
Abstract
Polyurethanes (PUs) pose great potential in diverse applications, including the functionalization of fabrics, owing to their promising mechanical characteristics and biological compatibility. Here, a green synthesis of PUs has been carried out using the dispersion polymerization technique. The process includes the production of soybean oil-polyol through epoxidation, the preparation of PUs through the formation of polyurethane pre-polymer using an aromatic diisocyanate, i.e., toluene-2,4-diisocyanate, and different mole ratios of sodium alginate used as chain extender. Polyurethane dispersions were obtained by adding specific amount of distilled water. Further, structural attributes of soybean-sodium alginate-based waterborne polyurethanes (SO-SA-PUAro) were identified using an ATR-based Fourier Transform Infrared (FTIR) spectrophotometer, and physico-chemical properties like solid contents, physical appearance, and dispersion stability were observed and co-related with other outcomes. Moreover, synthesized polyurethane dispersions were applied to the plain weaved polyester-cotton blended textile substrate using pad-dry-cure methods. Significant gradual improvements in textile attributes like tensile strengths (from 873 N/m to 989 N/m on warp and 639 N/m to 729 N/m on warp and weft side of fabrics respectively), rubbing fastness (from 3 to 4/5 and 2 to 4 for dry and wet rubbing tests respectively), washing fastness (from 3–4 to 4–5), anti-pilling property (from 2–3 to 4–5), crease recovery angle (from 1.96 cm to 2.74 cm and 1.95 cm to 2.62 cm on warp and weft sides of the fabrics respectively), bending lengths (from 98° to 122° and 88° to 114° on warp and weft sides of the fabrics respectively), and anti-microbial tests (26 mm for E.coli and 27 mm for Cocci bacterial strains) were observed with increasing moles of sodium alginate as chain extender. However, a decreasing trend in tear strengths (from 12.89 lbf/in to 11.65 lbf/in and 10.12 lbf/in to 8.92 lbf/in on warp and weft sides of the fabrics) of the treated fabrics was noted. The outcomes of the study demonstrated that polyurethanes synthesized through bio-based polyol and chain extender have considerably enhanced the textile attributes of fabric and are good alternatives to replace petroleum-based raw materials.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Polyurethanes are one of the most attractive among synthetic polymers because of their ability to get molded, injected, extruded, and recycled according to requirements [1]. Therefore, these are interesting options for a variety of industrial applications [2, 3] such as textile coatings, floor coatings, foams, adhesives, etc. In [4,5,6,7,8,9], a very small quantity of organic solvent is involved in their synthesis process, i.e., approximately 3% by weight. Consequently, such a small quantity of organic solvent emissions is comparable to the emissions of organic solvents involved in powder coatings, i.e., 0.1 to 4% by weight [10]. Polyols are an important part of waterborne PUs and are generally extracted from non-renewable petroleum-based resources. Moreover, the non-degradability of these materials impart some environmental concerns [5, 11]. It negates the concepts of environmental sustainability, green development in society, and eco-friendliness. The development of sustainable products using renewable resources and replacements of petroleum-based raw materials is presently one of the primary issues being faced by the polymer industry. Hence, in recent years, vegetable oils have become an abundant renewable bio-based resource, which has drawn much attention because of their low price, diverse functional range, environment-friendly character, potential bio-degradability, and, most importantly, growing apprehension about the depletion of worldwide crude oil stocks. Bio-based polymers have various advantages over crude oil-based polyols because of several properties like abundance, ease of availability, natural renewability, and encouraging nature [9, 12,13,14].
Currently, vegetable oils (VO) are known as fascinating renewable sources for raw materials [3, 15] and can contribute toward diminishing unnatural climate change due to their biodegradability, inexhaustible accessibility, environmental friendliness, and affordable procedures [9, 16], which leads them to be considered an important natural raw material from an industrial point of view [17]. Vegetable oils for example linseed, sunflower, and soybean oils are some of the most significant sources for the synthesis of biopolymers [18]. Vegetable oils mainly contain triglyceride molecules from many plants (soybean, palm, rapeseed, etc.), where three hydroxyl functional groups present in glycerin are esterified with the fatty acids [19] and are among most significant sources of biopolymers [18].
Unsaturated plant oils contain mono, di, and triglyceride oils, which are very helpful in creating the products of invention [20]. In addition to vegetable oils, other bio-renewable sources of triglycerides like fish oil can be used to prepare polyurethane coatings [21]. These oils can be easily transformed to the respective polyols by one of the following methods:
-
The straight oxidation of vegetable oil via epoxidation of olefins and then ring-opening substitution.
-
The hydro-formylation /reduction of double bonds present between two adjacent carbon atoms in the fatty acid backbone [19, 20].
-
Ozonolysis / reduction of the carbon—carbon double bonds, resulting in primary and short alcohols [19, 22, 23].
-
Trans-esterification/amidation of triglycerides with glycols or diethanolamine [24].
Various polyols, having different molecular structure, position and number of hydroxyl functional groups, can be obtained using different synthesis approaches and the structure of triglyceride. Soybean oil (SO) is mainly consumed as cooking oil and is also used to synthesize printing inks, paints, dispersions, and so forth. Among other vegetables oils, SO has attracted significant industrial interest since it can be easily customizable to get desired products [25].
Alginates are polysaccharide carbohydrates. These are extracted from brown seaweeds and consist of α-l-guluronic acids and β-d-mannuronic acids. Alginates are a well-recognized, easily available source of a natural polymer with good biodegradability and low cost [26, 27]. Alginates are polysaccharide carbohydrates. These are extracted from brown seaweeds and consist of α-l-guluronic acids and β-d-mannuronic acids. Alginates are a well-recognized, easily available source of a natural polymer with good biodegradability and low cost [28]. Alginates, being non-toxic, are used in fields such as medicine, food packaging, dietology, biomedicine, and the pharmaceutical industry [29]. Besides, these can easily be incorporated into other synthetic polymers to develop other polymeric materials with properties of choice [30]. The hydrophilic groups present in alginates provide good miscibility among waterborne PUs and natural polysaccharides. Therefore, it is very easy to synthesize alginate-based PUs and their polymeric hydrogels, wherein physical cross-link networks can effortlessly be broken and recovered. Further, blends of PUs with natural polysaccharides show fair biodegradability [31, 32].
The alginate-based polyurethane are also utilized to devise pH and temperature-responsive compositions with variable viscosities to get the required thickness of water-based polyurethane [33] aiming to get good mechanical strength and thermo-stability [34]. Alginates have received a lot of attention for their important uses as thickening and gelling agents in the textile and food industries, film coaters, stabilizers, etc. Naturally, in various water bodies, many alginates are found abundantly, comprising exceptional chelating properties, fair biocompatibility, and biodegradability [35]. Possessing these attributes, alginates can be found in many textile applications. These textile items have a vital use in hospitals as pathogens have the ability to adhere to the surface of the fabrics [36].
In this research work, the green synthesis of polyurethane dispersions (PUDs) has been realized by using SO-based polyols as an alternative to petrochemicals. The PUDs were functionalized with sodium alginate to induce bioactivity. Textile fabrics were functionalized using the bio-active PUDs to induce the same attributes in fabrics. The finished fabrics showed excellent fastness characteristics and also exhibited potential as a good alternative to conventional, non-biodegradable, and formaldehyde-based carcinogenic fixing agents.
2 Materials and Synthesis
Toluene-2,4-diisocyanate (TDI) was purchased from Alfa Aesar, Thermofisher, Germany. 1,4-butanediol (BDO), sodium alginate (SA), acetic acid (AA), triethylamine (TEA), dimethylol propionic acid (DMPA), and di-butyltin di-laurate (DBTDL) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Soybean oil having a molecular weight of 1100 g/mol and an OH value of 250 mg KOH/g of SO was supplied by Azhar Corporation Pvt. Ltd., Faisalabad, Punjab, Pakistan. DMPA was dried at 80 °C in a vacuum overnight before use to ensure the removal of water vapors and avoid interaction during isocyanate reactions. For confirmation of the molecular weight of polyol, the ASTM D-4274C method was used. Further, analytical grade reagents were utilized in the study and were used as received. Crescent Textile Mills, Ltd., Faisalabad, Pakistan, provided polyester-cotton textile substrate (having a 48/52 polyester-cotton blend ratio) that was mill de-sized, scoured, bleached, and plain woven. The features related to the construction of fabric were 76 × 56 (the number of threads used in the warp and weft directions in one inch area of fabric) and 30 × 30 (count of threads). Before being subjected to the waterborne polyurethane dispersions, fabric was decontaminated by washing it at 100 °C for 60 min with a solution containing 1 g/L of a non-ionic surfactant (Triton-X-100, a product of BASF) and 2 g/L of Na2CO3. After that, 20 g of the fabric was thoroughly rinsed with hot and cold water at a 1:30 w/v ratio with a hardness > 8 mg/L, followed by drying at ambient conditions, i.e., 37 °C in temperature and 40% in humidity index.
2.1 Synthesis of SO-SA-PUAro
The soybean oil-sodium alginate-based polyurethanes (SO-SA-PUAro) were synthesized by varying mole ratios of SA, while the amounts of remaining monomers were kept constant in terms of molar ratios.
2.1.1 Synthesis of Soybean Oil-Polyol (SO-PO)
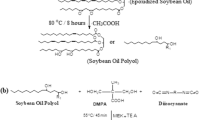
The raw SO was utilized after a few suitable modifications as an obligatory requirement to make the carbon–carbon unsaturated bonds present in fatty acids react with other molecules to get cross-linking after curing for thermosetting of the final polymer. Figure 1 shows the scheme in which the process of epoxidation of SO is illustrated. The type of polymerization mainly determines the degree of unsaturation of fatty acids [37]. Conversion of SO into polyol was carried out through the process of formic acid-based epoxidation assisted by hydrogen peroxide to generate performic acid, followed by treatment with butanol, hydrochloric acid, methanol, or acetic acid. For this purpose, 250 g vegetable oil, and 85–100 g hydrogen peroxide (30%) were mixed with 38 g of formic acid (88%) in a round bottom flask attached to the magnetic stirrer and obtained material was subjected to continuous stirring at 25 °C for 21 h on a water bath to form epoxidized soybean oil (ESO). Ethyl acetate and 0.5 M aqueous sodium chloride solution were used to wash the ESO which was filtered with the help of vacuum filtration and subjected to a rotary evaporator at 38 °C for 1 h, followed by drying at 70 °C in the oven for overnight [38].

Process of epoxidation of SO
Figure 2 represents the step in which ESO was converted into SO-PO. For this purpose, acetic acid (AA) was used to convert ESO into the respective polyol (SO-PO) by opening oxirane rings. Carefully weighed 120 g of acetic acid and 40 g of ESO were poured into a double-necked reaction flask fitted with a reflux condenser to complete the reaction for 8 h at 80 °C with continuous stirring. The workup followed the procedures as described for methoxylated polyols [39].

Conversion of epoxidized soybean oil into the polyol
2.1.2 Synthesis of Waterborne Polyurethane Dispersions
Figure 3 demonstrates the reaction in which, synthesis of polyurethane pre-polymer is defined. First of all, a polyurethane pre-polymer based on dimethylol propionic acid (DMPA), SO, and a diisocyanate was prepared as per the previously reported method [11]. For this purpose, SO as a polyol, dimethylol propionic acid (DMPA), and toluene diisocyanate were poured into a reaction flask equipped with a reflux condenser with a nitrogen gas inlet and outlet system. The temperature was carefully monitored on a thermometer, and stirring was also carried out using a mechanical stirrer. The reaction was conducted at 90 °C for 45 min with strong stirring in the presence of a catalyst (1 to 2 drops of DBTDL). The temperature of the reaction mixture was lower up to 70–80 °C for the next 2 h with continuous stirring to synthesize the hydrophilic –NCO terminated SO -based polyurethane pre-polymer. A little amount of MEK was also introduced to decrease the viscosity of the prepared PU pre-polymer. After that, neutralization of –COOH group was performed with triethylamine (TEA) at 55 °C for 45 min. to neutralize carboxylate groups present in SO-PO molecules.

Synthesis of NCO- terminated SO-based pre-polymer of polyurethane
Figure 3 identifies the synthesis of -NCO terminated SO-based pre-polymer of polyurethane. After the completion of the synthesis of polyurethane pre-polymer, chain extension step was carried out at 25 °C for 30 min with sodium alginate and 1,4-butanediol to react with the remaining isocyanates of polyurethane pre-polymer by constant stirring. After that, the required quantity of distilled water was introduced dropwise in the presence of a dispersing agent. The dispersion was prepared at room temperature for 1 h, and various dispersions were created by varying mole ratios of sodium alginate and 1,4-butanediol. The preparation of water born polyurethane has been given in Fig. 4.
In view of above-mentioned scheme, a series of bio-based PUDs were produced by varying the mole ratios of sodium alginate and 1,4-butane diol as chain extenders using toluene diisocyanate. The formulations of different PUDs are illustrated in Table 1.
The OH concentration include OH from SO polyol (functionality/OH value: 5), sodium alginate (functionality/OH value: 02), and DMPA (functionality/OH value: 02) as per their functionalities and stoichiometric balance.
2.2 Characterization and Evaluation of PUDs
Physical characterization like molecular characterization with FTIR, dispersion stability, solid contents, and physical appearances were recorded to correlate with other properties.
2.2.1 Molecular Characterization
Molecular scans of the different steps associated with the synthesis of PUDs were obtained with the help of ATR-based Fourier Transform Infrared (FTIR) spectroscopy in transmission mode [40] using Bruker, Alpha-compact-II FT-IR spectrometer (Ettlingen, Germany) in the region of 400–4,000 cm−1 wavenumber and each scan is average of 26 repeated measurements at a resolution of 4 cm−1.
2.2.2 Solid Contents
Samples were heated at 80 °C for the measurement of the solid contents of SO-SA-PUAro. The extra water and organic solvents were removed at this temperature. The difference in weights before and after heating will determine the weight of the sample. After completion of the drying process, the solid contents were calculated by following the mathematical relation [41].
where,
x = weight (mg) of empty cup of aluminum foil,
y = weight (mg) of aluminum foil and sample
z = weight (mg) of aluminum foil and dried sample.
2.3 Treatment of Fabrics with SO-SA-PUAro
The plain weaved polyester-cotton blended dyed fabrics were treated with a dilution of 20 g/L (2%) and 40 g/L (4%) of SO-SA-PUAro using the pad-dry-cure technique. The treated samples were dried at 100 °C for 3 min followed by a curing process at 140 °C for 5 min to obtain a uniform polymeric polyurethane network on the fabric surface with the help of a laboratory scale stenter machine.
2.4 Textile Performance
All the prepared SO-SA-PUAro dispersions were applied to dyed textile fabrics, i.e., polyester-cotton fabrics, for evaluation of textile performances following the reported methods [42, 43] i.e., tear strengths (ASTM standard method No. D-1424/BS EN ISO 13937–2), the tensile strengths (ISO No. 13934–2), rubbing fastness (ISO-105-X12), washing fastness (ISO-105-C06) and, pilling testing (ISO 12945–2).
2.5 Evaluation of Anti-microbial Activities
For the evaluation of antimicrobial performance, two different concentrations of the synthesized PUDs were prepared, i.e., 2% and 4%, and then applied to the fabrics as an antibacterial finish using TDI as aromatic diisocyanate. All the treated fabric samples were incubated at 37 °C for 24 h in an agar medium. The bacterial growth was analyzed by measuring zones of inhibition. Antimicrobial activities of different SO-SA-PUAro treated fabrics were determined by the agar well diffusion method [43, 44] against two bacterial strains gram-positive (Cocci) and gram-negative (E.coli).
2.6 Statistical Analysis of Results
Each test was performed in triplicate in order to ensure repeatability and reproducibility of the results. The results of different tests performed during the study were analyzed with the help of an analysis of variance (ANOVA) test using XLSTAT software. The results were presented as the mean + standard deviations. The mean values of the results were compared using the Tukey test, and the obtained values are considered statistically significant with (p ≤ 0.05).

Synthesis of waterborne SO-SA-PUAro dispersion
3 Results and Discussion
3.1 FTIR Characterization
The FT-IR scans of all the monomers and products of various steps involved during synthesis of SO-SA-PUAro are illustrated in the Fig. 5. The FTIR Spectrum of SO displayed the characteristic peaks at 3011.57 cm−1 (C=C) [40], 723.95 cm−1 (methylene groups in-phase rocking), 957.64 cm−1, 1017.38 cm−1, 1105.54 cm−1 (ether groups, anti-symmetric stretching), 1163.21 cm−1, 1239.97 cm−1 (ester groups, anti-symmetric stretch), 1378.62 cm−1 (methyl groups symmetric deformation), 1463.85 cm−1 (methyl groups anti-symmetric deformation) and 1745.65 cm−1 (esters groups, aliphatic C–O stretch) [45] as indicated in Fig. 5a. After conversion from SO into respective epoxide, the characteristic band of epoxy group was noted (doublet peaks) at 818.39 cm−1 and 823.58 cm−1 [45,46,47,48] (Fig. 5b). The epoxide of SO was then converted into polyols and it was well indicated in the FT-IR spectrum as C=C double bond band at 3011.57 cm–1 disappeared, and presence of aldehyde bands at 2,701.17 cm–1 (broad, weak) and 1728.36 cm–1 (shoulder, strong) as referred in the Fig. 5c. These bands disappeared after complete hydrogenation of epoxide of SO and a wide band related to the hydroxyl group emerged at 3462.16 cm−1,which is clear evidence of the formation of a polyol of SO [48, 49].

FTIRs of a SO b ESO c SO-PO d dimethylol propionic acid e toluene diisocyanate f polyurethane pre-polymer g sodium alginate h SO-SA-PUAro
FT-IR analysis of polyurethane pre-polymer showed a characteristic peak around 2267.46 cm−1 that was conforming to –N=C=O groups incorporated on terminals of polyurethane pre-polymer as shown in Fig. 5d [50]. Further, vanishing of bands owing to –OH groups that were formerly positioned in FT-IR spectra of soybean oil polyol at 3462.16 cm−1 and in DMPA at 3559.56 cm−1 indicated that these groups were effectively utilized during the synthesis of PU pre-polymer and emergence of a tiny peak around 3328.36 cm−1 representing -NH stretching vibrations band was a clear evidence of initiation of urethane linkage [32]. Other very significant peak appeared from 1653.61 to 1705.95 cm−1was due to stretching of C=O in ester and urethane linkages (-NHCOO-), peaks from 2930.37 to 2870.48 cm−1 were related to the C−H (methylene) stretching and the C − H (methylene) symmetrical bending at 1448.13 cm−1. The C−O stretching vibrations were shown in a peak at 1019.47 cm−1 in FT-IR spectrum of PU pre-polymer (refer to the Fig. 5f) [51].
The Fig. 5h demonstrates that the peak previously observed at 2267.65 cm−1 due to –NCO group in polyurethane pre-polymer has been consumed during the formation of final PU by emergence of a band around 3337.65 cm−1 which represents -NH groups in urethane linkages and a crest at 1531.84 cm−1 was attributed to NHCO bonds [43, 52]. Further, the spikes from 1500 cm−1 to 1600 cm−1 were attributed to the C=C stretching of aromatic rings present in TDI and two clear absorption vibration at 2854.79–2927.15 cm−1 were due to stretching of C–H bond in-CH2 and -CH groups.
A prominent band observed at 1711.83 cm−1 was assigned to carbonyl (–C=O) groups present in urethane and ester linkages. The absorptions around 1240.18 cm−1and 1051.67 cm−1 were noted because of the symmetric stretching of C–O and C–O–C functional groups in the final PUs.
3.2 Physical Appearance of PUDs
The physical appearances of all the samples were noted carefully, and a gradual change in color from translucent yellow to translucent brown was observed. The color or appearance of the PUDs having 0 mol of sodium alginate was found to be translucent light yellow, whereas the appearances of PUDs with 0.25 and 0.5 mol of SA were found to be translucent dark yellow and translucent brownish yellow, respectively, whereas the PUDs having 0.75 mol and 1 mol of SA were brownish in color. This trend was due to the gradual increase in the contents of sodium alginate in PUDs [42, 43]. All the samples were not totally transparent in appearance, and the transparency of PUDs was found to decrease with increasing mole ratios of SA [42]. The results can be verified in Table 2 (column 3).
3.3 Stability of PUDs
Investigations for the stability of synthesized polyurethane dispersions were also carried out, and the synthesized samples were found stable due to the presence of DMPA, which acted as an internal emulsifier and played a key role by aiding the formation of smooth and uniform dispersions due to -COOH groups, and TEA, which neutralized the carboxylic groups in the PUs [53]. The synthesized samples of polyurethane dispersions were placed in an incubator without distressing at standard temperature and pressure to investigate their stability, and all the dispersions were found to be stable for more than 12 months [42, 44, 54] as mentioned in Table 2 (column 4).
3.4 Solid Contents of PUDs
Dry mass or solid contents of the synthesized PUDs were carefully noted and found to increase gradually from 32.3 to 41.6% with increasing the mole ratio of SA and solid contents during the progression of PUDs. Since the molecular weight (MW) of SA is greater than that of BDO, the increment in dry mass might be due to the increase in SA contents and decrease in mole ratios of BDO as chain extenders, which resulted in a gradual increment in the solid contents on increasing mole ratios of SA from 0 to 1 mol [55] as shown in Table 2 (column 5).
3.5 Tear Strength of the Treated Fabrics
The evaluation of tear strengths of treated fabrics was carried out according to the ASTM standard method No. D-1424/BS EN ISO 13937–2 [42], and results of the measurements are reported for untreated fabrics and those treated with synthesized polyurethane (SO-SA-PUAro) with dispersion concentrations of 20 g/L and 40 g/L, respectively. The PUDs were applied to plain-weaved polyester-cotton blended fabrics. The results thereof were recorded carefully, compared, and presented in Fig. 5. As a general assessment, on increasing the concentration of dispersion, a decline in tear strengths on warp and weft sides of fabrics was observed. It was also noted that on increasing the molar concentrations of sodium alginate from 0 to 1 mol in PUD, the tear strengths decreased from 12.89 to 11.65 lbf/in on the warp and 10.12–8.92 lbf/in on the weft sides of the fabrics treated with dispersion of 20 g/L strength. When the fabric samples were treated with the strength of 40 g/L of PU dispersions, the same trend was observed, i.e., the tear strengths decreased progressively from 11.29 to 10.11 lbf/in on the warp and 9.51–8.39 lbf/in on the weft sides of the fabrics. The detailed results are presented in Fig. 6.

Evaluation of tear strength along a warp and b weft of fabrics treated with PUDs
The trend of decrease in tear strengths on the warp and weft of treated fabrics may be due to the higher content of the chain extender and its interaction with fibers. As the dispersion is applied to the fabric, it binds the fibers together firmly due to various physical interactions like hydrogen bonding, van der Waals forces, etc. that hinders the sliding movements of fibers of the fabric over each other [43]. For the dispersion to be beneficial for the tear strength, it should not decrease the flexibility of the yarn and should enhance the sliding movement of the fibers. Besides, factors hindering the sliding movement of the fibers over each other decrease the tear strength. Due to this reason, the tear strengths of treated fabric decreased as compared to untreated fabric. Therefore, sliding movement decreased with increasing the contents of SA in the sample, which ultimately resulted in decreasing the tear strengths [56].
3.6 Tensile Strength of Treated Fabrics
Tensile strengths of untreated and treated textile fabrics were measured following the standard method ISO No. 13934–2 [57] and are presented in Fig. 7. The values of the tensile strengths (N/m) of the fabric treated with SO-SA-PUAro were found in increasing order. It was further observed that on increasing the mole ratio of SA in PUDs, the value of tensile strength also increased, and the same trend was further noted on increasing the mole contents of SA (1 mol) in PUDs, i.e., from 873.77 + 0.01 N/m to 989.94 + 0.03 N/m on warp and 639.32 + 0.03 N/m to 720.85 + 0.01 N/m on weft sides of fabrics using 20 g/L of dispersion dilution with the maximum number of moles of SA (1 mol). When using 40 g/L of dispersion dilution, the same trend was noticed, i.e., from 986.93 + 0.02 N/m to 1158.68 + 0.02 N/m on the warp and from 729.58 + 0.02 N/m to 809.36 + 0.01 N/m on the weft sides of fabrics when the maximum molar concentration of SA (1 mol) was used in PUD.

Tensile strengths along a warp and b weft of polyurethane treated fabrics treated
From different studies, it has been observed that there are many reasons that may affect the outcomes of tensile strengths, like the adherence of PU to the fabric, the level of cross-linking, intermolecular forces, hard and soft segments of PU, and the concentration of PUDs applied to the fabric [46, 58]. All these features remained in increasing order when mole ratios of SA were increased in the PUs, strengthening bonding between fabric and dispersion coating [49]. The hydrogen bonding and van der Waals forces enhance the formation of cross-linking of PUs with fabric and enable them to bind to the fabric firmly. These factors collectively increased the tensile strength of fabrics[59].
3.7 Crease Resistance of Treated Fabrics
The crease recovery angle of the treated fabrics was evaluated with the PUDs prepared at different conditions. It has been observed that the crease recovery angle of fabrics increases both along the warp and weft upon treatment with polyurethane, as illustrated in Fig. 8. Polyurethane generates cross-linking with the hydroxyl groups of cellulose through hydrogen bonding. Hydroxyl groups in cellulose are responsible for creasing in fabrics. PUDs limit the availability of hydroxyl groups for creasing phenomenon. Thus, the crease recovery angle of fabric increases.

Crease recovery angle along a warp and b weft of polyurethane treated fabrics
3.8 Bending Length of Treated Fabrics
Bending length is the indicator of fabric softness and stiffness. Bending length of fabric was evaluated after treatment with PUDs. Bending length of the treated fabrics, both along warp and weft, is higher as compared to untreated fabric. Furthermore, it increases with the increase in SA (Fig. 9). Polyurethane binds with the yarns of fabric to decrease their mobility. This phenomenon increases the fabric stiffness.

Bending lengths along a warp and b weft of polyurethane treated fabrics
3.9 Evaluation of Color Fastness Properties of Treated Fabrics
The resistance of a textile article to resist the color change when it is passed through the various testing procedure is called its "fastness properties" [60, 61]. To evaluate the influence of different synthesized PUDs on the textile substrate, different textile testing procedures were employed i.e. ISO-105-CO6 (for washing fastness), ISO 105 X12 for (rubbing fastness), and results were compared with untreated fabrics (Table 3).
3.9.1 Rubbing Fastness of Treated Fabrics
"Crocking or rubbing fastness" is a well-established industrial parameter that refers to the transfer of color from the fabric to another substrate through rubbing. The assessment of the rubbing test describes the resistance offered by the color of the textile item when getting rubbed with other materials, i.e., furniture, textile products, miscellaneous items, etc. The comparison of dry and wet rubbing fastness properties of both treated and untreated fabric samples is depicted in Table 3. The ratings of the tested fabrics revealed that treatment of PUDs has a prominent impact on the rubbing fastness characteristics of the treated fabric when compared to the untreated ones. Untreated fabric showed dry and wet rubbing test ratings of 3 and 2, respectively, while all the fabrics treated with PUDs with a dilution of 20 g/L displayed dry rubbing ratings from 3/4 to 4/5, whereas wet rubbing test ratings were found ranging from 2/3 to 3/4. A similar trend was observed when 40 g/L dilution PUDs were used, i.e., dry rubbing ratings were found from 3/4 to 4/5, whereas, wet rubbing test ratings were observed from 2/3 to 4. From the results, it appears that the PU coating significantly enhanced the resistance to rubbing impacts.
3.9.2 Washing Fastness of Treated Fabrics
Colorfastness during the washing of clothes is a major concern for the textile industry. The color of a textile item must withstand repeated washings throughout its existence without bleeding or staining when washed with other clothes. Washing with detergent determines the strength of textile colors in` commercial or domestic laundering dealings. The two accepted standard procedures for washing textile articles with detergent are ISO-105-C06 or AATCC 61. The ISO-105-C06 testing method was used, wherein fabric was tested at 40 °C with a detergent. The washing fastness of all fabrics treated with PUDs with dilutions of 20 g/L and 40 g/L of dilution displayed good ratings, i.e., 4–4/5 and 4–5, respectively, while the rating of untreated fabric was found to be 3/4.
It is crystal clear from the outcomes depicted in Table 3 that all the treated fabric samples have shown fair to excellent fastness properties when compared with untreated fabrics. Further, it has also been observed that on increasing the mole ratios of SA in the PU, the fastness attributes of fabrics improved gradually. This may be attributed to the formation of a continuous and tough coating of polymer layer on treated fabrics because, with increasing contents of SA in PUDs, molecular weight and polar sites in PUs increased. Consequently, the coating of PU on the surface of the fabric becomes denser, and the interaction between polar groups in the polyurethane polymer and the fabric is strengthened. This outcome is very appealing because it displays the correlation of structural properties. It appears that on increasing the SA contents in the polyurethane, the number of polar groups, i.e., carboxylic groups (-COO) and hydroxyl groups (-OH), increased in the polymer chain. This provides the maximum opportunity for inter-chain and polymer to substrate interactions that provides the better fastness properties [23, 43]. Hence, the amalgamation of both of these reasons ultimately resulted in better resistance to color fastness.
3.10 Pilling Resistance of Treated Fabrics
Pilling appears due to the unwanted filaments protruding from the surface of the fabric. It is a very handy tool to indicate the quality of fabric and a key point of concern for the end-user as it disturbs the quality of textile items and consequently decreases their worth. The industrialist always pays keen attention and takes remedial measures to reduce this factor because fabric appears fuzzy and rough with a high rating of pilling. Pilling appears to be a combined result of the development of pills at diverse points on the surface of fabrics [62]. Likewise, the size, height, and thickness of the pills were determined for rating the pilling strength. For the determination of pills, ASTM testing method No. D-3514–02 was adopted [44].
The gradual and fair improvement in pilling resistance was recorded parallel to the increment in the contents of SA in the PUs, i.e., from 3/4 to 4/5 when fabric samples were treated with PUDs dispersions of 20 g/L and from 3/4 to 5 when fabric samples were treated with PUDs dispersions of 40 g/L, as depicted in Table 3. This may be attributed to the development formation of a persistent and tough coating of polymer layers on the treated fabrics because, with increasing contents of SA, molecular weight and polar sites in PUs increased. Consequently, the coating of PU on the surface of the fabric becomes denser, and interaction between polar groups in polyurethane polymer efficiently entangles the protruding filament on the surface of the fabric through hydrogen bonding and other inter-molecular interactions to produce a smooth and intact layer of polymer network on the surface of the fabric by sticking the yarns together tightly. Consequently, a maximum rating of "5" was observed when maximum mole ratios of SA were used in the PU [44].
3.10.1 Evaluation of the Anti-microbial Activity of Treated Fabrics
Very distinguishable anti-microbial activities of the treated samples were noticed against both strains of the bacteria, i.e., gram-positive (Cocci) and gram-negative (E.coli) [63]. The sample treated with SO-SA0-PUAro exhibited the least inhibition zone, i.e., 26 mm for E.coli bacteria and 27 mm for Cocci bacteria using 20 g/L solution, and with 40 g/L solutions, a bacterial growth inhibition zone of 28 mm for both E.coli and Cocci was recorded. The sample treated with SO-SA0.25-PUAro showed increments in the bacterial inhibition zone, i.e., 31 mm for E.coli bacteria and 32 mm for Cocci bacteria using a 20 g/L dilution, and for a 40 g/L PUD dilution, a bacterial growth inhibition zone of 34 mm for E.coli, 33 mm for Cocci were observed. The inhibition zone for bacterial growth improved upon increasing the contents of SA in the PUs. As a result, the highest bacterial growth inhibition zones were recorded when PUD with maximum contents of SA was used, i.e., 40 mm for E.coli, 39 mm for Cocci bacteria using 20 g/L of dilution, and for 40 g/L of PUD dilution, a bacterial growth inhibition zone of 42 mm for E.coli, 43 mm for Cocci as shown by the data presented in Fig. 10. In light of the above discussion, it is confirmed that the occurrence of hydroxyl groups in the PUs plays a key role in the antimicrobial activity because, as the contents of SA in the PUs increased, a noticeable increment against bacterial growth in the treated fabric samples was observed. Consequently, the highest antibacterial activity was recorded when a sample was treated with PU having maximum contents of SA (SO-SA1-PUAro [42]. Therefore, it is suggested that SO-SA-PUAro are promising candidates for antibacterial textile coatings.

Antibacterial properties of PUDs against gram negative (E.coli) and gram positive (Cocci) bacterial strains
4 Conclusion
This study demonstrates the synthesis of water-based PUDs using renewable SO polyols. The polyurethane pre-polymer was functionalized with SA to impart bioactive properties. The prepared compounds exhibited excellent dispersal stability even after being stored for over 12 months, indicating their potential for commercial applications. Fabrics treated with SO-SA-PUAro displayed promising antibacterial activity against E.coli and Cocci, making them suitable for healthcare applications. Increasing the SA content resulted in improved tensile strength and pilling resistance due to enhanced binding of fibers and yarn, while tear strength decreased due to increased fabric stiffness. Additionally, the color fastness to washing and rubbing increased with higher SA content, as the SO-SA-PUAro formed a protective coating on the fabric surface. This development offers an eco-friendly alternative to petrochemical-based polyols for PUD synthesis.
Change history
16 November 2023
A Correction to this paper has been published: https://doi.org/10.1007/s13369-023-08424-1
References
Zia, K.M.; Bhatti, H.N.; Bhatti, I.A.: A review of Methods for polyurethane and polyurethane composites, recycling and recovery. React. funct. polym. 67(8), 675–692 (2007)
Kaur, R.; Kumar, M.: Function of silicon oil in the castor oil based rigid polyurethane foams. J. Polym. Eng. 33(9), 857–861 (2013)
Aggarwal, A.; Kaur, R.; Walia, R.S.: PU foam derived from renewable sources: perspective on properties enhancement: an overview. Eu. Polym. J. 95, 255–274 (2017)
Akindoyo, J.O.; Beg, M.D.; Ghazali, S.; Islam, M.; Jeyaratnam, N.; Yuvaraj, A.: Polyurethane types, synthesis and applications–a review. Rsc. Advs. 6(115), 114453–114482 (2016)
Howarth, G.: Polyurethanes, polyurethane dispersions and polyureas: Past, present and future. Surf. Coat. Int. Part B Coat. Trans. 86(2), 111–118 (2003)
Meng, Q.B.; Lee, S.-I.; Nah, C.; Lee, Y.S.: Preparation of waterborne polyurethanes using an amphiphilic diol for breathable waterproof textile coatings. Prog. Org. Coat. 66(4), 382–386 (2009)
Orgilés-Calpena, E.; Arán-Aís, F.; Torró-Palau, A.M.; Orgilés-Barceló, C.; Martín-Martínez, J.M.: Effect of annealing on the properties of waterborne polyurethane adhesive containing urethane-based thickener. Int. J. Adhes. Adhes. 29(8), 774–780 (2009)
Agrawal, A.; Kaur, R.; Walia, R.: Development of vegetable oil-based conducting rigid PU foam. E-Polym 19(1), 411–420 (2019)
Kaur, R.; Kumar, M.: Function of silicon oil in the castor oil based rigid polyurethane foams. J. Polym. Eng. 33(9), 875–880 (2013)
Garrison, T. F.: (2013). Synthesis and characterization of vegetable oil-based polyurethane dispersions. In Digit. Repos. Iowa State University
Madbouly, S.A.; Otaigbe, J.U.: Recent advances in synthesis, characterization and rheological properties of polyurethanes and POSS/polyurethane nanocomposites dispersions and films. Prog. in Polym. Sci. 34(12), 1283–1332 (2009)
Bullermann, J.; Friebel, S.; Salthammer, T.; Spohnholz, R.: Novel polyurethane dispersions based on renewable raw materials—Stability studies by variations of DMPA content and degree of neutralisation. Prog. Org. Coat. 76(4), 609–615 (2013)
Gurunathan, T.; Arukula, R.: High performance polyurethane dispersion synthesized from plant oil renewable resources: a challenge in the green materials. Polym. Degrad. Stab. 150, 122–132 (2018)
Liang, B.; Li, R.; Zhang, C.; Yang, Z.; Yuan, T.: Synthesis and characterization of a novel tri-functional bio-based methacrylate pre-polymer from castor oil and its application in UV-curable coatings. Ind. Crops. Prod. 135, 170–178 (2019)
Moreno, V.C.; Russo, V.; Tesser, R.; Di Serio, M.; Salzano, E.: Thermal risk in semi-batch reactors: the epoxidation of soybean oil. Process Saf. Environ. Prot. 109, 529–537 (2017)
Chavan, V.P.; Patwardhan, A.V.; Gogate, P.R.: Intensification of epoxidation of soybean oil using sonochemical reactors. Chem. Eng. Process 54, 22–28 (2012)
de Souza, V.H.R.; Silva, S.A.; Ramos, L.P.; Zawadzki, S.F.: Synthesis and characterization of polyols derived from corn oil by epoxidation and ozonolysis. J. Am. Chem. Soc. 89(9), 1723–1731 (2012)
Nagy, T.; Róth, G.; Kuki, Á.; Zsuga, M.; Kéki, S.: Mass spectral filtering by mass-remainder analysis (MARA) at high resolution and its application to metabolite profiling of flavonoids. Int. J. Mol. 22(2), 864 (2021)
Desroches, M.; Escouvois, M.; Auvergne, R.; Caillol, S.; Boutevin, B.: From vegetable oils to polyurethanes: synthetic routes to polyols and main industrial products. Polym. Rev. 52(1), 38–79 (2012)
Khan, M. L.; Tomkinson, J.; Fitchett, C. S.; Black, M. J.: Oxidative cleavage of unsaturated oils and products obtained therefrom. In: Google Patents (2004)
Athawale, V.D.; Nimbalkar, R.V.: Polyurethane dispersions based on sardine fish oil, soybean oil, & their interesterification products. J. Dispers. Sci. Technol. 32(7), 1014–1022 (2011)
Petrović, Z.S.: Polyurethanes from vegetable oils. Polym. Rev. 48(1), 109–155 (2008)
Petrović, Z.S.; Zhang, W.; Javni, I.: Structure and properties of polyurethanes prepared from triglyceride polyols by ozonolysis. Biomacromol 6(2), 713–719 (2005)
Lyon, C.; Garrett, V.H.; Goldblatt, L.A.: Solvent-blown rigid urethane foams from castor-based polyols. J. Am. Chem. Soc. 38(5), 262–266 (1961)
Ivanov, D.S.; Lević, J.D.; Sredanović, S.A.: Fatty acid composition of various soybean products. Food Res. Int. 37(2), 65–70 (2010)
Comaposada, J.; Gou, P.; Marcos, B.; Arnau, J.: Physical properties of sodium alginate solutions and edible wet calcium alginate coatings. Food Sci. Technol. 64(1), 212–219 (2015)
Wallen, L. L.: Type reactions in fermentation chemistry (Vol. 71): US Government Printing Office (1959)
Pawar, S.N.; Edgar, K.J.: Alginate derivatization: a review of chemistry, properties and applications. Biomaterials 33(11), 3279–3305 (2012)
Sellimi, S.; Younes, I.; Ayed, H.B.; Maalej, H.; Montero, V.; Rinaudo, M.; Nasri, M.: Structural, physicochemical and antioxidant properties of sodium alginate isolated from a Tunisian brown seaweed. Int. J. Biol. Macromol. 72, 1358–1367 (2015)
Zhou, X.; Li, Y.; Fang, C.; Li, S.; Cheng, Y.; Lei, W.; Meng, X.: Recent advances in synthesis of waterborne polyurethane and their application in water-based ink: a review. J. Mater. Sci. Technol. 31(7), 708–722 (2015)
Travinskaya, T.; Savelyev, Y.V.: Aqueous polyurethane–alginate compositions: Peculiarities of behavior and performance. Eur. Polym. J. 42(2), 388–394 (2006)
Zia, K.M.; Zia, F.; Zuber, M.; Rehman, S.; Ahmad, M.N.: Alginate based polyurethanes: A review of recent advances and perspective. Int. J. Biol. Macromol. 79, 377–387 (2015)
Travinskaya, T.; Savelyev, Y.; Mishchuk, E.: Waterborne polyurethane based starch containing materials: preparation, properties and study of degradability. Polym. Degrad. Stab. 101, 102–108 (2014)
Solanki, A.; Das, M.; Thakore, S.: A review on carbohydrate embedded polyurethanes: An emerging area in the scope of biomedical applications. Carbohydr. Polym. 181, 1003–1016 (2018)
Mørch, ÝA.; Donati, I.; Strand, B.L.; Skjåk-Bræk, G.: Effect of Ca2+, Ba2+, and Sr2+ on alginate microbeads. Biomacromol 7(5), 1471–1480 (2006)
Sun, G.: Antimicrobial finishes for improving the durability and longevity of fabric structures. In Antimicrobial Textiles (pp. 319–336). Elsevier(2016)
Adekunle, K.: A review of vegetable oil-based polymers: synthesis and applications. Open J. Polym. Chem. 5, 34–40 (2015)
Clark, A.J.; Hoong, S.S.: Copolymers of tetrahydrofuran and epoxidized vegetable oils: application to elastomeric polyurethanes. Polym. 5(9), 3238–3244 (2014)
Garrison, T.F.; Kessler, M.R.; Larock, R.C.: Effects of unsaturation and different ring-opening methods on the properties of vegetable oil-based polyurethane coatings. Polym. 55(4), 1004–1011 (2014)
Uprety, B.K.; Reddy, J.V.; Dalli, S.S.; Rakshit, S.K.: Utilization of microbial oil obtained from crude glycerol for the production of polyol and its subsequent conversion to polyurethane foams. Bioresour. Technol. 235, 309–315 (2017)
Mumtaz, F.; Zuber, M.; Zia, K.M.; Jamil, T.; Hussain, R.: Synthesis and properties of aqueous polyurethane dispersions: Influence of molecular weight of polyethylene glycol. Korean J. Chem. Eng. 30(12), 2259–2263 (2013)
Misbah; Bhatti, I.A.; Zia, K.M.; Bhatti, H.N.; Shahid, M.: Synthesis, biological efficiency evaluation and application of sodium alginate-based polyurethane dispersions using cycloaliphatic isocyanate, as antibacterial textile coating. J. Ind. Tex. 50(10), 1625–1642 (2019)
Tabasum, S.; Zuber, M.; Jabbar, A.; Zia, K.M.: Properties of the modified cellulosic fabrics using polyurethane acrylate copolymers. Carbohydr. Polym. 94(2), 866–873 (2013)
Tabasum, S.; Zuber, M.; Jamil, T.; Shahid, M.; Hussain, R.: Antimicrobial and pilling evaluation of the modified cellulosic fabrics using polyurethane acrylate copolymers. Int. J. Biol. Macromol. 56, 99–105 (2013)
Alagi, P.; Ghorpade, R.; Jang, J.H.; Patil, C.; Jirimali, H.; Gite, V.; Hong, S.C.: Controlled hydroxyl functionality of soybean oil-based polyols for polyurethane coatings with improved anticorrosion properties. Macromol. Res. 26(8), 696–703 (2018)
Zuber, M.; Shah, S.A.A.; Jamil, T.; Asghar, M.I.: Performance behavior of modified cellulosic fabrics using polyurethane acrylate copolymer. Int. J. Biol. Macromol. 67, 254–259 (2014)
Muzaffar, S.; Bhatti, I.A.; Zuber, M.; Bhatti, H.N.; Shahid, M.: Synthesis and characterization of aqueous chitosan-polyurethanes dispersion for textile applications with multipurpose performance profile. Fibers Polym. 19(3), 587–598 (2018)
Lee, K.H.; Kim, B.K.: Structure-property relationships of polyurethane anionomer acrylates. Polymers 37(11), 2251–2257 (1996)
Nagle, D.J.; Celina, M.; Rintoul, L.; Fredericks, P.M.: Infrared microspectroscopic study of the thermo-oxidative degradation of hydroxy-terminated polybutadiene/isophorone diisocyanate polyurethane rubber. Polym. Degrad. Stab. 92(8), 1446–1454 (2007)
Wang, C.C.; Zhao, Y.; Purnawali, H.; Huang, W.M.; Sun, L.: Chemically induced morphing in polyurethane shape memory polymer micro fibers/springs. React. Funct. Polym. 72(10), 757–764 (2012)
Sidra; Tabasum, S.; Zia, K.M.; Parveen, B.; Shahid, M.: Polyurethane dispersions prepared from vegetable oil and their application as textile finishes. Tex. Res. J. 92, 4639 (2022)
Sultan, M.; Zia, K.M.; Bhatti, H.N.; Jamil, T.; Hussain, R.; Zuber, M.: Modification of cellulosic fiber with polyurethane acrylate copolymers. Part I: Physicochem. Properties. Carbohydr. Polym. 87(1), 397–404 (2012)
Dieterich, D.: Aqueous emulsraions, dispersions and solutions of polyurethanes; synthesis and properties. Prog. Org. Coat. 9(3), 281–340 (1981)
Tabasum, S.; Zia, K.M.; Parveen, B.; Hussain, M.T.: A novel water borne green textile polyurethane dispersions finishes from cotton (Gossypium arboreum) seed oil based polyol used in modification of cellulosic fabrics. Carbohydr. Polym. Technol. Appl. 2, 100170 (2021)
Man, L.; Feng, Y.; Hu, Y.; Yuan, T.; Yang, Z.: A renewable and multifunctional eco-friendly coating from novel tung oil-based cationic waterborne polyurethane dispersions. J. Clean. Prod. 241, 118341 (2019)
Eryuruk, S.H.; Kalaoğlu, F.: The effect of weave construction on tear strength of woven fabrics. Autex Res. J. 15(3), 207–214 (2015)
Bakare, I.O.; Pavithran, C.; Okieimen, F.E.; Pillai, C.: Synthesis and characterization of rubber-seed-oil-based polyurethanes. J. Appl. Polym. Sci. 109(5), 3292–3301 (2008)
Lee, K.Y.; Mooney, D.J.: Alginate: properties and biomedical applications. Progress Polym. Sci. 37(1), 106–126 (2012)
Huang, K.S.; Wu, W.J.; Chen, J.B.; Lian, H.S.: Application of low-molecular-weight chitosan in durable press finishing. Carbohydr. Polym. 73(2), 254–260 (2008)
Muzaffar, S.; Abbas, M.; Siddiqua, U.; Arshad, M.; Tufail, A.; Ahsan, M.; Iqbal, M.: Enhanced mechanical, UV protection and antimicrobial properties of cotton fabric employing nanochitosan and polyurethane based finishing. J. Mater. Res. Technol. 11, 946–956 (2021)
Naveed, T.; Babar, A.A.; Rashdi, S.Y.; Rehman, F.; Naeem, M.A.; Wang, W.; Ramzan, M.B.: Dyeing and colorfastness properties of tencel fabric treated with natural dye extracted from orange peel. Surf. Rev. Lett. 28(03), 2050055 (2021)
Jasińska, I.: Assessment of a fabric surface after the pilling process based on image analysis. Fibres Text. East. Eur. 17(2), 73 (2009)
Yagci, M.; Bolca, S.; Heuts, J.; Ming, W.; De With, G.: Self-stratifying antimicrobial polyurethane coatings. Prog. Org. Coat. 72(3), 305–314 (2011)
Acknowledgements
Authors are thankful to the Department of Applied Chemistry, GCUF, Pakistan for providing facilities to achieve the present project successfully.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Asghar, T., Fazal-ur-Rehman, Javid, A. et al. Green Synthesis of Polyurethanes Using Soybean Oil-Based Polyols for Bioactive Functional Fabrics. Arab J Sci Eng 49, 531–545 (2024). https://doi.org/10.1007/s13369-023-08276-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13369-023-08276-9