
Abstract
Background
Ovarian cancer is one of the most common lethal gynecological malignancies world-wide. Despite an initial 70–80 % response rate, most patients relapse within 1–2 years and develop chemo-resistance. Hence, the identification of novel drugs or the repositioning of known drugs to re-sensitize ovarian cancer cells to existing chemotherapy regimens is needed. Here, we evaluated the effect of metformin (an anti-diabetic drug) on ovarian cancer cells, based on its putative effect on other solid tumors.
Methods
Primary cultures of epithelial ovarian cancer cells established from ascitic fluids of untreated ovarian cancer patients and the SKOV-3 ovarian cancer-derived cell line were used. The respective cells were treated with metformin, carboplatin and paclitaxel alone and its various combinations and their effects, including the ability to induce apoptosis, were examined. Concomitantly, the cells were assessed for the expression of several apoptosis-related mRNAs and proteins using quantitative real time PCR, flowcytometry and Western blotting.
Results
We found that metformin induced apoptosis in the ovarian cancer cells tested, and provoked a cell cycle arrest in the G0/G1 and S-phase. Metformin induced apoptosis by down-regulating Bcl-2 and Bcl-xL expression, and up-regulating Bax and Cytochrome c expression. We also found that the apoptosis induction by metformin could be enhanced by a combinatorial use of carboplatin and/or paclitaxel.
Conclusions
Our data indicate that metformin can induce apoptosis in both primary ovarian cancer cells and in SKOV-3 cells. When metformin was combined with carboplatin or paclitaxel, an increased apoptotic activity was observed, implicating a chemo-adjuvant potential.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ovarian cancer has emerged as one of the most common lethal gynecological malignancies. As per statistics of GLOBOCAN 2012 and the International Agency for Research on Cancer (IARC), ovarian cancer ranks as the seventh most common cancer, with a high mortality rate in women globally (http://globocan.iarc.fr/). In India, ovarian cancer is the fourth leading cause of cancer amongst women, trailing behind cervix, breast and colorectal cancer. A major cause of its high mortality rate is the fact that the disease is usually clinically silent until an advanced stage has been reached, and that the symptoms are usually vague and non-specific. Most patients diagnosed with advanced disease undergo remission after optimal surgical cytoreduction and platinum- and taxane-based chemotherapy. Epithelial ovarian cancer exhibits intra- and inter-tumor heterogeneity, leading to widely variable clinical phenotypes even amongst patients of the same stage and histological subtype. Despite an initial 70–80 % response rate, it has been found that in the majority of the cases the disease ultimately recurs within 1 to 2 years, with a 5 year survival rate of 30–50 % [1–4]. Thus, the identification of novel prognostic and therapeutic factors is needed to improve the patient’s survival.
Here, we evaluated the effect of metformin on epithelial ovarian cancer. Normally, metformin is the drug of choice for the treatment of type-2 diabetes mellitus. Recent interest in metformin as an agent for cancer prevention and treatment reflects the convergence of several areas of research [5]. Exciting preclinical studies have, for example, shown that metformin can inhibit the growth of cancer cells both in vitro and in vivo. Evidence has been put forward that metformin results in (a) the initiation of a LKB1-mediated AMPK-dependent energy stress response that can adversely affect the survival of cancer cells and (b) the inhibition of PI3K/AKT/mTOR signaling that can lead to a reduced proliferation of cancer cells. These observations have provided a molecular basis for a direct, insulin-independent, anti-tumor effect and underscore a rationale for evaluating metformin in cancer-associated clinical trials [6, 7].
An association of metformin with a reduced cancer mortality that has been noted in three studies [8–10]. In support of this notion, a retrospective study in breast cancer patients showed that diabetic patients treated with metformin and neo-adjuvant chemotherapy exhibited a higher complete response rate than the respective control groups [11]. Additional promising trends were noted on overall survival in other specific cancer types, particularly pancreatic and hepatocellular cancers and, to a lesser extent, colon cancer. Together, these findings warrant further research on the therapeutic potential of metformin [12].
It has been shown that metformin can induce a significant growth inhibition in OVCAR-3 and OVCAR-4 ovarian cancer-derived cells in a time- and dose-dependent manner. Next to the observation that metformin may exhibit anti-neoplastic activity as a single agent, its co-incubation with cisplatin revealed an increase in cytotoxicity compared to each agent alone [13]. Additionally, it has been found that metformin treatment can (a) significantly inhibit the proliferation of various chemo-responsive and -resistant ovarian cancer-derived cell lines (i.e., A2780, CP70, C200, OV202, OVCAR-3, SKOV3ip, PE01 and PE04) and (b) cause cell cycle arrest in conjunction with decreased Cyclin D1 and increased p21 protein expression [14]. Also, an in vivo potential of metformin, and its synergism with cisplatin, in reducing ovarian cancer tumor burden, angiogenesis and metastatic potential have been reported [15].
Clinically, metformin exhibits a good safety profile and is well-tolerated by patients. The main limited side effect is transient gastrointestinal distress, such as nausea and diarrhea. In only a few cases discontinuation of the drug is required. Metformin may also promote hirsuitism and can cause vitamin B12 mal-absorption during long-term therapy. No teratogenic effects or adverse fetal outcomes have been reported. The only potential major adverse effect of metformin therapy is lactic acidosis. This condition is rare and is primarily confined to patients with concomitant renal and hepatic disorders [16]. Metformin may exhibit two potential anti-neoplastic effects: indirectly by reducing circulating insulin levels and directly by inhibiting growth through the AMPK-LKB1 pathway.
Here, we set out to evaluate whether, in addition to its anti-proliferative effect, metformin can induce apoptosis in human ovarian cancer cells. We also sought to identify the pathways underlying this putative effect. Apoptosis plays an important role in chemotherapy-induced cell death. A failure of the apoptotic machinery can lead to resistance towards chemotherapeutic agents (i.e., chemo-resistance). Bcl-2 family member proteins represent crucial players in apoptosis induction, mainly via the mitochondrial pathway, which is frequently triggered in response to chemotherapeutic agents. Elevated levels of Bcl-2 may contribute to chemo-resistance by stabilizing the mitochondrial membrane against apoptotic insult [17–19]. As a consequence Bcl-2, or its family member Bcl-xl, may serve as therapeutic targets. We have established primary cultures of ovarian cancer cells from ascitic fluids of 15 patients, treated these with metformin, carboplatin and paclitaxel alone or in combination, and assessed the subsequent apoptotic effects. We also studied the effects in SKOV-3 ovarian cancer-derived cells, which are p53 deficient [20]. Since more than 95 % of primary ovarian cancers carry p53 mutations, SKOV-3 serves as a suitable model for studying apoptotic effects in ovarian cancer.
2 Materials and methods
2.1 Cell cultures
All ovarian cancer ascitic samples for establishing primary cell cultures were obtained after signed informed consent and approval from the IRCH AIIMS ethical committee. The ovarian cancer-derived cell line SKOV-3 was obtained from the National Centre for Cell Science (NCCS), Pune, India, and was grown in DMEM medium supplemented with heat inactivated 10 % fetal bovine serum (FBS), 2 mM glutamine and 10 μg/ml gentamicin. The cells were routinely passaged every 5–7 days. All cells were maintained at 37 °C in a 5 % CO2, 95 % air atmosphere incubator. The respective assays were performed in medium containing 1 % FBS. Metformin was obtained from Sigma-Aldrich, USA (cat#D150959) and kept as a stock solution of 1 M in DMEM without serum. Carboplatin and paclitaxel were obtained from the hospital pharmacy department.
2.2 Chemicals and antibodies
All cell culture materials (disposables) were obtained from Sigma Aldrich, USA. The anti-Bax, anti-Bcl-2, anti-Bcl-xL, anti-Cyt C, anti-Survivin and anti-β-actin antibodies, and the alkaline phosphatase-conjugated anti-rabbit IgG and anti-mouse IgG reagents, were purchased from Santa Cruz, USA.
2.3 Establishment of primary cultures
Primary cultures of epithelial ovarian cancer cells were established from ascitic fluids collected from untreated ovarian cancer patients (see above). Ascitic fluids were collected under sterile conditions and the subsequent establishment of cultures was initiated only when grape-like clusters of cells, characteristic of ovarian cancer cells, were observed under the microscope. MCDB 105 medium and Medium 199 (Sigma Aldrich, USA) were used in the ratio 1:1, supplemented with 10 % FCS and antibiotics (100 units/ml penicillin, 100 μg/ml streptomycin), to culture the ovarian cancer cells. The ascitic fluids (200 ml) were centrifuged at 8000 rpm for 10 min at 4 °C. Next, the pellets were suspended in 15 ml ascitic fluid and 15 ml MCDB 105/199 medium and incubated in 75 mm2 flasks in a humidified atmosphere of 5 % CO2 at 37 °C. After 4 days the media were removed and the cells were washed in PBS, while the clusters of ovarian cancer cells remained attached to the flask surface. The latter cells were maintained in culture until they reached 80 % confluency. After this, the cells were trypsinized and sub-cultured [21].
2.4 MTT assay
The cytotoxic effects of metformin, carboplatin and paclitaxel were assessed using a MTT assay (Sigma-Aldrich). Briefly, 5 × 104 cells/well were seeded in 96-well culture plates. After overnight incubation, the cells were treated with varying concentrations of the drugs for 24, 48 and 72 h. After this, the cells were incubated with 100 μl of 5 mg/ml MTT for 4 h at 37 °C. The formazan crystals thus formed were dissolved in DMSO and the absorbance was measured at 570 nm, and 620 nm as a reference wavelength, using an ELISA reader. The doses of 15 mM metformin (m), l00 μg/ml carboplatin (c), 5 μg/ml paclitaxel (p) for 48 h alone, or in the combinations metformin (15 mM) + carboplatin(100 μg/ml) (mc), metformin(15 mM) + paclitaxel(5 μg/ml) (mp), carboplatin(100 μg/ml) + paclitaxel(5 μg/ml) (cp), metformin(15 mM) + carboplatin(100 μg/ml) + paclitaxel (5 μg/ml) (mcp) were used for the treatment of SKOV-3 and ascitic ovarian cancer cells after standardization, respectively.
2.5 Flowcytometry
SKOV-3 cells were treated with 15 mM metformin, l00 μg/ml carboplatin, 5 μg/ml paclitaxel alone or in its respective combinations for 48 h, and then harvested. After this, the adherent cells were collected using trypsin EDTA while floating cells were collected by centrifugation. The cells were combined and washed twice with ice-cold PBS. To determine the percentage of apoptotic cells and to carry out cell cycle analyses, the cells were fixed in 70 % ethanol after collection and washing. Next, the cells were washed twice with ice-cold PBS and resuspended in propidium iodide buffer (PBS, 0.1 % Triton X-100, 0.1 mM EDTA, 0.05 mg/ml ribonuclease A, and 50 mM propidium iodide). After incubation for 30 min at room temperature, the cell cycle distribution was determined by flowcytometry (BD Facs, USA) using Win Mdi 2.9 software.
2.6 Quantitative RT-PCR
Total cellular RNA was harvested from ovarian cancer cells using Trizol reagent (Sigma Aldrich, St. Louis USA) according to the manufacturer’s instructions and quantified by spectrophotometric absorbance at 260 nm. One μg of total RNA was reverse-transcribed into cDNA for quantitative real-time PCR (RT-PCR) using Revert Aid M-Mul-V Reverse transcriptase (MBI Fermentas) according to the manufacturer’s instructions. RT-PCR reactions were carried out using gene-specific primers (listed in Table 1) in a volume of 20 μl in an ABI 7500 Thermocycler using Biotool Taq Polymerase (B&M Labs, S.A., Spain ) under the following conditions: denaturation at 94 °C for 10 s; annealing (as per primer) for 20 s; extension at 72 °C for 5 s. The respective mRNA expression levels were normalized using β-actin. The comparative Ct method was used to evaluate differential expression levels in SKOV-3 cells. The expression of each target gene relative to β-actin was determined using the following equation 2−∆∆Ct, where ∆∆Ctgene= [Ctgene – Ctβactin] treated - [Ctgene – Ctβactin] Control. For each target gene we performed three replicates of quantitative RT-PCR.
2.7 Protein extraction and Western blotting
Cells were lysed in RIPA buffer (25 mM Tris–HCl pH 7.6, 150 mM NaCl, 1 % NP-40, 0.25 % sodium deoxycholate, 0.1 % SDS) supplemented with protease inhibitor cocktail tablets (G Biosciences, USA). Determination of the total protein content was performed according to the Bradford method. Briefly, clarified protein lysates (60–100 μg) were resolved electrophoretically in 10–15 % denaturing SDS-polyacrylamide gels and transferred to nitrocellulose membranes. After blocking in 5 % non-fat dry milk, the membranes were probed with primary antibodies directed against Bcl-2, Bcl-xL, Bax, Cytochrome c, Survivin and β-actin. The immunoblotted proteins were visualized using alkaline phosphatase-conjugated secondary antibodies and detected using BCIP/NBT substrate (Promega, USA). Appropriate positive and negative controls were run simultaneously. The protein bands were analyzed and quantified using an Alphaimager scanning densitometer (Alpha Innotech, USA), and the protein expression levels were expressed in Relative Units (RU). The control was taken as 1 and the treatment results were expressed relative to the control.
2.8 Statistical analyses
All values are expressed as mean ± SEM (standard error of mean). For multiple comparisons, data were analyzed using the one-way ANOVA test followed by the Post hoc Bonferronis test. P < 0.05 was considered significant. All statistical analyses were carried out using Graph Pad Prismv.5 software.
3 Results
3.1 Metformin, carboplatin and paclitaxel inhibit ovarian cancer cell viability
Out of the 20 ascitic fluid samples collected, 15 primary cultures of ovarian cancer cells could be established. To examine the effects of metformin, carboplatin and paclitaxel on cancer cell growth, we treated primary ovarian cancer cells and SKOV-3 ovarian cancer-derived cells with the respective agents. Cell viability was determined by MTT assay using increasing doses of these agents for 48 h. We found that metformin, carboplatin and paclitaxel all inhibited cell viability compared to the control (Fig. 1). Metformin inhibited cell viability in doses ranging from 15 to 30 mM, reaching a plateau at 35 mM in both SKOV-3 and primary ovarian cancer cells (Fig. 1a). Similarly, carboplatin and paclitaxel inhibited cell viability in doses ranging from 100–200 μg/ml to 2–10 μg/ml in SKOV-3 and primary ovarian cancer cells, respectively (Fig. 1b, c).

a, b, c Cell viability measured by MTT assay of primary ovarian cancer cell cultures and SKOV-3 ovarian cancer-derived cells treated with increasing doses of metformin, carboplatin and paclitaxel, respectively, for 48 h. d Flowcytometric cell cycle analyses of control and drug-treated primary ovarian cancer and e SKOV-3 cells, respectively. Cells were treated for 48 h with the respective (combinations of) drugs: m metformin, c carboplatin, p paclitaxel, mc metformin + carboplatin, mp metformin+paclitaxel, cp carboplatin+paclitaxel, mcp metformin+carboplatin+paclitaxel
When lower doses of metformin were used in combinatorial studies, no synergistic effects were observed (data not shown). Doses of 15 mM metformin (m), l00μg/ml carboplatin (c), 5 μg/ml paclitaxel (p) for 48 h alone, and in the combinations metformin(15 mM) + carboplatin(100 μg/ml) (mc), metformin(15 mM) + paclitaxel(5 μg/ml) (mp), carboplatin(100 μg/ml) + paclitaxel(5 μg/ml) (cp), metformin(15 mm) + carboplatin(100 μg/ml) + paclitaxel (5 μg/ml) (mcp) were used for the treatment of SKOV-3 and ascitic ovarian cancer cells after standardization.
3.2 Metformin, carboplatin and paclitaxel induce ovarian cancer cell cycle arrest
To further evaluate the mechanism underlying the growth inhibition by metformin, carboplatin and paclitaxel, cell cycle profiles were analyzed by flowcytometry after treatment of primary ovarian cancer cells and SKOV-3 cells with the respective agents alone or its combinations (Fig. 1d, e). In the control situation, the majority of the cells were found to be in the G0/G1-phase of the cell cycle, a small fraction in the G2/M-phase and the rest in the S-phase. Subsequent metformin, carboplatin and paclitaxel treatment led to increases in G0/G1-phase, S-phase and G2/M-phase sub-populations in primary ovarian cancer cell and SKOV-3 cell cultures, respectively. However, metformin in combination with carboplatin, paclitaxel or carboplatin + paclitaxel resulted in increases in G0/G1 and S-phase cells compared to carboplatin and paclitaxel only treated cells. From these results we conclude that, at the cellular level, metformin interferes with the cell cycle leading to G0/G1-phase or S-phase arrest.
3.3 Metformin, carboplatin and paclitaxel induce apoptosis in ovarian cancer cells
To assess apoptosis induction, control and treated primary ovarian cancer cells were analyzed by flowcytometry (Fig. 2a). As evident from the bar diagrams (Fig. 2b), there was a wide variation in response of the individual primary cultures to treatment with the different agents used. The mean percentage of apoptosis in untreated control cells was 4.9 %. Upon treatment with metformin this percentage was 15.4 % (range 9–16.8 %), with carboplatin 19.2 % (range 9.9–20 %) and with paclitaxel 16.6 % (range 10–20.5 %). Upon treatment with metformin + carboplatin (mc) a percentage of 40.2 % apoptosis was seen (range 12–42.4 %), with metformin + paclitaxel (mp) 32.4 % (range 18.2–39 %), with carboplatin + paclitaxel (cp) 32.2 % (range 16–37.3 %) and with metformin + carboplatin + paclitaxel (mcp) 55.4 % (range 46–55 %).

a Representative experiment showing flowcytometric analyses of control and drug-treated primary ovarian cancer cells. Treatments: m metformin, c carboplatin, p paclitaxel, mc metformin + carboplatin, mp metformin+paclitaxel, cp carboplatin+paclitaxel, mcp metformin+carboplatin+paclitaxel. The horizontal lines drawn from the Y-axis to the peak represent the % of apoptotic cells. b Mean percentages of apoptosis induced in control and drug-treated primary ovarian cancer cells as measured by flowcytometry. The diagram represents means ± SEM of 15 primary cultures
Similarly, logarithmically growing SKOV-3 cells were treated with metformin, carboplatin and paciltaxel. The mean percentage apoptosis in untreated SKOV-3 (control) cells was 5.1 %. Upon treatment with metformin this percentage was 14.5 %, with carboplatin 17.2 % and with paclitaxel 12.06 %, whereas with metformin + carboplatin (mc) this was 35.6 %, with metformin + paclitaxel (mp) 28.2 %, with carboplatin + paclitaxel (cp) 43.7 % and with metformin + carboplatin + paclitaxel (mcp) 50.8 % (Fig. 3a, b).

a Representative experiment showing flowcytometric analyses of control and drug-treated SKOV-3 ovarian cancer-derived cells. The horizontal lines drawn from the Y-axis to the peak represent the % of apoptotic cells. b Mean percentages of apoptosis induced in control and drug-treated SKOV-3 ovarian cancer cells as measured by flowcytometry. The diagram represents means ± SEM of three independent experiments
As evident from the above results, metformin by itself was as good as carboplatin or paclitaxel in inducing apoptosis in both the primary ovarian cancer cells and the SKOV-3 cells. However, the combination of metformin and paclitaxel (mp) increased apoptosis, the combination of metformin and carboplatin (mc) was equally effective as carboplatin and paclitaxel, and the combination of metformin, carboplatin and paclitaxel turned out to be the best combination with the highest apoptotic percentage. Together, we again conclude that at the cellular level, metformin interferes with the cell cycle, leading to G0/G1 and/or S-phase arrest. Besides its anti-proliferative capacity as a single agent metformin showed, in conjunction with carboplatin and paclitaxel, an increased cytotoxic effect compared to each agent alone.
3.4 Metformin, carboplatin and paclitaxel affect the expression of pro-apoptotic and anti-apoptotic genes in SKOV-3 cells
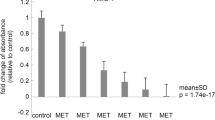
Next, we set out to elucidate the pathway involved in apoptosis induction by metformin. To do so, we measured mRNA expression levels of Bcl-2 family members in the presence of metformin, carboplatin and paclitaxel and its combinations. We found that, upon treatment, the expression levels of the anti-apoptotic Bcl-2, Survivin and Bcl-xL mRNAs were all down-regulated in SKOV-3 cells (Fig. 4a). The mRNA expression level of Bcl-2 decreased 1.35, 1.41, 1.16, 2.5, 1.7, 2.3 and 3.4 fold after treatment with metformin, carboplatin, paclitaxel, metformin + carboplatin (mc), metformin + paclitaxel (mp), carboplatin + paclitaxel (cp) and metformin + carboplatin + paclitaxel (mcp), respectively. Similarly the mRNA expression level of Bcl-xL decreased 1.49, 1.5, 1.47, 3.5, 2, 2.8 and 5 fold after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively, whereas the mRNA expression level of Survivin decreased 1.29, 1.52, 1.49, 1.61, 1.51, 1.69 and 1.75 fold after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively.

a Bar diagram representing relative fold mRNA expression levels of Bcl-2, Bcl-xL and Survivin normalized to β-actin in control and drug-treated SKOV-3 ovarian cancer-derived cells measured by quantitative RT-PCR. Results represent the mean ± SEM of three independent experiments. *P < 0.05 versus metformin, # P < 0.05 versus carboplatin, + P < 0.05 versus paclitaxel, $ P < 0.05 versus control. b Bar diagram representing relative fold mRNA expression levels of Bax and Cytochrome c normalized to β-actin in control and drug-treated SKOV-3 ovarian cancer cells as measured by quantitative RT-PCR. Results represent the mean ± SEM of three independent experiments. *P < 0.05 versus metformin, # P < 0.05 versus carboplatin, + P < 0.05 versus paclitaxel. $ P < 0.05 versus control
We observed a significant decrease in Bcl-2 mRNA expression level in SKOV-3 cells after treatment with metformin + carboplatin (mc) (p < 0.05) compared to metformin or carboplatin alone. Moreover, we found that paclitaxel did not significantly decrease Bcl-2 expression, either as a single agent or in combination with metformin. Similarly, we found a significant decrease in Bcl-2 mRNA expression after treatment with carboplatin+ paclitaxel (cp) (p < 0.05) compared to carboplatin or paclitaxel alone. Similar results were obtained for Bcl-xL mRNA expression. In contrast, no significant difference in Survivin mRNA expression was observed in any of the treatment regimens used.
In analogy, we measured mRNA expression levels of several pro-apoptotic Bcl-2 family members. We found that the mRNA levels of both Bax and Cytochrome c (Cyt c) were up-regulated in SKOV-3 cells (Fig. 4b). Increases of 1.2, 2.3, 1.92, 5.5, 2.8, 5.8 and 8.8 fold in Bax mRNA expression were observed after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively. Similarly, increases of 1.2, 1.5, 1.3, 3.8, 1.66, 3.5 and 6.2 fold in Cytochrome c mRNA expression were observed after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively. Significant increases in Bax and Cytochrome c mRNA expression levels after treatment with metformin + carboplatin (mc) (p < 0.05) and carboplatin + paclitaxel (cp) (p < 0.05) were observed compared to the respective agents alone.
3.5 Metformin, carboplatin and paclitaxel affect the expression of pro-apoptotic and anti-apoptotic proteins in SKOV-3 cells
Next, we measured the levels of several pro-apoptotic and anti-apoptotic proteins using Western blotting (Fig. 5a). We found that after the various treatment regimens the levels of Bcl-2, Survivin and Bcl-xL were down-regulated in SKOV-3 cells (Fig. 5b). The protein expression of Bcl-2 decreased by 1.1, 1.21, 1.1, 2, 1.4, 2.1 and 3.3 fold after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively. Similarly, the protein expression of Bcl-xL decreased by 1.14, 1.12, 1.1, 2.3, 1.8, 2.4 and 3.4 fold after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp respectively. In contrast, and in accordance with the mRNA data, no significant changes in Survivin protein expression levels were observed after any of the treatment regimens used.

a Effect of metformin, carboplatin and paclitaxel on the expression of anti-apoptotic and pro-apoptotic proteins (Bcl-2, Bcl-xL, Bax, Cytochrome c, Survivin and control(β-Actin)) in SKOV-3 ovarian cancer-derived cells treated with 15 mM metformin and/or 100 μg/ml carboplatin and 5 μg/ml paclitaxel for 48 h. Cell lysates were subjected to Western blotting (one representative example out of three is shown). b Densitometric analyses of Bcl-2, Bcl-xL and Survivin protein expression in control and drug-treated SKOV-3 cells as measured by Western blotting. The bar diagram represents the mean ± SEM of three independent experiments. *P < 0.05 versus metformin, #P < 0.05 versus carboplatin, +P < 0.05 versus paclitaxel, $ P < 0.05 versus control. c Densitometric analyses of Bax and Cytochrome c protein expression in control and drug-treated SKOV-3 cells as measured by Western blotting. The diagram represents the mean ± SEM of three independent experiments. *P < 0.05 versus metformin, # P < 0.05 versus carboplatin, + P < 0.05 versus paclitaxel, $ P < 0.05 versus control
The protein levels of both Bax and Cytochrome c were found to be up-regulated in SKOV-3 cells after application of the various treatment regimens (Fig. 5c). Bax protein increases of 1.2, 1.5, 1.94, 3.6, 2.4, 3.3 and 3.2 fold were observed after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively. Similarly, Cytochrome c protein increases of 1.2, 1.3, 1.39, 1.9, 1.4, 2.1 and 1.8 fold were observed after treatment with metformin, carboplatin, paclitaxel, mc, mp, cp and mcp, respectively. Overall, our Western blotting results were in concordance with our RT-PCR results, with significant increases in pro-apoptotic proteins and significant decreases in anti-apoptotic proteins after treatment of SKOV-3 cells with metformin + carboplatin(mc) and carboplatin + paclitaxel(cp) compared to the respective agents alone.
4 Discussion
Currently, the mortality rate of ovarian cancer is high, which is primarily due to the fact that75% of the patients present with an advanced stage of the disease (stage >III) at diagnosis, and that the available therapies for primary and recurrent ovarian cancers exhibit limited efficacies. Consequently, there is a great need for improved upfront and salvage therapies for ovarian cancer. Novel therapies may originate from the designing of new drugs. Although promising, this approach carries the limitation of being tremendously expensive and time consuming [22]. Therefore, alternative approaches need to be explored. One such approach is drug repositioning, in which existing drugs are used to treat new diseases. This approach carries the promise of accelerating the translation of research into clinical practice in a relatively short time frame [23]. Metformin, which is derived from the French Lilac plant (Galega officinalis), is one of the most commonly used medications for diabetes worldwide. Previously, it has been shown that metformin may also exhibit anti-proliferative effects in ovarian cancers, both in vitro and vivo [13]. Similar observations from others have suggested anti-cancer effects in prostate [24], colon [25], pancreas [26] and brain [27] tumors. As yet, very few studies have been performed to assess the apoptosis-inducing potential of metformin. Moreover, it has been suggested that deregulated apoptosis is a major contributor to the development of acquired resistance to anticancer therapies. Enhancement of this process, either by directly activating apoptosis or by lowering the threshold for its initiation by cytotoxic drugs, represents an attractive therapeutic strategy [28]. Therefore, therapeutic manipulation of the apoptotic pathway(s) may represent a way to improve the clinical response of ovarian cancer patients. Apoptosis is a complex process and involves a number of mitochondria-associated proteins including Bcl-2 family members such as Bcl-xL, Bad and Bax. The Bcl-2 protein is located at the outer mitochondrial membrane and protects cells against apoptosis by blocking the release of Cytochrome c. Cells that over-express Bcl-2 resist apoptosis induced by anticancer drugs. As a corollary, suppression of Bcl-2 expression promotes apoptosis induced by a number of stimuli, including anticancer drugs.
Here, we addressed the question whether metformin can induce apoptosis of ovarian cancer cells. We hypothesized that metformin might be used in conjunction with carboplatin and/or paclitaxel (standard chemotherapies) to increase the chemo-sensitivity of ovarian cancer cells. Upon treatment with standard doses of the chemotherapeutic agents and metformin alone or in combination, we found variable responses in independent primary ovarian cancer cell cultures. Each primary ovarian cancer cell culture was found to be unique in nature, with different responses to metformin and its combinations with carboplatin and/or paclitaxel. Primary cell cultures more closely mimic the physiological state of the cancer cells in vivo and, therefore, they generate more relevant data. As such, they provide a model that allows a vast spectrum of cell types from a larger number of patients to be considered, in the absence of artificially induced genetic mutations. Nevertheless, primary epithelial cell cultures are technically challenging and time consuming. In our study, the primary cultures posed a limitation on the detection of apoptotic responses to chemotherapeutic drugs in conjunction with the assessment of (changes in) mRNA and protein expression levels of various apoptotic factors. We evaluated the effects of metformin on cell viability, cell cycle distribution and progression. We found that metformin exhibited anti-proliferative effects, and that the ovarian cancer cells were blocked in S and G0/G1-phases of the cell cycle upon metformin exposure. One way by which metformin may induce cell cycle arrest may be the activation of AMP-activated protein kinase (AMPK), which leads to down-regulation of Cyclin D1. This down-regulation may result in a release of sequestered cyclin-dependent kinase (CDK) inhibitors such as p27Kip1 and p21Cip1. The released CDK inhibitors can bind to and inhibit Cyclin E/CDK2, thereby preventing cell cycle progression from the G1 to the S-phase and, thus, causing cell cycle arrest and decreasing proliferation [29]. Differences exist between studies regarding the effect of metformin on cell cycle distribution. One possible explanation for these variations may be the occurrence of polymorphisms in the metformin transporter, OCT1 (organic cation transporter-1) [30]. The role of OCT1 in metformin uptake by ovarian cancer cells is currently under investigation.
The ovarian cancer-derived SKOV-3 cells were similarly treated with metformin, carboplatin and paclitaxel and similar findings were obtained as for the primary ovarian cancer cell cultures. We found that metformin by itself was as good as carboplatin or paclitaxel in inducing apoptosis, and that combinations of carboplatin + paclitaxel (cp), metformin + carboplatin (mc) and metformin + paclitaxel (mp) modestly increased apoptosis compared to the respective agents alone. As yet, data on the effect of metformin on apoptosis in cancer cells are limited and somewhat inconsistent. It has e.g. been reported that metformin can block the cell cycle at the G0/G1-phase in prostate cancer cells without inducing apoptosis [24]. Similarly, it has been found that breast cancer cells do not undergo apoptosis in response to metformin [5]. In contrast, metformin has been shown to stimulate apoptotic cell death in pancreatic cancer cells [31]. These discrepancies may result from variations in experimental conditions and/or cell type-specific functions.
We set out to confirm our initial findings by evaluating the putative down-regulation of anti-apoptotic and up-regulation of apoptotic factors. To this end, we tested the effect of metformin on various pro-and anti-apoptotic Bcl-2 family members. Our results revealed a decreasing trend in the expression of the anti-apoptotic Bcl-2, Bcl-xL mRNA and protein levels in cells treated with metformin, carboplatin and paclitaxel, and its various combinations. Significant decreases were found with combinations of metformin + carboplatin (mc) and carboplatin + paclitaxel (cp). Concomitantly, we found that the pro-apoptotic factors Bax and Cytochrome c were induced in these cells. However, we did not observe any significant down-regulation of Survivin, either alone or in combination with carboplatin or paclitaxel. Moreover, we did not find a significant up-regulation of pro-apoptotic or down-regulation of anti-apoptotic factors at either the mRNA or protein levels after treatment with combinations of metformin + paclitaxel (mp) compared to metformin or paclitaxel alone.
We found not only an additional pro-apoptotic effect to the previously described anti-proliferative effect of metformin, but also a beneficial effect of combining metformin with the cytotoxic drug carboplatin, which is commonly used for the treatment of ovarian cancer. These effects were observed using flowcytometry, quantitative RT-PCR and Western blotting. Taken together, our results suggest that there is an additive effect of carboplatin + metformin (mc) on ovarian cancer cells. The combinations of the metformin + carboplatin (mc) and carboplatin + paclitaxel (cp) induced a stronger inhibition of the anti-apoptotic proteins and a higher induction of the pro-apoptotic proteins tested, suggesting a cell type-specific effect of the combined drugs.
Although the doses used in this study are similar to those used in most in vitro and pre-clinical studies reported in the literature (ranging from 1 to 100 mM), it may be argued that these doses are still supra-physiological. Indeed, metformin concentration in the blood of type 2 diabetic patients treated with this drug approximates 50 μM [32], indicating that we used a 200-fold excess over these therapeutic levels. However, it has been reported that metformin accumulates in tissues, where it can reach concentrations at which our in vitro observations might occur [33]. As such, targeted inhibition of Bcl-2 and Bcl-xL has the potential to facilitate tumor cell apoptosis. Here we report correlations between metformin treatment, the inhibition of anti-apoptotic Bcl-2 family proteins, and increases in apoptosis when metformin is combined with known cytotoxic drugs. As such, metformin appears to increase the effectiveness of standard chemotherapy and may, thus, act as chemo-adjuvant. This in vitro study offers a basis for pre-clinical studies on the benefits of metformin for the treatment ovarian cancer. A detailed understanding of the apoptotic mechanisms and factors that can compromise them is critical to the design of more potent, specific and effective ovarian cancer therapies.
5 Conclusion
Besides its anti-proliferative activity as a single agent, metformin together with carboplatin and paclitaxel showed increased apoptotic activities compared to the respective agents alone, thus demonstrating a chemo-adjuvant potential for epithelial ovarian cancer.
References
J.R. Chien, G. Aletti, D.A. Bell, G.L. Keeney, V. Shridhar, L.C. Hartmann, Molecular pathogenesis and therapeutic targets in epithelial ovarian cancer. J. Cell. Biochem. 102, 1117–1129 (2007)
Y. Li, K. Wang, Y.-Z. Jiang, X.-W. Chang, C.-F. Dai, J. Zheng, 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibits human ovarian cancer cell proliferation. Cell. Oncol. 37, 429–437 (2014)
M. Momeni, T. Kalir, S. Farag, L. Chuang, D. Fishman, D.E. Burstein, Expression of H1.5 and PLZF in granulosa cell tumors and normal ovarian tissues: a short report. Cell. Oncol. 37, 229–234 (2014)
J. Di, T. Duiveman-de Boer, P.L.M. Zusterzeel, C.G. Figdor, L.F.A.G. Massuger, R. Torensma, The stem cell markers Oct4A, Nanog and c-Myc are expressed in ascites cells and tumor tissue of ovarian cancer patients. Cell. Oncol. 36, 363–374 (2013)
I.N. Alimova, B. Liu, Z. Fan, S.M. Edgerton, T. Dillon, S.E. Lind, A.D. Thor, Metformin inhibits breast cancer cell growth, colony formation and induces cell cycle arrest in vitro. Cell Cycle 8, 909–915 (2009)
M. Zakikhani, R. Dowling, I.G. Fantus, N. Sonenberg, M. Pollak, Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 66, 10269–10273 (2006)
G. Zhou, R. Myers, Y. Li, Y. Chen, X. Shen, J. Fenyk-Melody, M. Wu, J. Ventre, T. Doebber, N. Fujii, N. Musi, M.F. Hirshman, L.J. Goodyear, D.E. Moller, Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 108, 1167–1174 (2001)
S.L. Bowker, S.R. Majumdar, P. Veugelers, J.A. Johnson, Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 29, 254–258 (2006)
G.W.D. Landman, N. Kleefstra, K.J.J. van Hateren, K.H. Groenier, R.O.B. Gans, H.J.G. Bilo, Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care 33, 322–326 (2010)
G. Libby, L.A. Donnelly, P.T. Donnan, D.R. Alessi, A.D. Morris, J.M.M. Evans, New users of metformin are at low risk of incident cancer A cohort study among people with type 2 diabetes. Diabetes Care 32, 1620–1625 (2009)
S. Jiralerspong, S.L. Palla, S.H. Giordano, F. Meric-Bernstam, C. Liedtke, C.M. Barnett, L. Hsu, M.-C. Hung, G.N. Hortobagyi, A.M. Gonzalez-Angulo, Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J. Clin. Oncol. 27, 3297–3302 (2009)
A. DeCensi, M. Puntoni, P. Goodwin, M. Cazzaniga, A. Gennari, B. Bonanni, S. Gandini, Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev. Res. 3, 1451–1461 (2010)
W.H. Gotlieb, J. Saumet, M.-C. Beauchamp, J. Gu, S. Lau, M.N. Pollak, I. Bruchim, In vitro metformin anti-neoplastic activity in epithelial ovarian cancer. Gynecol. Oncol. 110, 246–250 (2008)
R. Rattan, S. Giri, L.C. Hartmann, V. Shridhar, Metformin attenuates ovarian cancer cell growth in an AMP-kinase dispensable manner. J. Cell. Mol. Med. 15, 166–178 (2011)
R. Rattan, R.P. Graham, J.L. Maguire, S. Giri, V. Shridhar, Metformin suppresses ovarian cancer growth and metastasis with enhancement of cisplatin cytotoxicity in vivo. Neoplasia 13, 483–491 (2011)
C.J. Bailey, R.C. Turner, Metformin. N. Engl. J. Med. 334, 574–579 (1996)
K. Mohankumar, S. Pajaniradje, S. Sridharan, V.K. Singh, L. Ronsard, A.C. Banerjea, B.C. Selvanesan, M.S. Coumar, L. Periyasamy, R. Rajagopalan, Apoptosis induction by an analog of curcumin (BDMC-A) in human laryngeal carcinoma cells through intrinsic and extrinsic pathways. Cell. Oncol. 37, 439–454 (2014)
V.V. Dhawan, G.V. Joshi, A.S. Jain, Y.P. Nikam, R.P. Gude, R. Mulherkar, M.S. Nagarsenker, Apoptosis induction and anti-cancer activity of LeciPlex formulations. Cell. Oncol. 37, 339–351 (2014)
T. Nakaoka, A. Ota, T. Ono, S. Karnan, H. Konishi, A. Furuhashi, Y. Ohmura, Y. Yamada, Y. Hosokawa, Y. Kazaoka, Combined arsenic trioxide-cisplatin treatment enhances apoptosis in oral squamous cell carcinoma cells. Cell. Oncol. 37, 119–129 (2014)
O.N. Ikediobi, H. Davies, G. Bignell, S. Edkins, C. Stevens, S. O’Meara, T. Santarius, T. Avis, S. Barthorpe, L. Brackenbury, G. Buck, A. Butler, J. Clements, J. Cole, E. Dicks, S. Forbes, K. Gray, K. Halliday, R. Harrison, K. Hills, J. Hinton, C. Hunter, A. Jenkinson, D. Jones, V. Kosmidou, R. Lugg, A. Menzies, T. Mironenko, A. Parker, J. Perry, K. Raine, D. Richardson, R. Shepherd, A. Small, R. Smith, H. Solomon, P. Stephens, J. Teague, C. Tofts, J. Varian, T. Webb, S. West, S. Widaa, A. Yates, W. Reinhold, J.N. Weinstein, M.R. Stratton, P.A. Futreal, R. Wooster, Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol. Cancer Ther. 5, 2606–2612 (2006)
L.D. Dunfield, T.G. Shepherd, M.W. Nachtigal, Primary culture and mRNA analysis of human ovarian cells. Biol. Proced. Online. 4, 55–61 (2002)
E.L. Tobinick, The value of drug repositioning in the current pharmaceutical market. Drug News Perspect. 22, 119–125 (2009)
C. Campas, Drug repositioning summit: finding new routes to success. Drug News Perspect. 22, 126–128 (2009)
I. Ben Sahra, K. Laurent, A. Loubat, S. Giorgetti-Peraldi, P. Colosetti, P. Auberger, J.F. Tanti, Y. Le Marchand-Brustel, F. Bost, The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 27, 3576–3586 (2008)
M. Buzzai, R.G. Jones, R.K. Amaravadi, J.J. Lum, R.J. DeBerardinis, F. Zhao, B. Viollet, C.B. Thompson, Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 67, 6745–6752 (2007)
K. Kisfalvi, G. Eibl, J. Sinnett-Smith, E. Rozengurt, Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 69, 6539–6545 (2009)
A. Isakovic, L. Harhaji, D. Stevanovic, Z. Markovic, M. Sumarac-Dumanovic, V. Starcevic, D. Micic, V. Trajkovic, Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell. Mol. Life Sci. 64, 1290–1302 (2007)
Z. Yuan, K. Cao, C. Lin, L. Li, H.-y. Liu, X.-y. Zhao, L. Liu, H.-x. Deng, J. Li, C.-I. Nie, Y.-Q. Wei, The p53 upregulated modulator of apoptosis (PUMA) chemosensitizes intrinsically resistant ovarian cancer cells to cisplatin by lowering the threshold set by Bcl-x(L) and Mcl-1. Mol. Med. 17, 1262–1274 (2011)
Y. Zhuang, W.K. Miskimins, Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J. Mol. Signal. 3, 18 (2008)
H. Takane, E. Shikata, K. Otsubo, S. Higuchi, I. Ieiri, Polymorphism in human organic cation transporters and metformin action. Pharmacogenomics 9, 415–422 (2008)
L.-W. Wang, Z.-S. Li, D.-W. Zou, Z.-D. Jin, J. Gao, G.-M. Xu, Metformin induces apoptosis of pancreatic cancer cells. World J. Gastroenterol. 14, 7192–7198 (2008)
B. Martin-Castillo, A. Vazquez-Martin, C. Oliveras-Ferraros, J.A. Menendez, Metformin and cancer: doses, mechanisms and the dandelion and hormetic phenomena. Cell Cycle 9, 1057–1064 (2010)
M.R. Owen, E. Doran, A.P. Halestrap, Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348, 607–614 (2000)
Acknowledgments
We gratefully acknowledge all patients and IRCH without whose co-operation this study would have been impossible.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Patel, S., Kumar, L. & Singh, N. Metformin and epithelial ovarian cancer therapeutics. Cell Oncol. 38, 365–375 (2015). https://doi.org/10.1007/s13402-015-0235-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-015-0235-7