
Abstract
Purpose
The 4,5-diarylimidazole brimamin is an analog of the natural vascular-disrupting agent combretastatin A-4 (CA-4) with improved water solubility, tolerance by animals and efficacy in multidrug-resistant tumors. Here, we aimed at identifying the major mechanisms underlying the in vitro and in vivo actions of brimamin on endothelial and carcinoma cells, including vascularization.
Methods
The contribution of specific signaling kinases to the effects of brimamin on cytoskeleton organization and the viability and differentiation of endothelial cells was assessed by MTT and tube formation assays in the presence or absence of specific kinase inhibitors. Changes in DNA affinity and expression of NF-κB in endothelial and carcinoma-derived cells and their solid tumors (xenografts) treated with brimamin were ascertained by electrophoretic mobility shift assays and Western blotting. The anti-vascular effect of brimamin in solid tumors was verified by CD31 immunostaining.
Results
We found that brimamin can inhibit tubulin polymerization and cause a reorganization of F-actin in Ea.hy926 endothelial cells. Its inhibitory effect on tube formation was found to depend on functional Rho kinase and JNK. JNK inhibition was found to suppress the induction of endothelial cell apoptosis by brimamin. In CA-4-refractory human BxPC-3 pancreas carcinoma-derived and triple-negative MDA-MB-231 breast carcinoma-derived cells brimamin was found to inhibit growth and to induce apoptosis at low nanomolar concentrations by blocking NF-κB activation in a dose-dependent manner. Brimamin was also found to reduce the in vivo growth rate and vascularization of MDA-MB-231 xenografts in mice. Residual tumor cells of these treated xenografts showed a relatively low expression of the p65 subunit of NF-κB.
Conclusions
Our data indicate that cellular JNK and Rho kinase activities are crucial for the cytotoxic and cytoskeleton reorganizing effects of brimamin on endothelial cells. In addition, we found that in resistant carcinoma cells and xenografts brimamin can induce down-regulation of anti-apoptotic NF-κB expression and signaling. Its chemical properties and efficacy against clinically relevant cancer entities make brimamin a promising candidate vascular-disrupting agent.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The vascular-disrupting agent (VDA) combretastatin A-4 (CA-4, Fig. 1) was first isolated from the bark of the South African Eastern Cape bushwillow tree (Combretum caffrum) [1]. Fosbretabulin (CA-4P), its water-soluble phosphate pro-drug, has been found to selectively destroy the tumor vasculature in clinical trials [2]. Since CA-4 is insufficiently cytotoxic in vivo it requires combination regimens with other drugs. In a phase 1b trial, combinations of CA-4P with carboplatin or paclitaxel were found to be efficacious in CA-4 pre-treated patients with advanced cancer [3]. Treatment with CA-4 alone often resulted in residual colonies of peripheral cancer cells that quickly re-vascularized, leading to recurrence of the disease [4]. The related catechol combretastatin A-1 and its bisphosphate (OXi4503) are also potent VDAs by which complete tumor regression can be achieved [5]. They are more cytotoxic than CA-4, possibly due to catechol redox-cycling and the generation of reactive oxygen species (ROS) or quinoid intermediates [5, 6]. Chemically stable derivatives of CA-4, which tends to isomerize to a biologically inactive E-alkene [7–9], were obtained by bridging the Z-alkene as in imidazoles, oxazoles, isoxazolines, pyridines or triazoles [4]. Wang et al. reported the synthesis of a N-methylimidazole-bridged derivative that retained the tubulin affinity of its parental combretastatin, while exhibiting improved water solubility and pharmacokinetic characteristics [9]. This compound is, however, inferior to CA-4 with respect to cytotoxicity. We recently reported the preparation of brimamin (Fig. 1), an imidazole derivative bearing a 3-bromo-4,5-dimethoxyphenyl A-ring [10]. We found that this compound can inhibit the growth of various tumor cells, including combretastatin-resistant HT-29 colon cancer-derived cells, at distinctly lower IC50 concentrations than Wang’s 3,4,5-trimethoxyphenyl analog. In addition, we found that this compound could induce extensive xenograft regression of the highly vascularized cisplatin-resistant 1411HP testicular germ cell tumor-derived cell line, while being well-tolerated by tumor-bearing mice, even at high doses [10]. We also found that brimamin [11], like CA-4P [12], can induce blebbing of endothelial cells and can lead to disruption of tubular endothelial cellular networks via a reorganization of the actin cytoskeleton. It has been reported that the survival of drug-resistant breast carcinoma and pancreas carcinoma cells, frequently observed after treatment with combretastatins, can be promoted by activated NF-κB [13–15]. There is a connection between Rho signaling and NF-κB [16–18], and there is also evidence indicating that microtubules are required for the translocation of NF-κB from the cytoplasm to the nucleus [19]. On the other hand, tubulin polymerization inhibitors such as vinblastine can activate NF-κB in epithelial cells [20]. Gemcitabin, clinically applied to pancreas cancer, has been found to induce activation of NF-κB in BxPC-3 pancreas carcinoma-derived cells, which may explain the poor response of some of the patients [15].

Structures of the vascular-disrupting combretastatins A-4 (CA-4) and A-1 (CA-1), their phosphate pro-drugs CA-4P and OXi4503, and brimamin
The present study aims at shedding light on the mode of action of brimamin, compared to that of CA-4, on endothelial and carcinoma-derived cells. In vitro experiments were carried out to identify key proteins that are involved in endothelial differentiation and cancer growth and that are also responsible for the more efficacious anticancer effects of brimamin in vivo. We assessed the interaction of brimamin with tubulin, the dependency of its effects on various protein kinases (including Rho kinase, PI3K, ERK1/2 p38, and JNK) and its influence on NF-κB, which is involved in tumor angiogenesis and therapy resistance [21, 22]. Finally, cytotoxic and vascular-disrupting efficacies of brimamin in NF-κB-dependent pancreatic carcinoma and triple-negative breast carcinoma-derived cells were assessed in vitro and in vivo.
2 Materials and methods
2.1 Compounds and reagents
Brimamin (N-methyl-4-(3-bromo-4,5-dimethoxyphenyl)-5-(3-amino-4-methoxy-phenyl)-imidazole) was prepared as described before [10]. The kinase inhibitors HA-1077 (1-(5-isoquinolinesulfonyl)homopiperazine dihydrochloride salt), LY294002 (2-(4-morpholino)-8-phenyl-4H-1-benzopyran-4-one), PD98059 (2-amino-3-methoxyflavone), SB202190 (4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole) and SP600125 (anthra(1,9-cd)pyrazol-6(2H)-one) were purchased from LC Laboratories (Woburn, MA, USA) and used without further purification. Combretastatin A-4 (CA-4) was obtained from Sigma-Aldrich.
2.2 Docking calculations
Coordinate files of the ligand structures were generated using the GlycoBioChem PRODRG2 Server (http://davapc1.bioch.dundee.ac.uk/prodrg/submit.html) [23]. Molecular docking calculations were carried out using the Autodock Vina software package [24]. Gasteiger partial charges [25] were calculated on ligand atoms using Autodock Tools. The X-ray structure of the tubulin-colchicine complex (PDB accession code: 1SA0) was downloaded from the Protein Data Bank (http://www.rcsb.org) and later used for docking. As for the ligand, polar hydrogen atoms were added to the protein and Gasteiger partial charges were calculated using Autodock Tools. Water molecules, hetero-atoms and ligands were removed from the structure prior to docking calculations. Simulation boxes were centred on the originally crystallized ligand colchicine. Residues Lys254, Lys352, Asn101, Val318 and Ile378 were treated as flexible residues. An 18 × 22 × 20 Å simulation box was used in the docking calculations, applying an exhaustiveness option of 20 (average accuracy). For comparison, the docking was also performed for colchicine. Since the standard error of Autodock Vina is 2.85 kcal mol−1 [24], binding affinities cannot be predicted quantitatively. This problem is not limited to Vina software [26]. Figures were prepared with the program PYMOL [27].
2.3 Cell cultures
BxPC-3 pancreas carcinoma and MDA-MB-231 breast carcinoma-derived cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained in DMEM (Dulbecco’s Modified Eagle Medium; Life technologies, Carlsbad, CA, USA) supplemented with 10 % FBS, 100 U/ml penicillin and 100 μg/ml streptomycin in a 5 % CO2 atmosphere at 37 °C. The endothelial hybrid cell line Ea.hy926 (ATCC no. CRL-2922), derived from HUVEC (human umbilical vein endothelial cells), was cultured in DMEM containing 10 % FBS, 100 U/ml penicillin G, 100 μg/ml streptomycin, 0.25 μg/ml amphothericin B and 200 μg/ml gentamycine (all from Life Technologies).
2.4 Fluorescent staining assays
Fluorescent staining of microtubules and microfilaments was carried out essentially as reported before [28]. Ea.hy926 cells (100,000 cells/well) were grown on glass coverslips in 24-well plates for 24 h. Next, HA-1077 (10 μM) or SP600125 (20 μM) were added 2 h before incubation with 100 nM brimamin for an additional 12 h. After this, the cells were fixed in 4 % formaldehyde in PBS (20 min, room temperature, RT) and permeabilized (1 % BSA, 0.1 % Triton X-100 in PBS, 30 min, RT). For microtubule staining, coverslips were incubated with a primary antibody directed against α-tubulin (anti-alpha-tubulin, mouse mAb, Invitrogen; 5 μg/ml, 1 h at 37 °C in a humidified atmosphere) followed by incubation with a secondary antibody conjugated to DyLight 550 dye (goat anti-mouse IgG-DyLight 550 conjugate, Pierce/Thermo Scientific; 2.5 μg/ml) for 1 h at RT in the dark. For visualization of filamentous actin (F-actin), fixed and permeabilized cells were incubated with AlexaFluor 488-conjugated phalloidin (Cell Signaling Technologies; 1 h at 37 °C in a humidified atmosphere). All coverslips were mounted in Mowiol 4-88-based mounting medium containing 2.5 % (w/v) DABCO and 1 μg/ml DAPI (4′,6-diamidino-2-phenylindole; all from Carl Roth) for counterstaining the nuclei. Fluorescence microscopy analyses were performed using an Axio Imager. A1 fluorescence microscope (40-fold magnification, ZEISS).
2.5 Tube formation assays
Tube formation assays were carried out essentially as reported before [29]. Glass coverslips (12 mm diameter) were coated with 10 μl of Matrigel™ basement membrane matrix (BD Biosciences; gelation for 30 min in a humidified atmosphere at 37 °C) and placed in 24-well plates. Next, Ea.hy926 cells were seeded (100,000 cells/well in serum-free DMEM) on the resulting thin Matrigel layers where they start to differentiate into blood vessel precursor-like networks through growth factor stimulation within the next 12 h. Rho kinase inhibitor HA-1077 (10 μM), PI3K inhibitor LY294002 (10 μM), or MAPK inhibitors SP600125 (JNK inhibitor, 20 μM) or PD98059 (ERK1/2 inhibitor, 10 μM) were added for 2 h followed by replacing the cell culture media with fresh DMEM containing final concentrations of 50 nM CA-4 or 100 nM brimamin. Finally, the cells were incubated for another 24 h at 37 °C and the effect of the different compounds on tube formation was documented through microscopy (100-fold magnification, Axiovert 135, ZEISS).
2.6 MTT growth assay
BxPC-3 cells (3,000 cells/well) and MDA-MB-231 cells (3,000 cells/well) were seeded in 96-well microplates. After overnight incubation, the medium was replaced with fresh medium containing DMSO (vehicle control) or different concentrations of brimamin diluted from a 10 mM stock. After 72 h of incubation, 25 μl 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (0.5 mg/ml in PBS) was added to each well to a final concentration of 0.05 mg/ml and incubated for another 2 h at 37 °C. The supernatants were aspirated and the formazan, formed by metabolically viable cells, was dissolved by adding isopropanol (100 μl). Finally, the plates were agitated on a gyratory shaker for 30 min and the absorbance at 595 nm was measured on an Ultra Multifunctional Microplate Reader (TECAN, Durham, NC). Each experiment comprised eight replicate wells and the amount of DMSO in the reaction mixtures never exceeded 1 %. Ea.hy926 cells were cultured and treated identically with minor alterations. Briefly, cells (10,000 cells/well) were cultured in 96-well microplates for 24 h and pre-treated with non-toxic concentrations of commercially available kinase inhibitors for 2 h: 10 μM HA-1077, 10 μM LY294002, 5 μM PD98059, 10 μM SB202190, or 20 μM SP600125 [12, 30]. Next, the cells were treated with CA-4 (10 nM) or brimamin (50 or 100 nM) for an additional 48 h before adding MTT to a final concentration of 0.05 mg/ml. After a 2 h incubation, cell lysis and formazan dissolution were achieved by adding 30 μl SDS-DMSO solution (10 % SDS, 0.6 % acetic acid in DMSO), and the formazan absorbance was measured at 570 and 630 nm (background) using a microplate reader (TECAN). Cell viability was calculated from the resulting absorbance values relative to DMSO controls set to 100 %.
2.7 Flow cytometry
For the quantification of early apoptosis, Ea.hy926 cells grown in 6-well plates (200,000 cells/well) were treated with DMSO (control), brimamin alone (50 nM, 24 h), or combinations of brimamin (50 nM) and various kinase inhibitors (10 μM HA-1077, 10 μM LY294002, 5 μM PD98059, 10 μM SB202190, 20 μM SP600125; 2 h pre-treatment in each case) and prepared for transferase-mediated dUTP-fluorescein nick end labeling (TUNEL) of DNA fragments according to the manufacturer’s protocol (FragEL DNA fragmentation detection kit, Calbiochem). Briefly, after TUNEL staining of fixed cells (4 % formaldehyde, 20 min, RT), 5,000 cells were analyzed for fluorescence intensity (530 nm emission wavelength, CXP software, Beckman Coulter). Gates defining the amounts of viable or apoptotic cells (%) were set relative to untreated (DMSO) controls. The increase of apoptotic fractions relates to controls with kinase inhibitor pre-treatment (<5 % apoptosis by inhibitor treatment; cf Fig. S1-2, Electronic supplementary material for original histograms).
2.8 Western blotting
For the detection of cellular NF-κB levels in Ea.hy926 cells the latter were grown in 24-well plates (50,000 cells/well), treated with CA-4 (10 nM, 50 nM) or brimamin (50 nM, 100 nM) for 2 h or 24 h, and harvested by trypsinisation. Cell lysates and nuclear protein fractions were prepared as previously described [14, 31]. In brief, cells were lysed in lysis buffer (20 mM Tris–HCl, 1 mM MgCl2, 2 mM EGTA, 0.5 % Triton X-100, pH 6.8) supplemented with a protease inhibitor solution (protease inhibitor cocktail III, Calbiochem) for 10 min on ice followed by centrifugation (800 g, 5 min, 4 °C) to obtain total cell lysates. For the preparation of cell nuclei, cell membranes were lysed in a mild nuclear extraction buffer (20 mM HEPES-KOH, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1.5 mM MgCl2, 1 mM DTT, pH 7.4) supplemented with a protease inhibitor solution for 20 min on ice. After addition of Triton X-100 to a final concentration of 0.1 % (2 min on ice) and centrifugation (3,000 g, 2 min), pelleted nuclei were lysed in 30 μl nuclear extraction buffer with 10 % glycerol and 0.1 % SDS. For Western blot analyses of NF-κB levels in vivo, cells of xenograft tissue samples were lysed in RIPA buffer (20 mM Tris–HCl, 137 mM NaCl, 1 % NP-40, 2 mM EDTA, 0.5 % sodium deoxycholate and 0.1 % SDS) containing a complete mini EDTA-free protease inhibitor cocktail (Roche, Indianapolis, IN, USA). The total protein concentration of each sample was measured using a BCA Protein Assay (Pierce, Rockford, IL, USA). 10 μg samples of total protein were separated through 12 % SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose or PVDF membranes. Primary antibodies (rabbit anti-p65 monoclonal antibody, Cell Signaling Technology; mouse anti-beta-actin monoclonal antibody, Sigma-Aldrich as a loading control) were added in appropriate concentrations followed by incubation with HRP-conjugated secondary antibodies. Finally, bands were visualized and documented by chemiluminescence (for original Western blot images, cf Fig. S4-S6, Electronic supplementary material).
2.9 ELISA apoptosis assay
The Cell Death Detection Kit (Roche, Palo Alto, CA, USA) was used to assess apoptosis in BxPC-3 and MDA-MB-231 cells according to the manufacturer’s instructions [14]. Briefly, cells were treated with test compounds for 72 h. After harvesting the cells by trypsinisation, lysates were prepared and separated into pellet and supernatant fractions by centrifugation (20,000 g, 10 min, RT). The supernatants represent the cytoplasmic fractions containing histone-associated DNA fragments (nucleosomes) that were subsequently detected by ELISA. The absorbance of the peroxidase-converted ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid)) reaction product was determined using an Ultra Multifunctional Microplate Reader (TECAN) at 405 nm.
2.10 Electrophoretic mobility shift assay
To evaluate the effect of brimamin on the DNA-binding ability of nuclear NF-κB in BxPC-3 and MDA-MB-231 cells, these cells were treated with vehicle or brimamin (10–400 nM) for 72 h. Next, nuclear cell lysates were prepared (see 2.8) and an electrophoretic mobility shift assay (EMSA) was carried out by incubation of 8 μg nuclear protein extract with IRDye-700-labeled oligonucleotides containing the NF-κB binding sequence (LI-COR, Lincoln, NE) and 2 μg of poly desoxyinosinic-desoxycytidylic acid (poly dI-dC, background reduction) oligonucleotides as described before [14, 32]. DNA-protein complexes that were formed during incubation were separated from free oligonucleotides by 8 % native PAGE. Finally, the gels were scanned using an Odyssey Infrared Imaging System (LI-COR, Inc.) and the detected bands were analyzed by densitometry using the ImageJ software package. Equal protein loading was ensured by immunoblotting of 10 μg of nuclear protein and probing with an anti-retinoblastoma (Rb) antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
2.11 In vivo studies
For xenografting, female homozygous ICR SCID mice, aged 4 weeks, were used. The experimental protocol employed was approved by the Committee on the Ethics of Animal Experiments of Wayne State University Institutional Users of Animal Care Committee. To initiate the xenografts, 5 × 106 MDA-MB-231 cells (in serum-free medium) were injected sub-cutaneously bilaterally in the flanks of the mice. The animals were examined thrice per week until they developed palpable tumors. Next, the animals were randomly divided into two groups of 6 animals each. Group I was assigned as control and received only sesame seed oil without brimamin, whereas group II mice were administered brimamin. Brimamin treatment was started once the tumors were detectable, and was administered intra-peritoneally at a dose of 20 mg/kg on day 1, day 9, day 14, day 20 and day 27. On day 30, when the tumors in the control group weighed ~2 g on average, all animals were sacrificed. The volumes of the tumors in either group were determined with a calliper every 4 days according to the formula ab2/2, wherein ‘a’ is the length and ‘b’ the cross-sectional diameter. Tumors were harvested from each animal and processed for molecular and histological analyses.
2.12 Statistical analyses
The experimental results represent three or more independent observations. The data are presented as mean values ± SD. Statistical comparisons between the groups were performed using one-way ANOVA. P-values < 0.05 were considered statistically significant. Individual p-values are reported, as appropriate. ImageJ software was used for densitometric analyses of the original images.
3 Results
3.1 Docking of the brimamin-tubulin interaction
The anticancer and vascular-disrupting effects of brimamin are likely related to its inhibitory effect on the polymerization of tubulin, as we reported previously [10]. For a deeper insight into its molecular interaction with tubulin we performed docking studies. The crystal structure geometry of dimeric bovine tubulin (PDB accession code 1SA0, 100 % sequence identity to human tubulin), with ligated colchicine and Mg2+-complexed GDP and GTP, was used as a starting point for docking brimamin in place of colchicine [33]. The stathmin-like domain (SLD) of RB3 was deleted for docking. These docking studies, performed with the Autodock Vina software package, predicted brimamin to bind in an orientation analogous to colchicine with the A-ring proximal to Cys241 (Fig. 2a; colchicine not shown). Although pointing towards the SH-group of Cys241, the 5-methoxy oxygen of ring A is probably too far off to establish a stable hydrogen bond. In contrast, the nitrogen atom N-1 of the imidazole ring is ideally positioned for entering into a hydrogen bond with the backbone amide hydrogen of Ala250. The NH2 group of the second phenyl ring B then locks the ligand by acting as a H-bridge donor over to the backbone carbonyl oxygen of Thr179. Unlike natural combretastatin analogs [34], the side chain ε-amino group of Lys352 points away from the B-ring and cannot establish any supporting interaction. An additional H-bond was calculated as plausible between the p-methoxy oxygen of ring B and the backbone amido NH of Val181. This p-methoxy group is also entangled in a Van-der-Waals interaction with a methyl group of the same Val181. In a similar manner the methoxy groups of the A ring are involved in attractive Van-der-Waals interactions with their neighbouring amino acid residues. Due to the conserved pattern of methoxy substituents on ring A this hydrophobic interaction network is very similar for brimamin and colchicine (docking results not shown). As a result, the calculated energy for the binding of brimamin to tubulin is ca. 80 % of that for colchicine (Table 1).

a Proposed binding mode of brimamin in the colchicine binding pocket within the β-tubulin subunit. Schematic representations of hydrogen bonds (dashed lines) and Van-der-Waals interactions (hatched lines) of brimamin with bovine tubulin with carbon colored orange, hydrogen white, nitrogen blue, oxygen red, bromine purple. b Immunostaining of microtubules in Ea.hy926 endothelial cells treated with brimamin (100 nM) for 12 h; α-tubulin stained in red and nuclei counterstained in blue with DAPI (merge). Scale bar: 100 μm (400-fold magnification)
3.2 Effects of brimamin on cytoskeletal reorganization in endothelial cells
Next we focused on the cytoskeletal reorganization that occurs in endothelial cells upon brimamin treatment and which eventually leads to the observed disruption of the tumor vasculature. Since CA-4P is known to cause early membrane blebbing in endothelial cells [12], we also assessed the morphological changes of microtubules and actin filaments of endothelial Ea.hy926 cells treated with brimamin. We found that exposure to 100 nM brimamin for 12 h led to an extensive disruption of the microtubular network followed by actin stress fiber formation (Figs. 2b and 3). This reorganization of the actin filaments in the wake of microtubule de-polymerization could be prevented by pre-treating the cells with the Rho kinase inhibitor HA-1077 prior to the addition of brimamin (Fig. 3). Apparently, brimamin induces an actin cytoskeleton response through activation of RhoA and Rho kinase as has previously been shown for CA-4P [12, 35].

Fluorescent staining of microfilaments in Ea.hy926 endothelial cells. Cells were treated either with brimamin alone (100 nM) or a combination of brimamin and Rho kinase inhibitor HA-1077 (10 μM) for 12 h. Filamentous actin (F-Actin) was visualized by staining with a fluorescent phalloidin conjugate (merge: green) and nuclei were counterstained with DAPI (merge: blue). Scale bar: 100 μm (400-fold magnification). Typical bundles of actin stress fibers are indicated by arrows
The loss of microtubule and actin cytoskeleton integrity initiated by brimamin translates into macroscopically observable vascular-disruptive effects, which were assessed at various stages of endothelial cell aggregation. In so-called tube formation assays Ea.hy926 cells were grown on Matrigel and allowed to form tubular structures mimicking blood vessels. When treated with brimamin, individual cells showed an increased contractility and looser cell-cell contacts, eventually leading to a breakup of the organized conduit system (Fig. 4).

Fluorescent tube formation assay in Ea.hy926 endothelial cells. Cells were grown on thin layers of basement membrane matrix (Matrigel) for 12 h and then treated with vehicle (DMSO, left column) or 100 nM brimamin (right column) for an additional 12 h followed by formaldehyde fixation and fluorescence staining of F-actin (phalloidin-AlexaFluor-488 conjugate). Nuclei were counterstained with DAPI (blue). a Overlay of actin (green) and DNA (blue) staining in 100-fold magnification (scale bar: 200 μm). b Fluorescent F-actin (gray) and overlay of F-actin with nuclear staining (merge) in 400-fold magnification (scale bar: 100 μm)
3.3 Contribution of kinase signaling to the cytotoxicity of brimamin
Certain protein kinase signaling pathways have been reported to play a crucial role in the survival of endothelial cells as well as in endothelial cell differentiation into blood vessel precursors. Therefore, we investigated the influence of the kinase inhibitors LY294002 (PI3K), PD98059 (ERK1/2), SB202190 (p38 MAPK) and SP600125 (JNK1/2) on the cytotoxicity of brimamin against Ea.hy926 cells and on its ability to induce apoptosis in these cells, compared to the lead compound CA-4. HA-1077, an inhibitor of Rho-associated kinase, is less selective and addresses some other kinases, too.
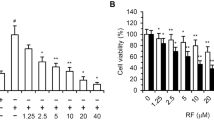
MTT assays were performed by co-incubation of Ea.hy296 cells with non-toxic concentrations of the individual kinase inhibitors and slightly toxic concentrations of either brimamin or CA-4. The addition of a PI3K or ERK inhibitor (i.e., LY294002 or PD98059, respectively) strongly sensitized these endothelial cells to brimamin and also CA-4, while combinations with either a JNK or SAPK inhibitor (i.e., HA-1077 or SP600125, respectively) led to a decreased cytotoxicity (Fig. 5a). The p38-MAPK inhibitor SB202190 did not affect the activity of brimamin or CA-4. These results were substantiated for brimamin by the measurement of late apoptosis induction in the endothelial cells after incubation with the respective inhibitors (Fig. 5b).

a Effects of kinase inhibitors on the viability of Ea.hy926 endothelial cells treated with CA-4 or brimamin. Cells were treated with CA-4 (10 nM) or brimamin (100 nM, 50 nM) for 48 h with or without pre-treatment with the kinase inhibitors HA-1077 (Rho-associated kinase), LY294002 (PI3K), PD98059 (ERK1/2), SB202190 (p38 MAPK) or SP600125 (JNK1/2). Cell viability scores (%) were obtained from three independent MTT assays, and error bars represent the standard deviation. b Effects of protein kinase inhibitors on the induction of apoptosis in Ea.hy926 endothelial cells by brimamin. Confluent monolayers of cells were treated with brimamin for 24 h with or without pre-treatment with the kinase inhibitors HA-1077, LY294002, PD98059, SB202190 or SP600125. Induction of apoptosis was assessed by TUNEL assays. Results represent the mean of two independent experiments, and error bars represent the standard deviation
We found that treatment with brimamin alone (50 nM) led to an increase in the cellular fraction with DNA strand breaks of about 10–15 %. Co-incubation with the PI3K inhibitor LY294002 and the ERK inhibitor PD98059 strongly enhanced DNA fragmentation. These results are consistent with our results obtained from the MTT assays. Since both PI3K and ERK are known to protect cells from triggering apoptosis as a consequence of a cellular stress response, inhibition of these two kinases might lead to an enhanced predisposition to microtubule disruption [36]. In contrast to the MTT results, we found that addition of HA-1077 sensitized the Ea.hy926 cells, resulting in a two-fold increase in apoptotic events. The attenuation of the growth inhibitory effect of brimamin by HA-1077, as observed in the MTT assays, might be a consequence of a retardation in cell cycle progression induced by this kinase inhibitor. This notion is supported by a slight increase in G1-phase cells observed by cell cycle analyses performed after 48 h (data not shown). At this early stage of brimamin treatment, Rho kinase inhibition may positively affect cell-protecting signaling pathways. After combination with the JNK inhibitor SP600125, we observed slightly decreased apoptotic rates consistent with the protective effect of SP600125 in the MTT assays. The activation of JNK by tubulin-binding agents such as vinblastine through prolonged G2-M arrest can eventually lead to apoptosis via Bcl-2 multi-site phosphorylation, as has been reported before [37]. Accordingly, inhibition of JNK by SP600125 may diminish apoptosis induction because of insufficient Bcl-2 degradation. We also found that inhibition of JNK can induce microtubule stabilization to a degree that overrides the contrary effect of brimamin (data not shown). So, active JNK signaling seems to be crucial for apoptosis induction after cellular sensing of cytoskeletal damages induced by brimamin.
3.4 Contribution of kinase signaling to the vascular-disrupting effect of brimamin
Next, we applied a tube formation assay to further investigate the influence of various kinases on endothelial cell differentiation. Ea.hy926 cells grown on thin Matrigel layers form tubular networks due to growth factor stimulation and adhesion to the basement membrane matrix. Within these networks the individual cells undergo massive morphological changes (Figs. 2, 3, 4 and 6). We found that brimamin, like the lead compound CA-4, induced cytoskeletal disorganization and morphological changes that impaired both the maintenance and the additional formation of tubular branches. Similar results were obtained when pre-formed tubular networks of Ea.hy926 cells were treated with a combination of brimamin or CA-4, and the Rho kinase inhibitor HA-1077. This inhibitor did not lead to the preservation of the tubular network, at least not at the non-toxic concentrations that we applied (Fig. 6, second row from top). Rho signaling and Rho-mediated actin stress fiber formation and subsequent actin-myosin contractility were, however, identified by other groups as crucial for the activity of a variety of other VDAs [38]. In contrast, we found that pre-incubation with the JNK inhibitor SP600125 led to a marked stabilization of cellular connections and a preservation of pre-formed tubes from disruption or increased cell contractility induced by brimamin or CA-4 (Fig. 6, bottom row).

Tube formation assays with Ea.hy926 endothelial cells. Cells were grown on thin layers of basement membrane matrix (Matrigel) for 12 h and then treated with vehicle (DMSO), CA-4 (50 nM) or brimamin (100 nM) alone or in combination with 10 μM HA-1077, 10 μM LY294002, 5 μM PD98059 or 20 μM SP600125 for an additional 12 h. The effects were monitored by light microscopy (100-fold magnification, scale bar: 200 μm)
Co-treatment with the ERK1/2 inhibitor PD98059 or the PI3K inhibitor LY294002 was found to sensitize Ea.hy926 cells for the vascular-disrupting effects of CA-4 or brimamin, and was found to lead to a complete disintegration of tubular aggregates. Taken together, our data indicate that kinase inhibitors that increase CA-4 or brimamin cytotoxicity in 2D cultures also interfere with endothelial cell network formation on membrane matrices. ERK and PI3K/Akt are known to promote pro-survival pathways and, therefore, may protect both proliferating and differentiating cells from apoptosis induced by microtubule damage. Rho and JNK signaling, on the other hand, are crucial for both CA-4 and brimamin cytotoxicity and their vascular-disrupting activity. In Table 2 the benefits and potential synergisms of combinations of brimamin and selective kinase inhibitors are summarized.
3.5 Inhibition of nuclear translocation of NF-κB by CA-4 and brimamin
The dual role of NF-κB signaling in tumor progression and angiogenesis has been extensively documented. Although down-regulation of NF-κB in tumor cells in vitro and in vivo results in a reduced expression of NF-κB-related, anti-apoptotic target genes and a better clinical outcome, its activation seems to be essential for the activity of anti-angiogenic agents [47, 48]. NF-κB activation has even a negative effect on tumor angiogenesis and affects pro-apoptotic pathways in endothelial cells. Here, we assessed the role of endothelial NF-κB signaling in the anti-vascular activity of brimamin. NF-κB activation is associated with its translocation from cytoplasmic complexes into the nucleus, where it acts as a transcription factor for various target genes. We found that a short incubation (~2 h) with either CA-4 or brimamin led to slightly increased nuclear NF-κB p65 subunit levels in the treated cells, which is in line with previously reported findings in HUVEC cells [47, 48] (Fig. 7a). However, after a 24 h incubation period, the nuclear p65 subunit level had dropped significantly. We hypothesize that this is a consequence of a progressing disruption of the microtubular cytoskeleton along which the translocation of proteins into the nucleus occurs. An effect on the total NF-κB level, and thus on its expression or degradation, was not detected at any point in time.

a Time-dependent nuclear NF-κB translocation in Ea.hy926 endothelial cells after treatment with CA-4 (10, 50 nM) or brimamin (50, 100 nM) for 2 or 24 h. NF-κB levels in total cell lysates and nuclear protein fractions are visualized by immunoblotting of the NF-κB p65 subunit. b NF-κB DNA-binding activity in nuclear protein extracts of MDA-MB-231 breast and BxPC-3 pancreas carcinoma-derived cells after treatment (72 h) with various concentrations of brimamin. Electrophoretic mobility shift assay (EMSA) revealing nuclear NF-κB-DNA complexes, visualized by labeled oligonucleotide probes
3.6 Growth inhibition and apoptosis induction in pancreas and breast carcinoma cells
In light of a potential druggability of brimamin, we set out to evaluate its cytotoxicity in highly resistant cancer-derived cell lines that depend on activated NF-κB signaling [41]. First, we tested brimamin by MTT assays and found this compound to be highly active in BxPC-3 pancreatic carcinoma (IC50 = 25 ± 2 nM) and triple-negative MDA-MB-231 breast carcinoma (IC50 = 350 ± 19 nM) derived cells upon a 72 h incubation. For comparison, the IC50 concentrations of CA-4 have previously been found to be 43 nM (MDA-MB-231) and 57 μM (BxPC-3), respectively [49, 50]. Next, the induction of apoptosis in BxPC-3 and MDA-MB-231 cells by brimamin was evaluated using histone/DNA ELISA assays (Fig. S3, Electronic supplementary material). By doing so, we found a strong effect at low nanomolar concentrations. Its magnitude correlated fairly well with the IC50 values obtained from the MTT tests.
3.7 NF-κB inactivation in pancreas and breast carcinoma cells
We found that brimamin significantly inhibited the in vitro DNA-binding of NF-κB at nanomolar concentrations in BxPC-3 pancreas carcinoma and MDA-MB-231 breast carcinoma-derived cells (Fig. 7b). As a consequence, brimamin may lead to a reduced activation of NF-κB target genes. The impact of brimamin on NF-κB was particularly strong in the BxPC-3 cells, which are known to respond to treatment with the approved anticancer drug gemcitabin by enhanced NF-κB activation [15]. Hence, a combination of brimamin with gemcitabin, which is clinically applied for pancreas cancer, appears to be a promising option.
3.8 In vivo effects of brimamin on breast carcinoma growth and NF-κB expression
The observed in vitro anticancer activity of brimamin was subsequently studied in vivo in an MDA-MB-231 breast cancer xenograft mouse model. When applied at doses of 20 mg/kg over a period of 4 weeks, the compound was well-tolerated and found to reduce the rate of tumor growth (Fig. 8a). Residual tumor cells of the so-treated xenografts were processed further and subjected to protein expression analysis by Western blotting. By doing so, we observed a down-regulation of the NF-κB p65 subunit (Fig. 8b). These results are in support of our in vitro observations described above, suggesting an effective inhibition of NF-κB signaling by brimamin. This inhibition, in turn, is likely to be causative for the observed biological effects in the carcinoma-derived cells tested. Through immunostaining of tissue sections of untreated control xenografts (C in Fig. 8c) and treated xenografts (B in Fig. 8c) for the endothelial cell and angiogenesis marker CD31, the strong in vivo vascular-disrupting effect of brimamin was visualized. Figure 8c also shows that brimamin gives rise to large necrotic areas within the tumors, presumably as a consequence of its high cytotoxicity and a compromised vascularization.

Brimamin retards the growth of MDA-MB-231 xenografts by inhibition of NF-κB expression. a MDA-MB-231 cells were injected subcutaneously in SCID mice (bilaterally) after which they were randomized into two groups (6 mice each): control and brimamin-treated. Tumor weight was evaluated upon application of brimamin on days 1, 9, 14, 20 and 27. b Expression of NF-κB subunit p65 in tissues of two representative tumors each from control and treated mice. β-actin, loading control; C, control; B, brimamin treated. The figures underneath the p65 blot represent densitometric values relative to the control band set to 1.00 and normalized for the respective β-actin bands. c Immunohistochemical staining for CD31 of the untreated control tumor (C) reveals many intact blood vessels (red arrows) as compared to an occasional intact blood vessel in the tumor treated with brimamin (B). Tissue section B also shows large areas of necrosis (double-headed arrows). Magnification: 40-fold
4 Discussion
Docking studies allowed us to identify H-bonds and Van-der-Waals interactions by which the new vascular-disrupting agent (VDA) brimamin may be anchored to amino acid residues in the colchicine binding site of tubulin. Although these interactions differ from those of combretastatins [34], the A- and B-ring motifs of brimamin are arranged in the same orientation. We found that, like CA-4, brimamin leads to microtubule disruption in endothelial cells. The ensuing formation of actin stress fibers was found to be dependent on Rho kinase activation, resulting in early membrane blebbing as has been reported for CA-4 [12]. This extensive cytoskeletal reorganization may also be responsible for the observed vascular-disrupting effect of brimamin. We found that the in vitro tube formation by endothelial cells was strongly impaired upon brimamin treatment. This impairment may be a consequence of cell retraction caused by dense and contractile actin stress fibers and by loss of integrity of the microtubule cytoskeleton [51].
Upon a closer inspection of the molecular mechanism underlying this effect, we found that functional JNK is crucial for the cytotoxic as well as the vascular-disrupting effects of CA-4 and brimamin, which is in keeping with reports on JNK dependence of other tubulin-binding agents [37, 52, 53]. When we added low concentrations of the JNK inhibitor SP600125 to Ea.hy926 endothelial cells in the presence of brimamin we observed a stabilization of the microtubules, i.e., an attenuation of the vascular-disrupting effect of brimamin. We presume that the inhibitor SP600125 interferes with the regulatory function of JNK in stathmin phosphorylation, which is a key event in interphase microtubule stabilization [54–56]. JNK seems to play another crucial role in the effect of tubulin binders. It has been reported that sustained JNK activation by prolonged G2-M cell cycle arrest, induced by drugs such as vinblastine, is associated with Bcl-2 multi-site phosphorylation, which eventually results in apoptosis induction [37, 45]. We found that co-treatment of endothelial cells with SP600125 led to a reduced brimamin cytotoxicity. Therefore, we conclude that active JNK is involved in apoptosis induction and, thus, is essential for the anticancer effects of both CA-4 and brimamin. Although SP600125 has been reported to re-sensitize doxorubicin-resistant cancer cells to chemotherapy [57], a therapeutic combination of this JNK inhibitor with vascular disrupting agents does not appear to be advisable based on our results with endothelial cells.
On the other hand, there is evidence indicating that ERK and PI3K signaling triggered by brimamin treatment may be responsible for the induction of pro-survival and anti-apoptotic pathways in endothelial cells. We found that combinations with specific ERK and PI3K inhibitors sensitized endothelial cells to brimamin-induced cytotoxicity and its anti-vascular activity in vitro. We also found indications for an involvement of anti-apoptotic NF-κB in the action of brimamin. In the past, NF-κB signaling has been implicated in tumor progression and angiogenesis. We found that in endothelial cells NF-κB is activated early upon brimamin and CA-4 treatment, as exemplified by the nuclear translocation of its cytosolic p65 subunit. However, after longer incubation periods, the nuclear level of the NF-κB p65 subunit dropped significantly. We hypothesize that this may be due to extensive cytoskeletal reorganization or microtubule disruption. It has been shown before that nuclear translocation of NF-κB in neuroblastoma cells depends on a functional cytoskeleton [19]. The effect of brimamin on the activity of NF-κB was also evaluated in pancreas carcinoma (BxPC-3) and triple-negative breast carcinoma (MDA-MB-231) derived cells. By doing so, we confirmed the connection between NF-κB down-regulation and microtubule disruption in these NF-κB expressing, clinically important cancer entities. We found that brimamin reduced the affinity of NF-κB for DNA in both cancer cell lines, and blocked the expression of p65 in cells of brimamin-treated MDA-MB-231 xenografts. Histologically, brimamin gave rise to an extensive necrosis and a disruption of blood vessels in these tumor xenografts. Since an activation and constitutive high expression of NF-κB has previously been observed by us [14] and others [58] in various resistant cancer types, including triple-negative breast carcinoma, brimamin may serve as a potential drug candidate for the treatment of these prognostically unfavorable cancers.
Others have reported that inhibition of tubulin polymerization by vinblastine or colchicine can block the formation of the tubulin-dynein-karyopherin α-p50 complex required for nuclear translocation of NF-κB in neuronal cells. It also led to a low nuclear NF-κB DNA-binding activity, to low nuclear levels of p65 and p50, and to a decreased transcription of NF-κB target genes [19]. In addition, NF-κB activation has been observed upon short incubations with vinblastine, colchicine or nocodazole [59], which is thought to be decisive for the inhibition of tumor angiogenesis in vivo [47, 48]. These observations point at paradoxical roles of NF-κB in proliferation and apoptosis, depending on the cell type and/or tissue context. It is not surprising that there is cross-talk between the NF-κB and Rho signaling pathways, as has been observed in inflammation processes, and that the down-regulation of Rho can cause an inactivation of NF-κB in particular cell types, including endothelial cells [16–18]. Proteins encoded by the NF-κB target genes may also inactivate JNK signaling [60]. Taken together, the negative effects observed of brimamin on NF-κB signaling, including reduced DNA affinity, lower nuclear translocation in vitro and reduced p65 expression in vivo, may indicate an improved efficacy against certain types of cancer. Considering the clinical relevance, it should be mentioned that pancreatic cancer patients are routinely treated with the nucleoside gemcitabin, which might be combined with brimamin in a synergistic regimen, since NF-κB signaling is strongly activated in gemcitabin-resistant pancreas carcinoma cells [41]. Aggarwal et al. reported, for example, growth inhibitory effects of the natural compound curcumin on pancreas carcinoma cells resulting from NF-κB down-regulation [61]. A combination of brimamin with proteasome inhibitors that block NF-κB by an alternative pathway, i.e., by inhibition of the degradation of the cellular NF-κB inhibitor IκB, would allow a further optimization of the anticancer efficacy and the dose schedule in clinical trials. A combination of these two drugs might lead to an improved outcome in patients suffering from pancreatic cancer. A Korean study from 2010 supports this assumption, i.e., it showed that microtubule disruption and NF-κB inactivation sensitized HeLa cells for apoptosis induced by DNA damage [59]. The influence of JNK and Rho signaling on the effect of brimamin in NF-κB-dependent cancer cells remains to be elucidated in more detail. However, the in vitro and in vivo efficacy of brimamin against multidrug-resistant cancer cells, its applicability [10], and its ability to inhibit tumor pro-survival pathways, turn this compound into a promising anticancer drug candidate.
References
G. Pettit, S. Singh, E. Hamel, C. Lin, D. Alberts, D. Garcia-Kendall, Isolation and structure of the strong cell growth and tubulin inhibitor combretastatin A-4. Experientia 45, 209–211 (1989)
C.J. Mooney, G. Nagaiah, P. Fu, J.K. Wasman, M.M. Cooney, P.S. Savvides, J.A. Bokar, A. Dowlati, D. Wang, S.S. Agarwala, A phase II trial of fosbretabulin in advanced anaplastic thyroid carcinoma and correlation of baseline serum-soluble intracellular adhesion molecule-1 with outcome. Thyroid 19, 233–240 (2009)
G.J. Rustin, G. Shreeves, P.D. Nathan, A. Gaya, T.S. Ganesan, D. Wang, J. Boxall, L. Poupard, D.J. Chaplin, M.R.L. Stratford, J. Balkissoon, M. Zweifel, A Phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer. Br. J. Cancer 102, 1355–1360 (2010)
G.C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca, A.A. Genazzani, Medicinal chemistry of combretastatin A4: present and future directions. J. Med. Chem. 49, 3033–3044 (2006)
S.E. Holwell, P.A. Cooper, M.J. Thompson, G.R. Pettit, L.W. Lippert 3rd, S.W. Martin, M.C. Bibby, Anti-tumor and anti-vascular effects of the novel tubulin-binding agent combretastatin A-1 phosphate. Anticancer Res. 22, 3933–3940 (2002)
L.K. Folkes, M. Christlieb, E. Madej, M.R.L. Stratford, P. Wardman, Oxidative metabolism of combretastatin A-1 produces quinone intermediates with the potential to bind to nucleophiles and to enhance oxidative stress via free radicals. Chem. Res. Toxicol. 20, 1885–1894 (2007)
G.R. Pettit, M.R. Rhodes, D.L. Herald, D.J. Chaplin, M.R. Stratford, E. Hamel, R.K. Pettit, J.C. Chapuis, D. Oliva, Antineoplastic agents 393. Synthesis of the trans-isomer of combretastatin A-4 prodrug. Anticancer Drug Des. 13, 981–993 (1998)
G.R. Pettit, B.E. Toki, D.L. Herald, M.R. Boyd, E. Hamel, R.K. Pettit, J.C. Chapuis, Antineoplastic agents. 410. Asymmetric hydroxylation of trans-combretastatin A-4. J. Med. Chem. 42, 1459–1465 (1999)
L. Wang, K.W. Woods, Q. Li, K.J. Barr, R.W. McCroskey, S.M. Hannick, L. Gherke, R.B. Credo, Y.-H. Hui, K. Marsh, R. Warner, J.Y. Lee, N. Zielinski-Mozng, D. Frost, S.H. Rosenberg, H.L. Sham, Potent, orally active heterocycle-based combretastatin A-4 analogues: synthesis, structure − activity relationship, pharmacokinetics, and in vivo antitumor activity evaluation. J. Med. Chem. 45, 1697–1711 (2002)
R. Schobert, B. Biersack, A. Dietrich, K. Effenberger, S. Knauer, T. Mueller, 4-(3-Halo/amino-4,5-dimethoxyphenyl)-5-aryloxazoles and -N-methylimidazoles that are cytotoxic against combretastatin A resistant tumor cells and vascular disrupting in a cisplatin resistant germ cell tumor model. J. Med. Chem. 53, 6595–6602 (2010)
K. Mahal, B. Biersack, H. Caysa, R. Schobert, T. Mueller, Combretastatin A-4 derived imidazoles show cytotoxic, antivascular, and antimetastatic effects based on cytoskeletal reorganisation. Investig. New Drugs 33, 541–554 (2015)
C. Kanthou, The tumor vascular targeting agent combretastatin A-4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood 99, 2060–2069 (2002)
B.B. Aggarwal, S. Shishodia, S.K. Sandur, M.K. Pandey, G. Sethi, Inflammation and cancer: how hot is the link? Biochem. Pharmacol. 72, 1605–1621 (2006)
A. Ahmad, S. Banerjee, Z. Wang, D. Kong, F.H. Sarkar, Plumbagin-induced apoptosis of human breast cancer cells is mediated by inactivation of NF-κB and Bcl-2. J. Cell. Biochem. 105, 1461–1471 (2008)
S. Ali, A. Ahmad, S. Banerjee, S. Padhye, K. Dominiak, J.M. Schaffert, Z. Wang, P.A. Philip, F.H. Sarkar, Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 70, 3606–3617 (2010)
Y. He, H. Xu, L. Liang, Z. Zhan, X. Yang, X. Yu, Y. Ye, L. Sun, Antiinflammatory effect of Rho kinase blockade via inhibition of NF-kappaB activation in rheumatoid arthritis. Arthritis Rheum. 58, 3366–3376 (2008)
H. Shimada, L.E. Rajagopalan, Rho kinase-2 activation in human endothelial cells drives lysophosphatidic acid-mediated expression of cell adhesion molecules via NF-kappaB p65. J. Biol. Chem. 285, 12536–12542 (2010)
S. Shimizu, M. Tahara, S. Ogata, K. Hashimoto, K. Morishige, K. Tasaka, Y. Murata, Involvement of nuclear factor-kappaB activation through RhoA/Rho-kinase pathway in LPS-induced IL-8 production in human cervical stromal cells. Mol. Hum. Reprod. 13, 181–187 (2007)
G.G. Mackenzie, C.L. Keen, P.I. Oteiza, Microtubules are required for NF-kappaB nuclear translocation in neuroblastoma IMR-32 cells: modulation by zinc. J. Neurochem. 99, 402–415 (2006)
V. Bourgarel-Rey, S. Vallee, O. Rimet, S. Champion, D. Braguer, A. Desobry, C. Briand, Y. Barra, Involvement of nuclear factor kappaB in c-Myc induction by tubulin polymerization inhibitors. Mol. Pharmacol. 59, 1165–1170 (2001)
T.F. Hansen, B.S. Nielsen, A. Jakobsen, F.B. Sorensen, Visualising and quantifying angiogenesis in metastatic colorectal cancer. A comparison of methods and their predictive value for chemotherapy response. Cell. Oncol. 36, 341–350 (2013)
B. Pula, M. Olbromski, A. Wojnar, A. Gomulkiewicz, W. Witkiewicz, M. Ugorski, P. Dziegiel, M. Podhorska-Okolow, Impact of SOX18 expression in cancer cells and vessels on the outcome of invasive ductal breast carcinoma. Cell. Oncol. 36, 469–483 (2013)
A.W. Schüttelkopf, D.M.F. van Aalten, PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 (2004)
O. Trott, A.J. Olson, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2009)
J. Gasteiger, M. Marsili, Iterative partial equalization of orbital electronegativity – a rapid access to atomic charges. Tetrahedron 36, 3219–3228 (1980)
G.L. Warren, C.W. Andrews, A.-M. Capelli, B. Clarke, J. LaLonde, M.H. Lambert, M. Lindvall, N. Nevins, S.F. Semus, S. Senger, G. Tedesco, I.D. Wall, J.M. Woolven, C.E. Peishoff, M.S. Head, A critical assessment of docking programs and scoring functions. J. Med. Chem. 49, 5912–5931 (2006)
W. DeLano, The PyMOL molecular graphics system (DeLano Sci, LLC San Carlos CA, 2003)
K. Mahal, S. Schruefer, G. Steinemann, F. Rausch, R. Schobert, M. Höpfner, B. Biersack, Biological evaluation of 4,5-diarylimidazoles with hydroxamic acid appendages as novel dual mode anticancer agents. Cancer Chemother. Pharmacol. 75, 691–700 (2015)
K. Mahal, M. Resch, R. Ficner, R. Schobert, B. Biersack, T. Mueller, Effects of the tumor-vasculature-disrupting agent verubulin and two heteroaryl analogues on cancer cells, endothelial cells, and blood vessels. ChemMedChem 9, 847–854 (2014)
G.M. Tozer, C. Kanthou, B.C. Baguley, Disrupting tumour blood vessels. Nat. Rev. Cancer 5, 423–435 (2005)
B. Biersack, K. Effenberger, R. Schobert, M. Ocker, Oxazole-bridged combretastatin a analogues with improved anticancer properties. ChemMedChem 5, 420–427 (2010)
S. Ali, Simultaneous targeting of the epidermal growth factor receptor and cyclooxygenase-2 pathways for pancreatic cancer therapy. Mol. Cancer Ther. 4, 1943–1951 (2005)
R.B.G. Ravelli, B. Gigant, P.A. Curmi, I. Jourdain, S. Lachkar, A. Sobel, M. Knossow, Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428, 198–202 (2004)
M. Botta, S. Forli, M. Magnani, F. Manetti, Molecular modeling approaches to study the binding mode on tubulin of microtubule destabilizing and stabilizing agents. Top. Curr. Chem. 286, 279–328 (2009)
C. Kanthou, G.M. Tozer, Tumour targeting by microtubule-depolymerising vascular disrupting agents. Expert Opin. Ther. Targets 11, 1443–1457 (2007)
H. Quan, Y. Xu, L. Lou, p38 MAPK, but not ERK1/2, is critically involved in the cytotoxicity of the novel vascular disrupting agent combretastatin A4. Int. J. Cancer 122, 1730–1737 (2007)
M. Fan, L. Du, A. Stone, K. Gilbert, T. Chambers, Modulation of mitogen-activated protein kinases and phosphorylation of Bcl-2 by vinblastine represent persistent forms of normal fluctuations at G2-M. Cancer Res. 60, 6403–6407 (2000)
C. Kanthou, G.M. Tozer, Microtubule depolymerizing vascular disrupting agents: novel therapeutic agents for oncology and other pathologies. Int. J. Exp. Pathol. 90, 284–294 (2009)
M. Chrzanowska-Wodnicka, K. Burridge, Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J. Cell Biol. 133, 1403–1415 (1996)
L.J. Williams, D. Mukherjee, M. Fisher, C.C. Reyes-Aldasoro, S. Akerman, C. Kanthou, G.M. Tozer, An in vivo role for Rho kinase activation in the tumour vascular disrupting activity of combretastatin A-4 3-O-phosphate: Rho kinase and tumour vascular targeting. Br. J. Pharmacol. 171, 4902–4913 (2014)
A. Arlt, A. Gehrz, S. Müerköster, J. Vorndamm, M.-L. Kruse, U.R. Fölsch, H. Schäfer, Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 22, 3243–3251 (2003)
M. Graupera, J. Guillermet-Guibert, L.C. Foukas, L.-K. Phng, R.J. Cain, A. Salpekar, W. Pearce, S. Meek, J. Millan, P.R. Cutillas, A.J.H. Smith, A.J. Ridley, C. Ruhrberg, H. Gerhardt, B. Vanhaesebroeck, Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature 453, 662–666 (2008)
J. Karar, A. Maity, PI3K/AKT/mTOR pathway in angiogenesis. Front. Mol. Neurosci. 4, 51 (2011)
G. Rajashekhar, M. Kamocka, A. Marin, M.A. Suckow, W.R. Wolter, S. Badve, A.R. Sanjeevaiah, K. Pumiglia, E. Rosen, M. Clauss, Pro-inflammatory angiogenesis is mediated by p38 MAP kinase. J. Cell. Physiol. 226, 800–808 (2011)
M. Kelkel, C. Cerella, F. Mack, T. Schneider, C. Jacob, M. Schumacher, M. Dicato, M. Diederich, ROS-independent JNK activation and multisite phosphorylation of Bcl-2 link diallyl tetrasulfide-induced mitotic arrest to apoptosis. Carcinogenesis 33, 2162–2171 (2012)
J.D. Orth, A. Loewer, G. Lahav, T.J. Mitchison, Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell 23, 567–576 (2012)
S.P. Tabruyn, A.W. Griffioen, A new role for NF-kappaB in angiogenesis inhibition. Cell Death Differ. 14, 1393–1397 (2007)
S.P. Tabruyn, S. Memet, P. Ave, C. Verhaeghe, K.H. Mayo, I. Struman, J.A. Martial, A.W. Griffioen, NF-kappaB activation in endothelial cells is critical for the activity of angiostatic agents. Mol. Cancer Ther. 8, 2645–2654 (2009)
M. Carr, L.M. Greene, A.J.S. Knox, D.G. Lloyd, D.M. Zisterer, M.J. Meegan, Lead identification of conformationally restricted β-lactam type combretastatin analogues: synthesis, antiproliferative activity and tubulin targeting effects. Eur. J. Med. Chem. 45, 5752–5766 (2010)
H. Wehbe, C.M. Kearney, K.G. Pinney, Combretastatin A-4 resistance in H460 human lung carcinoma demonstrates distinctive alterations in β-tubulin isotype expression. Anticancer Res. 25, 3865–3870 (2005)
E.C. Tampaki, L. Nakopoulou, A. Tampakis, K. Kontzoglou, W.P. Weber, G. Kouraklis, Nestin involvement in tissue injury and cancer - a potential tumor marker? Cell. Oncol. 37, 305–315 (2014)
D.O. Moon, M.O. Kim, C.H. Kang, J.D. Lee, Y.H. Choi, G.Y. Kim, JNK inhibitor SP600125 promotes the formation of polymerized tubulin, leading to G2/M phase arrest, endoreduplication, and delayed apoptosis. Exp. Mol. Med. 41, 665–677 (2009)
A.V. Singh, M. Bandi, N. Raje, P. Richardson, M.A. Palladino, D. Chauhan, K.C. Anderson, A novel vascular disrupting agent plinabulin triggers JNK-mediated apoptosis and inhibits angiogenesis in multiple myeloma cells. Blood 117, 5692–5700 (2011)
L. Ciani, P.C. Salinas, c-Jun N-terminal kinase (JNK) cooperates with Gsk3β to regulate dishevelled-mediated microtubule stability. BMC Cell Biol. 8, 27 (2007)
D.C.H. Ng, T.T. Zhao, Y.Y.C. Yeap, K.R. Ngoei, M.A. Bogoyevitch, c-Jun N-terminal kinase phosphorylation of stathmin confers protection against cellular stress. J. Biol. Chem. 285, 29001–29013 (2010)
Y.Y. Yip, Y.Y.C. Yeap, M.A. Bogoyevitch, D.C.H. Ng, Differences in c-Jun N-terminal kinase recognition and phosphorylation of closely related stathmin-family members. Biochem. Biophys. Res. Commun. 446, 248–254 (2014)
J.-H. Kim, T.H. Kim, H.S. Kang, J. Ro, H.S. Kim, S. Yoon, SP600125, an inhibitor of Jnk pathway, reduces viability of relatively resistant cancer cells to doxorubicin. Biochem. Biophys. Res. Commun. 387, 450–455 (2009)
X. Wang, K. Belguise, N. Kersual, K.H. Kirsch, N.D. Mineva, F. Galtier, D. Chalbos, G.E. Sonenshein, Oestrogen signalling inhibits invasive phenotype by repressing RelB and its target BCL2. Nat. Cell Biol. 9, 470–478 (2007)
H. Lee, J. Jeon, Y.S. Ryu, J.E. Jeong, S. Shin, T. Zhang, S.W. Kang, J.H. Hong, G.M. Hur, Disruption of microtubules sensitizes the DNA damage-induced apoptosis through inhibiting nuclear factor kappaB (NF-κB) DNA-binding activity. J. Korean Med. Sci. 25, 1574 (2010)
S. Papa, Linking JNK signaling to NF-kappaB: a key to survival. J. Cell Sci. 117, 5197–5208 (2004)
L. Li, B.B. Aggarwal, S. Shishodia, J. Abbruzzese, R. Kurzrock, Nuclear factor-kappaB and IkappaB kinase are constitutively active in human pancreatic cells, and their down-regulation by curcumin (diferuloylmethane) is associated with the suppression of proliferation and the induction of apoptosis. Cancer 101, 2351–2362 (2004)
Conflict of interests
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
The online version of this article contains supplementary material (determination of apoptosis in Ea.hy926 endothelial, resistant BxPC-3 pancreas, and MDA-MB-231 breast carcinoma cells; original Western blot images) which is available to authorized users.
ESM 1
(PDF 6051 kb)
Rights and permissions
About this article

Cite this article
Mahal, K., Ahmad, A., Sethi, S. et al. Role of JNK and NF-κB in mediating the effect of combretastatin A-4 and brimamin on endothelial and carcinoma cells. Cell Oncol. 38, 463–478 (2015). https://doi.org/10.1007/s13402-015-0243-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-015-0243-7