, 27641 (2016)
S.-H. Kim, A. Nagalingam, N.K. Saxena, S.V. Singh, D. Sharma, Benzyl isothiocyanate inhibits oncogenic actions of leptin in human breast cancer cells by suppressing activation of signal transducer and activator of transcription 3. Carcinogenesis 32, 359–367 (2010)
M. Higginbottom, A.V.-A. Horgan, J. Horton, I. Simpson, C. Tyzack, (Google Patents, 2009),
M. Fazeli, H. Zarkesh-Esfahani, Z. Wu, M. Maamra, M. Bidlingmaier, A.G. Pockley, P. Watson, G. Matarese, C.J. Strasburger, R.J. Ross, Identification of a monoclonal antibody against the leptin receptor that acts as an antagonist and blocks human monocyte and T cell activation. J. Immunol. Methods. 312, 190–200 (2006)
L. Zabeau, A. Verhee, D. Catteeuw, L. Faes, S. Seeuws, T. Decruy, D. Elewaut, F. Peelman, J. Tavernier, Selection of non-competitive leptin antagonists using a random nanobody-based approach. Biochem. J. 441, 425–434 (2012)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdiction-al claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ghasemi, A., Saeidi, J., Azimi-Nejad, M. et al. Leptin-induced signaling pathways in cancer cell migration and invasion. Cell Oncol. 42, 243–260 (2019). https://doi.org/10.1007/s13402-019-00428-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-019-00428-0