
Abstract
Purpose
Glioblastoma (GBM) ranks among the deadliest solid cancers worldwide and its prognosis has remained dismal, despite the use of aggressive chemo-irradiation treatment regimens. Limited drug delivery into the brain parenchyma and frequent resistance to currently available therapies are problems that call for a prompt development of novel therapeutic strategies. While only displaying modest efficacies as mono-therapy in pre-clinical settings, histone deacetylase inhibitors (HDACi) have shown promising sensitizing effects to a number of cytotoxic agents. Here, we sought to investigate the sensitizing effect of the HDACi trichostatin A (TSA) to the alkylating agent lomustine (CCNU), which is used in the clinic for the treatment of GBM.
Methods
Twelve primary GBM cell cultures grown as neurospheres were used in this study, as well as one established GBM-derived cell line (U87 MG). Histone deacetylase (HDAC) expression levels were determined using quantitative real-time PCR and Western blotting. The efficacy of either CCNU alone or its combination with TSA was assessed using various assays, i.e., cell viability assays (MTT), cell cycle assays (flow cytometry, FACS), double-strand DNA break (DSB) quantification assays (microscopy/immunofluorescence) and expression profiling assays of proteins involved in apoptosis and cell stress (Western blotting and protein array).
Results
We found that the HDAC1, 3 and 6 expression levels were significantly increased in GBM samples compared to non-neoplastic brain control samples. Additionally, we found that pre-treatment of GBM cells with TSA resulted in an enhancement of their sensitivity to CCNU, possibly via the accumulation of DSBs, decreased cell proliferation and viability rates, and an increased apoptotic rate.
Conclusion
From our data we conclude that the combined administration of TSA and CCNU eradicates GBM cells with a higher efficacy than either drug alone, thereby opening a novel avenue for the treatment of GBM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Glioblastoma (GBM) is the most prevalent and aggressive brain tumor in adults with an average survival rate of approximately 15 months, despite maximal therapeutic intervention [1]. A high degree of heterogeneity, a robust vasculature, as well as enduring resistance to current therapies are among the most prevalent hallmarks of GBM [2].
DNA methylation and histone modifications are common epigenetic changes that play central roles in many cellular processes, including proliferation, survival, angiogenesis, invasion and differentiation [3–6]. Functional interplays between histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate the chromatin state (relaxation and condensation, respectively) and its accessibility to DNA repair proteins upon induction of DNA damage [3, 7]. Epigenetic deregulation has long been suggested to play a role in gliomagenesis [8]. Class I HDACs have been found to be associated with the occurrence of de-differentiated, locally advanced and strongly proliferating tumors [9, 10], whereas class II HDACs have been found to be directly involved in tumorigenesis [11]. In the past, several HDAC inhibitors (HDACi) have been developed and US FDA-approved, such as vorinostat and romidepsin for the treatment of refractory cutaneous T-cell lymphoma [12]. Trichostatin A (TSA) is a pan-HDACi targeting class I/II HDACs and has been shown to sensitize GBM cells to radiation by inducing cell cycle arrest and differentiation [13–15].
GBMs are genetically highly unstable and exhibit a constitutive activation of DNA damage response pathways essential for their survival [16]. Based on this notion, agents preventing the repair of DNA damage in conjunction with chemo-irradiation have been developed for the treatment of GBM patients. One of these agents, lomustine (CCNU) is a highly lipid-soluble DNA alkylator that crosses the blood–brain barrier and has been shown to target proliferating GBM cells both in vitro and in vivo [17, 18]. It was also found, however, that CCNU treatment alone yielded only a modest improvement in GBM patient survival [19–21]. Thus, we hypothesized that pre-treatment of GBM cells with a HDACi (TSA) would result in chromatin restructuring and, thus, a limited access of the DNA repair machinery to DNA damage induced by CCNU, thereby increasing its therapeutic efficacy.
Here, we show that GBM cells exhibit elevated HDAC1, HDAC3 and HDAC6 expression levels compared to non-neoplastic brain tissues. Combined treatment of GBM cells with TSA and CCNU proved more efficient in impairing cell cycle progression and cell viability compared to either drug alone, which may at least in part be due to a reduced DNA repair capacity and a higher apoptotic rate. Our data indicate that the therapeutic efficacy of CCNU may be improved by co-administration of TSA, which warrants further pre-clinical evaluation of this combinational approach in the management of GBM.
2 Materials and methods
2.1 Primary cell cultures and reagents
GBM cell lines 017 (CPH017p4), 036 (CPH036p6), 047 (CPH047p3m1) and 048 (CPH048p6) have been described before [22]. GBM cell lines 4121 (GBM03), IN84 (GBM02), Xeus, T91, T115, IN326, 1587 and 1966 were obtained from The Danish Cancer Society Research Center and have also been described before [23, 24]. The GBM-derived cell line U87 MG (HTB-14) was purchased from the ATCC (VA, USA). For the in vitro studies GBM cells were maintained in Neurobasal®-A medium supplemented with N2, B27, bFGF (10 ng/ml), EGF (10 ng/ml), L-glutamine, penicillin (50 U/ml) and streptomycin (50 μg/ml) (Invitrogen, Taastrup, Denmark), and incubated in cell culture flasks in an incubator with 5 % CO2 at 37 °C. Twice a week fresh medium was added and spheres were dissociated at every passage. For the experiments, cells were dissociated, counted using a NucleoCounter® NC-200 (Chemometec, Allerod, Denmark), seeded in media with supplements and treated with trichostatin A (TSA), lomustine (CCNU) or a combination of these agents (Sigma-Aldrich, CA, USA). As a control, cells were treated with equal concentrations of DMSO.
2.2 Patient tissues
Tumor tissues were obtained from GBM patients during initial surgery at Rigshospitalet, Denmark with written consent. The tumor tissues were used according to the Declaration of Helsinki and the Danish legislation. Tumors were diagnosed as GBM according to the WHO 2000/2007 guidelines, and the use of patient-derived tissues was approved by the Danish Data Protection Agency (2006-41-6979) and the Scientific Ethical Committee for Copenhagen and Frederiksberg (KF-01-327718). At the Danish Cancer Society Research Center tissues for the generation of cell cultures were obtained in agreement with the Danish Ethical Committee guidelines and in all cases informed consent was obtained from the patients the day before surgery.
2.3 In silico analysis
For survival and HDAC expression analyses in normal brain tissues compared to gliomas (WHO grade II, III and IV) the Repository for Molecular Brain Neoplasia Data (Rembrandt) data set was used, available at the GlioVis website (http://gliovis.bioinfo.cnio.es/).
2.4 Western blotting
For the inhibitor studies, cells were pre-treated with TSA for 6 h followed by treatment with CCNU for an additional 24 h and harvested. Whole cell protein lysates were prepared from cell pellets by sonication in ice-cold modified RIPA buffer (50 mM Tris–HCl [pH 7.4], 1 % NP40, 0.25 % Na-deoxycholate, 150 mM NaCl, 1 mM EDTA) supplemented with protease and phosphatase inhibitor mixtures II and III (Millipore, Copenhagen, Denmark). Tissue protein lysates were extracted in the same buffer using a TissueLyser (Qiagen) and protein concentrations were determined using a BCA assay (Pierce Biotechnology, Rockford, IL). Next, the proteins were separated in 4–12 % NuPAGE Bis-Tris gels (Invitrogen) and electro-blotted onto nitrocellulose membranes (Invitrogen). Subsequently, the membranes were blocked for 1 h at room temperature (RT) and incubated with primary antibodies in 5 % non-fat milk overnight (ON) at 4 °C followed by incubation with horseradish peroxidase (HRP) conjugated secondary antibodies for 1 h at RT. Finally, the blots were developed using a SuperSignal West Dura Extended Duration Substrate (Pierce Biotechnology) after which the protein bands were captured using a Biospectrum Imaging System (UVP, Upland, CA, USA). The primary antibodies used are listed in Supplementary Table S1.
2.5 MTT assay
Cell viabilities were measured using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) assay (Sigma-Aldrich). To this end, GBM cells were dissociated and seeded at a density of 2 × 104 cells per well in 96-well plates and incubated ON. After this, the cells were pre-treated with TSA for 24 h followed by the addition of CCNU and incubated for another 72 h. After this treatment, 20 μl (5 mg/ml) MTT solution was added to each well and incubated for 4 h followed by the addition of 100 μl solubilization buffer (10 % SDS, 0.03 M HCl). The next day the absorbance was measured at 570 nm with 690 nm as background reference using a Synergy2 microplate reader in conjunction with Gen5, Microplate Data Collection and Analysis Software (Biotek, Winooski, VT, USA). Each experiment was performed in three replicate wells and repeated independently at least three times.
2.6 Quantitative real-time PCR
Total RNA was extracted from GBM spheres and cell pellets using a QIAshredder and RNeasy Mini kit (Qiagen). Total RNA from tumor tissue samples was extracted using TRIzol®Reagent (Gibco) and a Qiagen TissueLyser prior to purification using a RNeasy Mini kit. For cDNA synthesis and quantitative real-time PCR (qRT-PCR) amplifications a SuperscriptTM III Platinum® Two Step qRT-PCR kit with SYBR® Green (Invitrogen) was used. Gene expression levels were quantified using a comparative Ct method and normalized to the expression of three genes (TOP1, EIF4A2 and CYC1) included in the human geNorm house-keeping gene selection kit (Primerdesign, Southampton, UK). The primers used for qRT-PCR were: HDAC3 forward: 5′-TAG ACA AGG ACT GAG ATT GCC-3′; HDAC3 reverse: 5′-GTG TTA GGG AGC CAG AGC C-3′; HDAC1 forward: 5′-GGT CCA AAT GCA GGC GAT TCC T-3′; HDAC1 reverse: 5′-TCG GAG AAC TCT TCC TCA CAG G-3′; HDAC4 forward: 5′-AGG TGA AGC AGG AGC CCA TTG A-3′, HDAC4 reverse: 5′-GGT AGT TCC TCA GCT GGT GGA T-3′, HDAC6 forward: 5′-GCC TCA ATC ACT GAG ACC ATC C-3′; HDAC6 reverse: 5′-GGT GCC TTC TTG GTG ACC AAC T-3′ (TAG Copenhagen A/S, Denmark). The gene expression levels were related to those in normal brain (NB) obtained from Clontech (Takara), USA, cat. # 80151 and Ambion, USA, cat. # 7962 or normal human astrocytes (NHA) obtained from Lonza, Switzerland, cat. # CC-2565.
2.7 Immunofluorescence imaging
GBM cells (2 × 105 per condition) were seeded on coverslips pre-coated with Geltrex™ LDEV-Free Reduced Growth Factor Basement Membrane Matrix (Thermo Fisher Scientific). The next day, the cells were pre-treated with TSA for 6 h followed by treatment with CCNU for another 24 h. Next, the cells were washed in PBS, fixed in 4 % paraformaldehyde and immunostained using an anti-γH2AX Ser139 antibody (1:1000, Millipore # 05–636). Secondary detection was carried out using an Alexa Fluor 488 goat anti-mouse IgG (Invitrogen) and the nuclei of the cells were counter-stained with DAPI. Imaging was performed on a Zeiss LSM 700 Confocal Microscope and quantification was performed using a ScanR screening station (Olympus) by acquiring 100 non-overlapping images for each treatment condition. A minimum of 1000 cells was assessed and processed using the ScanR analysis software tool (Olympus). Each experiment was repeated independently at least two times.
2.8 Cell cycle analysis
For cell cycle analysis, GBM cells (2 × 105 per well) were seeded in 6-well plates and incubated ON. The next day, the cells were pre-treated with TSA for 6 h, followed by treatment with CCNU for another 24 h. During the last 20–60 min of this treatment, the cells were pulse-labelled with 10 μM 5-ethynyl-2′-deoxyuridine (EdU), after which the cells were collected and fixed in 4 % paraformaldehyde. Next, the cells were subjected to flow cytometry staining using a Click-iT EdU Alexa Flour 647 Flow Cytometry Assay Kit (Invitrogen) following the manufacturer’s instructions. Flow images were acquired on a FACS Verse Cell Sorter (BD Biosciences) and the data were analyzed using FlowJo software. Single cells were analyzed by gating out doublets and quantified as cells in the G1, S, or G2/M phase of the cell cycle. For each condition, 10.000 events were acquired and each experiment was repeated independently at least three times.
2.9 Mitotic index analysis
In order to assess cells arrested in mitosis we performed flow cytometry of cells positive for phospho-histone H3 Ser10 staining (i.e., cells in mitosis). GBM cells (2 × 105 per well) were seeded in 6-well plates and incubated ON. The next day, the cells were pre-treated with TSA for 6 h, followed by treatment with CCNU for another 24 h. Twelve hours before the end of this treatment, the cells were administered either nocadazole or DMSO. Next, the cells were collected, fixed in 4 % paraformaldehyde, stained for p-histone H3 Ser10 (Cell Signaling, #3377) and counterstained with Hoechst. Finally, images were acquired on a FACS Verse Cell Sorter (BD Biosciences) and the data were analyzed using FlowJo software.
2.10 Cell stress and apoptosis protein array
GBM cells (0.5–1.0 x 106 per condition) were pre-treated with TSA for 6 h, followed by treatment with CCNU for another 24 h. Next, the cells were harvested and lysed in a PathScan Sandwich ELISA Lysis Buffer (Cell Signaling Technology, MA, USA). The protein concentrations of the cell lysates were determined using a BCA assay. Subsequently, the cell lysates were transferred to a Pathscan Stress and Apoptosis Signaling Antibody Array Kit (Cell Signaling Technology) for the evaluation of 18 proteins involved in cell stress and apoptosis. In short, cell lysates were diluted to 0.4–0.8 μg/μl and applied to a nitrocellulose-coated glass slide with primary antibodies and incubated for 2 h. Next, the slide was washed and incubated with a detection antibody cocktail for 1 h. Subsequently, the slide was washed and incubated 30 min with a HRP-conjugated secondary antibody after which a LumiGLO/Peroxide solution (Cell Signaling Technology) was added. Finally, the slide was developed using a Biospectrum Imaging System (UVP) and the signal intensities of each spot (protein) were measured using an UVP system, normalized to intern α-tubulin levels and set relative to control (DMSO) treatment. Each experiment was performed on duplicate spots and repeated independently at least two times.
2.11 Statistics
Data are expressed as mean ± standard deviation (SD) or standard error of mean (SEM). Statistical analyses were performed using a two-tailed Student’s t-test. The software used for statistics and the generation of figures was Graphpad Prism 6.0 for windows (GraphPad, San Diego, CA, USA).
3 Results
3.1 HDAC 1, 3 and 6 are overexpressed in GBM
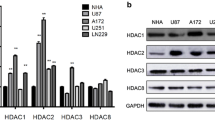
First, we sought to assess the expression of representative members of class I (HDAC1 and HDAC3) and class II (HDAC4 and HDAC6) HDACs in both GBM cells and non-neoplastic brain control cells (normal human astrocytes, NHA). We found that the expression of HDAC1, 3 and 6 were slightly and variably increased in GBM cells compared to NHA cells at both the protein (Fig. 1a) and the mRNA (Fig. 1b) level. In contrast, we found that the expression of HDAC4 was not increased in most of the GBM cells tested. Next, we set out to confirm our findings using a panel of primary GBM tissue samples, and found that the expression levels of HDAC1 and HDAC3 were markedly increased compared to those in normal brain tissues (Supplementary Fig. S1). These findings were substantiated by qRT-PCR-based expression analyses of HDAC1, 3, 4 and 6 in matched normal patient-derived tissue samples (Fig. 1c). Due to the limited sample size, we decided to subsequently validate our findings in silico using the REMBRANDT glioma dataset available through a GlioVis online application. Again, we found an increased HDAC1, 3 and 6 expression in GBM samples compared to non-neoplastic brain samples (Supplementary Fig. S2a). In addition, we found that the HDAC1 and 3 expression levels correlated with WHO tumor grades, with highest expression levels in GBM samples (Supplementary Fig. S2b). Additional Kaplan-Meier survival curve analyses revealed that HDAC3 expression is associated with a poor survival of GBM patients (Supplementary Fig. S2c).

Increased HDAC expression in GBM. a GBM cell panel and normal human astrocytes (NHA) assessed for HDAC1, HDAC3, HDAC4, HDAC6, acetylated histone H3 and total histone H3 expression by Western blotting. b Patient-derived cell cultures and (c) patient tissues assessed for HDAC1, HDAC3, HDAC4 and HDAC6 expression relative to NHA or normal brain (NB), respectively, by qRT-PCR. d U87, 047 and NHA cells treated with 0.5 μM TSA for 24 h assessed for acetylated histone H3 and total histone H3 expression using Western blotting. GAPDH was used as a loading control for the Western blots
3.2 GBM cells display differential sensitivities to TSA and CCNU
TSA, an inhibitor of class I and class II HDACs, has previously been shown to induce cell cycle arrest and apoptosis in GBM cells in vitro [13, 25]. To confirm its potency, we validated the induction of histone H3 acetylation by 0.5 μM TSA in both GBM (U87, 047) and NHA cells (Fig. 1d). Subsequently, we evaluated the sensitivity of three pre-selected GBM cell lines (017; 036; 4121) to TSA and CCNU alone. We found that the 017, 036 and 4121 cells exhibited different sensitivities to CCNU, with GI50 values of 8, 67 and 119 μM, respectively (Fig. 2a,c). Interestingly, we found that the GBM cell lines exhibited similar sensitivities to TSA, with GI50 values ranging from 0.65 to 1.19 μM (Fig. 2b,c). Moreover, we confirmed that a TSA concentration corresponding to the observed GI50 values (0.5–1.0 μM) was sufficient to induce acetylation of histone H3 and cleavage of caspase-3 in our model cell lines (Supplementary Fig. S3).

Differential inhibitory effects of TSA and CCNU on GBM cell viability. GBM cells (017, 036, 4121) were treated with increasing concentrations of a CCNU and b TSA for 72 h after which cell viabilities were assessed by MTT assay. c GI50 values calculated for each cell line treated with CCNU or TSA as indicated. Data are presented as mean ± SEM (n = 2)
3.3 Combined TSA-CCNU treatment results in enhanced induction of DNA damage and apoptosis, and a reduction in survival of GBM cells
Targeting HDACs has previously been shown to impair the capacity of cancer cells to repair DNA damage incurred by exogenous stimuli [23, 26]. Here, we evaluated the capacity of GBM cells to repair DNA damage using γH2AX (foci count and total intensity measurement) as a surrogate marker for DSBs [27]. GBM cells were treated with either TSA, CCNU or its combination (TSA+CCNU) at concentrations corresponding to their GI50 values. We found that single-agent treatment with either TSA or CCNU led to increased γH2AX foci counts and intensities compared to the respective untreated baseline levels (Fig. 3 and Supplementary Fig. S4). Importantly, we found that this effect was enhanced upon combined treatment with both TSA and CCNU (Fig. 3 and Supplementary Fig. S4). Additional results from a 18 protein array screen (Fig. 4a,b and Supplementary Table S2) revealed that the treatment effects on DSBs correlated with significant inductions of pro-apoptotic proteins, including phosphorylated Bad (pBad), cleaved PARP (cl.PARP) and cleaved caspase-3 (cl.Caspase-3). These results were further substantiated by independent Western blotting experiments conducted in the 017, 036 and 4121 cell lines (Fig. 4c,d) as well as in three other GBM cell lines (Supplementary Fig. S5). Despite the above mentioned combinatory effect on DNA damage induction, we failed to observe any enhanced activation of the Chk1 or Chk2 kinases upon TSA-CCNU treatment (Fig. 4c and Supplementary Fig. S5). Importantly, we found that the induction of apoptosis was associated with a decreased cell viability (Fig. 5a) for which a clear TSA-CCNU combinatory effect was observed compared to either drug alone. Subsequent cell cycle analyses (Fig. 5b, Supplementary Fig. S6 and S7) revealed that both TSA and CCNU treatment led to a reduction in actively proliferating cells (EdU-positive, S phase cells), which was further enhanced upon a combined TSA-CCNU administration. Taken together, we conclude that our results show an enhanced effect of a combined TSA and CCNU treatment on the induction of DNA damage and apoptosis, as well as on the reduction of survival of GBM cells, probably caused by an impaired capacity to repair DNA damage leading to an increased apoptotic rate.

TSA and CCNU treatment results in enhanced induction of DNA double stranded breaks. GBM cells (017, 036, 4121) were treated with the indicated GI50-derived concentrations of CCNU, TSA or its combination and subjected to immunofluorescence analyses. The cells were stained with an anti-γH2AX Ser139 antibody and counterstained with DAPI. a Representative images showing γH2AX staining in 036 cells treated with DMSO (control), TSA, CCNU or its combination. b Quantified total γH2AX intensities and c γH2AX foci counts obtained from 100 non-overlapping images in GBM cells treated with TSA, CCNU or its combination, relative to control (DMSO; black bar) treated cells. The γH2AX foci count could not be quantified in 017 cells due to both intense and diffuse signals in these cells. Data are presented as mean ± SEM (n = 2 for 017 and n = 3 for 036 and 4121 cells). * p < 0.05, ** p < 0.01

TSA and CCNU treatment results in apoptosis through the activation of pro-apoptotic proteins. GBM cells (017, 036, 4121) were treated with CCNU, TSA or its combination, harvested and submitted to protein array and Western blot analyses. a Representative image showing the levels of 18 phosphorylated proteins involved in cell stress and apoptosis, evaluated by array hybridization in 017 cells. Three major pro-apoptotic proteins (pBad, cleaved PARP and cleaved caspase-3) are marked 1, 2 and 3. b Quantitative analysis of the protein arrays showing fold differences in protein levels in cells treated with TSA, CCNU or its combination. The concentrations used of TSA and CCNU correspond to the designated concentrations in the Western blots under c. The quantified values were normalized to internal α-tubulin levels and set relative to the control (DMSO). Data are presented as mean ± SEM (n = 2 for 036 and n = 3 for 017 and 4121 cells). c GBM cell lysates submitted to Western blotting using anti-pChk1 (Ser345), anti-total Chk1, anti-pChk2 (T68), anti-total Chk2, anti-cleaved caspase-3, anti-total caspase-3, anti-cleaved/anti-total PARP and anti-acetylated histone H3 antibodies. GAPDH serves as loading control. d Semi-quantification of densitometry of cleaved PARP and cleaved caspase-3 levels deduced from Western blotting. The quantified expression levels were normalized to internal GAPDH levels and set relative to control (DMSO) levels. Data are presented as mean ± SEM (n = 3). * p < 0.05, ** p < 0.01, *** p < 0.001

TSA and CCNU treatment results in reduced cell viability and impairs cell cycle progression. a GBM cells (017, 036, 4121) were pre-treated with TSA for 24 h followed by administration of CCNU for another 72 h after which cell viabilities were measured by MTT assay. The results are shown relative (%) to control (DMSO; black bar) treated cells and presented as mean ± SEM (n = 3). b GBM cells (017, 036, 4121) were pre-treated with TSA for 6 h followed by administration of CCNU for another 24 h, fixed and submitted to FACS-based cell cycle analyses. Quantified cell cycle distributions (% G1, S, G2/M) are presented as mean ± SEM (n ≥ 3), * p < 0.05, ** p < 0.01
4 Discussion
GBM is among the deadliest of solid tumors for which currently available post-operative therapies (chemo-irradiation) offer only palliation [28]. Therefore, there is an urgent need for novel (targeted) therapies, which significantly improve GBM patient survival and abrogate commonly observed recurrences. Despite promising results from in vitro studies, mono-HDAC inhibitor (HDACi) therapies have only exhibited limited efficacies in clinical settings, and pre-clinical studies have encouraged the use of HDACi in combination with other anti-cancer drugs. HDACs have been found to be upregulated in various solid tumors and to play key roles in oncogenesis [10, 12]. Additionally, it has been found that in a number of malignancies such as prostate [29], colorectal [30], breast [31], lung [32], liver [33] and gastric [34] cancer, the expression of individual HDACs is inversely correlated with disease-free and overall survival rates, and is associated with a poor prognosis [12].
Based on aberrant activation of DNA damage responses in malignant gliomas [16, 23, 35] and marked resistances to DNA damaging therapies, it has been suggested that HDACi may be used as sensitizers to DNA damaging agents in GBM [36–38]. Moreover, the HDACi TSA has been found to sensitize GBM cells to ionizing radiation [15]. Hence, we hypothesized that TSA treatment may sensitize GBM cells to the alkylating agent CCNU by chromatin remodeling [39] leading to an impaired accessibility to DNA repair proteins.
A number of previous studies reported elevated expression levels of HDAC4 and HDAC6 in brain tumors [40, 41]. Here, we found that HDAC1, 3 and 6 are upregulated in GBM cells and in primary patient tissues compared to non-neoplastic brain controls at both the mRNA and protein levels. Despite the overall higher HDAC expression levels, we observed hyper-acetylation of histone H3 in GBM cells compared to normal human astrocytes. This phenomenon has previously also been observed by others [23, 41] and has been attributed to a significantly higher transcriptional activity commonly observed in GBM cells [41]. Our in silico analyses were consistent with our experimental findings and revealed increased expression levels of HDAC1, 3 and 6 in GBMs compared to non-neoplastic brain tissues and an inverse correlation of HDAC3 mRNA expression with GBM patient survival. Overall, these data indicate that both class I and class II HDACs are commonly overexpressed in GBMs and play important roles in the pathogenesis of these tumors.
We found that our GBM cell cultures displayed variable degrees of sensitivity to CCNU treatment, with GI50 values ranging from 8 to 119 μM, while the sensitivity to TSA treatment was rather consistent (GI50; 0.5 to 1.0 μM). The differential sensitivity of GBM cells to CCNU may be attributed to cell-specific resistance mechanisms such as high expression levels of multidrug resistance proteins and/or slow cell cycle kinetics. Indeed, our unpublished results revealed significantly longer cell doubling times in 017 and 036 cells compared to 4121 cells (data not shown).
TSA has previously been shown to induce apoptosis through caspase-3 activation and induction of the pro-apoptotic protein Bad [42, 43]. Our findings indicate that both TSA and CCNU alone can upregulate the expression of several intrinsic pro-apoptotic proteins (including Bad, PARP, caspase-3 and caspase-7) and that upon TSA-CCNU combination treatment this effect is enhanced, suggesting a more profound induction of apoptosis. Increased apoptosis was found to be associated with decreased cell viability. Intriguingly, besides a reduction in S phase cells (commonly observed upon combination treatment in all GBM cell lines used in our study), we did not observe a uniform response with respect to G1 or G2/M cell cycle arrest. These different responses in cell cycle dynamics and checkpoint activation may be attributed to inherent cell-specific characteristics such as p53 status (wild-type versus mutated) or expression of other cell cycle regulating proteins (i.e., cyclins, cyclin-dependent kinases etc.). To assess whether GBM cells arrest at the G1 or G2/M checkpoint upon CCNU and TSA treatment, we employed FACS analysis in conjunction with p-histone H3Ser10, a mitotic index (MI) marker, after nocodazole (NOC) treatment, which normally blocks cells in the G2/M phase. We did, however, not observe any increase in MI upon NOC treatment neither in single-agent nor in combo-agent treated GBM cells. This indicates that the cells either arrest at G1 (017 cells) and do not enter the S phase or, alternatively, die via apoptosis induction at the G2/M phase prior to cell division (036 and 4121 cells). Previously, it has been shown that both TSA and CCNU treatment can reduce the number of actively proliferating cells, a phenotype often accompanied by G2/M cell cycle phase arrest [25, 44, 45]. Cornago et al. reported that HDACi can impair G2 checkpoint activation and, thereby, promote premature entry of GBM cells into mitosis (i.e., before DNA damage is repaired), resulting in cell death via a process called mitotic catastrophe [46].
Depletion of HDAC1-3 has been shown to sensitize tumor cells to DNA damaging agents as a result of a reduction in DNA damage repair capacity [47, 48]. HDACi leads to hyper-acetylation of histones resulting in loose, transcriptionally active chromatin, making the DNA more prone to damage by additional agents such as CCNU. Indeed, we found that the co-administration of TSA and CCNU resulted in increased γH2AX foci formation and intensity compared to either of the drugs alone. The combined treatment in 017 cells resulted in intense and diffuse γH2AX signals, indicative of apoptosis [49, 50]. Our findings are consistent with previously published data showing enhanced γH2AX induction in erythroleukemic cells when TSA treatment was combined with ionizing radiation [36]. Moreover, it has been reported that the HDACi SAHA can cause a delay in DNA damage repair after radiation treatment [51] and sensitize GBM cells to PARP inhibition in GBM [23], breast cancer [52], prostate cancer [53] and ovarian cancer [54].
Currently, no HDACi has been US FDA approved for the treatment of GBM as mono-therapy, but several clinical trials are underway investigating HDACi in combination with other cytotoxic drugs such as temozolomide, bevacizumab and/or radiation therapy in GBM (https://clinicaltrials.gov). Considering the number of ongoing clinical trials and the finding that transformed cells are more sensitive to HDACi treatment than normal cells [55], there is a rationale for a combinational targeted approach using HDACi.
In summary, we found a higher efficacy of a combined treatment of GBM cells with TSA and CCNU compared to either of the drugs alone. Our data further indicate that this efficacy is due to an impaired DNA repair capacity, which subsequently leads to cell death via apoptosis. We conclude that our findings and those of others [13, 23, 26, 36, 38, 43, 46] warrant a further pre-clinical investigation and a thorough evaluation of combined HDACi applications for the treatment of GBM.
Abbreviations
- GBM:
-
glioblastoma
- TSA:
-
trichostatin A
- HDACi:
-
histone deacetylase inhibitor
- CCNU:
-
lomustine
- DSB:
-
double-stranded break
References
R. Stupp, W.P. Mason, M.J. van den Bent, M. Weller, B. Fisher, M.J. Taphoorn, K. Belanger, A.A. Brandes, C. Marosi, U. Bogdahn, J. Curschmann, R.C. Janzer, S.K. Ludwin, T. Gorlia, A. Allgeier, D. Lacombe, J.G. Cairncross, E. Eisenhauer, R.O. Mirimanoff, Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New Engl. J. Med. 352, 987–996 (2005)
P.O. Sakariassen, H. Immervoll, M. Chekenya, Cancer stem cells as mediators of treatment resistance in brain tumors: status and controversies. Neoplasia 9, 882–892 (2007)
J.P. Hamilton, Epigenetics: principles and practice. Dig. Dis. 29, 130–135 (2011)
M.B. Dhiab, S. Ziadi, S. Mestiri, R.B. Gacem, F. Ksiaa, M. Trimeche, DNA methylation patterns in EBV-positive and EBV-negative Hodgkin lymphomas. Cell. Oncol. 38, 453–462 (2015)
X. Xu, X. Chang, Z. Li, J. Wang, P. Deng, X. Zhu, J. Liu, C. Zhang, S. Chen, D. Dai, Aberrant SOX11 promoter methylation is associated with poor prognosis in gastric cancer. Cell. Oncol. 38, 183–194 (2015)
R.P. Nagarajan, J.F. Costello, Epigenetic mechanisms in glioblastoma multiforme. Semin. Cancer Biol. 19, 188–197 (2009)
C. Cortes, S.C. Kozma, A. Tauler, S. Ambrosio, MYCN concurrence with SAHA-induced cell death in human neuroblastoma cells. Cell. Oncol. 38, 341–352 (2015)
M. Maleszewska, B. Kaminska, Is glioblastoma an epigenetic malignancy? Cancers 5, 1120–1139 (2013)
M.L. Di, G. Canettieri, P. Infante, A. Greco, A. Gulino, Protected from the inside: endogenous histone deacetylase inhibitors and the road to cancer. Biochim. Biophys. Acta 1815, 241–252 (2011)
W. Weichert, HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 280, 168–176 (2009)
B. Barneda-Zahonero, M. Parra, Histone deacetylases and cancer. Mol. Oncol. 6, 579–589 (2012)
A.C. West, R.W. Johnstone, New and emerging HDAC inhibitors for cancer treatment. J. Clin. Invest. 124, 30–39 (2014)
K. Bajbouj, C. Mawrin, R. Hartig, J. Schulze-Luehrmann, A. Wilisch-Neumann, A. Roessner, R. Schneider-Stock, P53-dependent antiproliferative and pro-apoptotic effects of trichostatin A (TSA) in glioblastoma cells. J. Neurooncol. 107, 503–516 (2012)
F.D. Sassi, L. Caesar, M. Jaeger, C. Nor, A.L. Abujamra, G. Schwartsmann, C.B. de Farias, A.L. Brunetto, P.L. Lopez, R. Roesler, Inhibitory activities of trichostatin A in U87 glioblastoma cells and tumorsphere-derived cells. J. Mol. Neurosci. 54, 27–40 (2014)
J.H. Kim, J.H. Shin, I.H. Kim, Susceptibility and radiosensitization of human glioblastoma cells to trichostatin A, a histone deacetylase inhibitor. Int. J. Radiat. Oncol. Biol. Phys. 59, 1174–1180 (2004)
J. Bartkova, P. Hamerlik, M.T. Stockhausen, J. Ehrmann, A. Hlobilkova, H. Laursen, O. Kalita, Z. Kolar, H.S. Poulsen, H. Broholm, J. Lukas, J. Bartek, Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene 29, 5095–5102 (2010)
K. A. Harvey, Z. Xu, M. R. Saaddatzadeh, H. Wang, K. Pollok, A. A. Cohen-Gadol, R. A. Siddiqui, Enhanced anticancer properties of lomustine in conjunction with docosahexaenoic acid in glioblastoma cell lines. J. Neurosurg. 122, 547-556 (2015)
S.H. Yang, Y.K. Hong, S.S. Jeun, I.S. Kim, J.T. Hong, J.H. Sung, B.C. Son, S.W. Lee, M.C. Kim, K.S. Lee, Assessment of cetuximab efficacy by bioluminescence monitoring of intracranial glioblastoma xenograft in mouse. J. Neurooncol. 95, 23–28 (2009)
W. Taal, H.M. Oosterkamp, A.M. Walenkamp, H.J. Dubbink, L.V. Beerepoot, M.C. Hanse, J. Buter, A.H. Honkoop, D. Boerman, F.Y. de Vos, W.N. Dinjens, R.H. Enting, M.J. Taphoorn, F.W. van den Berkmortel, R.L. Jansen, D. Brandsma, J.E. Bromberg, I. van Heuvel, R.M. Vernhout, B. van der Holt, M.J. van den Bent, Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): a randomised controlled phase 2 trial. Lancet Oncol. 15, 943–953 (2014)
A.A. Brandes, M. Bartolotti, A. Tosoni, E. Franceschi, Nitrosoureas in the management of malignant gliomas. Curr. Neurol. Neurosci. Rep. 16, 13 (2016)
W. Wick, V.K. Puduvalli, M.C. Chamberlain, M.J. van den Bent, A.F. Carpentier, L.M. Cher, W. Mason, M. Weller, S. Hong, L. Musib, A.M. Liepa, D.E. Thornton, H.A. Fine, Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J. Clin. Oncol. 28, 1168–1174 (2010)
M.T. Stockhausen, H. Broholm, M. Villingshoj, M. Kirchhoff, T. Gerdes, K. Kristoffersen, M. Kosteljanetz, M. Spang-Thomsen, H.S. Poulsen, Maintenance of EGFR and EGFRvIII expressions in an in vivo and in vitro model of human glioblastoma multiforme. Exp. Cell Res. 317, 1513–1526 (2011)
R.D. Rasmussen, M.K. Gajjar, K.E. Jensen, P. Hamerlik, Enhanced efficacy of combined HDAC and PARP targeting in glioblastoma. Mol. Oncol. 10, 751–763 (2016)
P. Hamerlik, J.D. Lathia, R. Rasmussen, Q. Wu, J. Bartkova, M. Lee, P. Moudry, J. Bartek Jr., W. Fischer, J. Lukas, J.N. Rich, J. Bartek, Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J. Exp. Med. 209, 507–520 (2012)
M. Wetzel, D.R. Premkumar, B. Arnold, I.F. Pollack, Effect of trichostatin A, a histone deacetylase inhibitor, on glioma proliferation in vitro by inducing cell cycle arrest and apoptosis. J. Neurosurg. 103, 549–556 (2005)
M. Koprinarova, P. Botev, G. Russev, Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair 10, 970–977 (2011)
A.S. Balajee, C.R. Geard, Replication protein A and gamma-H2AX foci assembly is triggered by cellular response to DNA double-strand breaks. Exp. Cell Res. 300, 320–334 (2004)
C. Adamson, O.O. Kanu, A.I. Mehta, C. Di, N. Lin, A.K. Mattox, D.D. Bigner, Glioblastoma multiforme: a review of where we have been and where we are going. Expert Opin. Investig. Drugs 18, 1061–1083 (2009)
W. Weichert, A. Roske, V. Gekeler, T. Beckers, C. Stephan, K. Jung, F.R. Fritzsche, S. Niesporek, C. Denkert, M. Dietel, G. Kristiansen, Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 98, 604–610 (2008)
W. Weichert, A. Roske, S. Niesporek, A. Noske, A.C. Buckendahl, M. Dietel, V. Gekeler, M. Boehm, T. Beckers, C. Denkert, Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin. Cancer Res. 14, 1669–1677 (2008)
C.A. Krusche, P. Wulfing, C. Kersting, A. Vloet, W. Bocker, L. Kiesel, H.M. Beier, J. Alfer, Histone deacetylase-1 and −3 protein expression in human breast cancer: a tissue microarray analysis. Breast Cancer Res. Treat. 90, 15–23 (2005)
Y. Minamiya, T. Ono, H. Saito, N. Takahashi, M. Ito, S. Motoyama, J. Ogawa, Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumour Biol. 31, 533–539 (2010)
T. Rikimaru, A. Taketomi, Y. Yamashita, K. Shirabe, T. Hamatsu, M. Shimada, Y. Maehara, Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 72, 69–74 (2007)
W. Weichert, A. Roske, V. Gekeler, T. Beckers, M.P. Ebert, M. Pross, M. Dietel, C. Denkert, C. Rocken, Association of patterns of class I histone deacetylase expression with patient prognosis in gastric cancer: a retrospective analysis. Lancet Oncol. 9, 139–148 (2008)
M. Venere, P. Hamerlik, Q. Wu, R.D. Rasmussen, L.A. Song, A. Vasanji, N. Tenley, W.A. Flavahan, A.B. Hjelmeland, J. Bartek, J.N. Rich, Therapeutic targeting of constitutive PARP activation compromises stem cell phenotype and survival of glioblastoma-initiating cells. Cell Death Differ. 21, 258–269 (2014)
T.C. Karagiannis, K.N. Harikrishnan, A. El-Osta, The histone deacetylase inhibitor, Trichostatin A, enhances radiation sensitivity and accumulation of gammaH2A.X. Cancer Biol. Ther. 4, 787–793 (2005)
B. Sarcar, S. Kahali, P. Chinnaiyan, Vorinostat enhances the cytotoxic effects of the topoisomerase I inhibitor SN38 in glioblastoma cell lines. J. Neurooncol. 99, 201–207 (2010)
C.H. Ryu, W.S. Yoon, K.Y. Park, S.M. Kim, J.Y. Lim, J.S. Woo, C.H. Jeong, Y. Hou, S.S. Jeun, Valproic acid downregulates the expression of MGMT and sensitizes temozolomide-resistant glioma cells. J. Biomed. Biotechnol. 2012(987495) (2012)
A. Ferraro, Altered primary chromatin structures and their implications in cancer development. Cell. Oncol. 39, 195–210 (2016)
A.J. de Ruijter, A.H. van Gennip, H.N. Caron, S. Kemp, A.B. van Kuilenburg, Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem. J. 370, 737–749 (2003)
A.K. Lucio-Eterovic, M.A. Cortez, E.T. Valera, F.J. Motta, R.G. Queiroz, H.R. Machado, C.G. Carlotti Jr., L. Neder, C.A. Scrideli, L.G. Tone, Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC Cancer 8, 243 (2008)
V. Medina, B. Edmonds, G.P. Young, R. James, S. Appleton, P.D. Zalewski, Induction of caspase-3 protease activity and apoptosis by butyrate and trichostatin A (inhibitors of histone deacetylase): dependence on protein synthesis and synergy with a mitochondrial/cytochrome c-dependent pathway. Cancer Res. 57, 3697–3707 (1997)
H. Sawa, H. Murakami, Y. Ohshima, T. Sugino, T. Nakajyo, T. Kisanuki, Y. Tamura, A. Satone, W. Ide, I. Hashimoto, H. Kamada, Histone deacetylase inhibitors such as sodium butyrate and trichostatin A induce apoptosis through an increase of the bcl-2-related protein Bad. Brain Tumor Pathol. 18, 109–114 (2001)
G. Aquilina, M. Crescenzi, M. Bignami, Mismatch repair, G(2)/M cell cycle arrest and lethality after DNA damage. Carcinogenesis 20, 2317–2326 (1999)
Z. Shinwari, H. Al-Hindi, E. Al-Shail, Y. Khafaga, A. Al-Kofide, N. El-Kum, A. Aboussekhra, Response of medulloblastoma cells to vincristine and lomustine: role of TRKC, CTNNB1 and STK15. Anticancer Res. 31, 1721–1733 (2011)
M. Cornago, C. Garcia-Alberich, N. Blasco-Angulo, N. Vall-Llaura, M. Nager, J. Herreros, J.X. Comella, D. Sanchis, M. Llovera, Histone deacetylase inhibitors promote glioma cell death by G2 checkpoint abrogation leading to mitotic catastrophe. Cell Death Dis. 5, e1435 (2014)
K.M. Miller, J.V. Tjeertes, J. Coates, G. Legube, S.E. Polo, S. Britton, S.P. Jackson, Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 17, 1144–1151 (2010)
S. Bhaskara, B.J. Chyla, J.M. Amann, S.K. Knutson, D. Cortez, Z.W. Sun, S.W. Hiebert, Deletion of histone deacetylase 3 reveals critical roles in S phase progression and DNA damage control. Mol. Cell 30, 61–72 (2008)
E.P. Rogakou, W. Nieves-Neira, C. Boon, Y. Pommier, W.M. Bonner, Initiation of DNA fragmentation during apoptosis induces phosphorylation of H2AX histone at serine 139. J. Biol. Chem. 275, 9390–9395 (2000)
A. Duensing, X. Teng, Y. Liu, M. Tseng, N. Spardy, S. Duensing, A role of the mitotic spindle checkpoint in the cellular response to DNA replication stress. J. Cell. Biochem. 99, 759–769 (2006)
E. Diss, N. Nalabothula, D. Nguyen, E. Chang, Y. Kwok, F. Carrier, Vorinostat promotes hyper-radiosensitivity in wild type p53 human glioblastoma cells. J. Clin. Oncol. Res. 2, 1–9 (2014)
A. Min, S.A. Im, D.K. Kim, S.H. Song, H.J. Kim, K.H. Lee, T.Y. Kim, S.W. Han, D.Y. Oh, T.Y. Kim, M.J. O’Connor, Y.J. Bang, Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 17, 33 (2015)
O.S. Chao, O.B. Goodman Jr., Synergistic loss of prostate cancer cell viability by coinhibition of HDAC and PARP. Mol. Cancer Res. 12, 1755–1766 (2014)
P.A. Konstantinopoulos, A.J. Wilson, J. Saskowski, E. Wass, D. Khabele, Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol. Oncol. 133, 599–606 (2014)
A. Kretsovali, C. Hadjimichael, N. Charmpilas, Histone deacetylase inhibitors in cell pluripotency, differentiation, and reprogramming. Stem Cells Int. 2012, 1–10 184154 (2012)
Acknowledgments
This work was supported by grants from The Danish Cancer Research Foundation (Dansk Kræftforsknings Fond), the Danish Cancer Society and the Faculty of Health and Medical Sciences, University of Copenhagen, Denmark.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1755 kb)
Rights and permissions
About this article

Cite this article
Staberg, M., Michaelsen, S.R., Rasmussen, R.D. et al. Inhibition of histone deacetylases sensitizes glioblastoma cells to lomustine. Cell Oncol. 40, 21–32 (2017). https://doi.org/10.1007/s13402-016-0301-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-016-0301-9