
Abstract
The current research was aimed to examine the NLO properties of novel benzodithiophene-based donor–acceptor (D-A)-type compounds (BDTD1–BDTD8) via structural modulation of reference compound (BDTR). Optimization of the reference compound (BDTR) along with eight derivatives was accomplished at M06/6-311G(d,p) level. Subsequently, the optimized geometries were further employed to execute additional analyses: UV–Vis absorption, natural population analysis (NPA), NLO, frontier molecular orbitals (FMOs) and natural bond orbital (NBO) properties at the above-mentioned functional. All the tailored compounds (BDTD1–BDTD8) were reported with less energy difference in comparison with BDTR. The descending order of compounds accordant to Egap values was found to be as BDTR > BDTD1 > BDTD8 > BDTD4 > BDTD2 > BDTD5 > BDTD7 > BDTD6 > BDTD3. Furthermore, assisted by FMOs analysis, density of states (DOS) computations demonstrated significant charge mobility from HOMO towards LUMO in the derivatives. Global reactivity descriptors correspond to Egap values, BDTD3 with least energy difference accompanied with less hardness (1.028 eV) and highest softness (0.486 eV) among all the derivatives. BDTD3 exhibited the highest value of λmax (717.211 nm) in all the designed compounds relative to BDTR (576.161 nm). For all fabricated chromophores, the βtot values are presented in the decreasing order: BDTD3 > BDTD7 > BDTD6 > BDTD5 > BDTD2 > BDTD4 > BDTD1 > BDTD8 > BDTR. Interestingly, the highest values as 2.324 × 10–27 and 4.302 × 10–32 esu of βtot and γtot, respectively, were indicated by BDTD3 compound. Valuable NLO findings explored that our derivatives might be significant candidates for nonlinear optics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Nowadays, NLO materials are considered to be one of the most proficient materials due to their fascinating characteristics, i.e., data transformation [1], fibre optics [2], photonic laser [3], data retrieval in wireless communication sector [4] and electro-optics [3, 5]. These materials are very useful for optical computation [6], telecommunications [1], optoelectronics [7], laser technology [8] and photonic devices [9]. The majority of NLO materials exhibit a direct correlation among optical characteristics and the intensity of incident light [10]. On both experimental and theoretical grounds, the developments of NLO species have become a region of advance research [11]. Enormous efforts have resulted in designing diverse materials comprising synthetic and natural nano-materials, polymer systems, molecular dyes, organic and inorganic semiconductors [9, 10] that exhibit effective NLO responses. A comparison of inorganic and organic nonlinear compounds demonstrates that organic chromophores offer numerous advantages then inorganic systems, including a faster response time, greater molecular flexibility, superior processing capability and a higher nonlinear polarization rate [11, 12]. Moreover, organic-based substances have many benefits such as easy fabrication, low cost, tunable absorption wavelengths and structural modulation using appropriate substituents [13, 14].
In the π-bonded system of organic dyes, the electronic charge distribution is delocalized and the second-order NLO outcomes have been attributed to intramolecular charge transfer (ICT), which occurs via π-spacers from donor towards acceptor [18]. Some effective approaches for organic-based NLO chromophores are: extensive π-electron schemes, planar (D-π-A) model, greater push–pull effects and integration of alkali metal into the organic compounds [19]. According to the literature survey, D and A molecules are liable for contributing fundamental ground-state charge asymmetry, while π-conjugated moieties provide a path for the transference and rearrangement of charges in an electric field [17, 18]. Along with a variety of π-conjugated scaffolds, fullerene-based chromophores indicate a rationally great NLO outcome [22] and considered as good candidates in the NLO field [23]. Fullerenes show an appropriate NLO characteristics due to π-conjugation, charge delocalization [24], significant characteristics and accomplishment in the area of organic photo-electronics [25]. Despite their enormous success, fullerene-based acceptor moieties have been replaced with non-fullerene organic acceptor moieties, and this concept has reinforced the field of optoelectronics in the recent years [26]. In contrast to fullerene acceptors, non-fullerene acceptors emerged as a new research area due to their tunable energy levels as well as planar structure. In addition, non-fullerene acceptors (NFAs) have revealed significant stability as compared to fullerene acceptor moieties [27]. At present, the centre of attention of experimenters is transferred towards non-fullerene compounds owing to their efficient adaptation to broad electron affinities, chemical structures, facile synthesis and tunable energy levels [28]. Review of existing research reveals that the synthesis of NLO compounds based on NFAs is emerged as an innovation in materials sciences [25, 26]. The extent of π-conjugation as well as substituent’s nature significantly manipulates the molecular NLO properties. ICT is accomplished by utilizing π-bridges among the donor and acceptor moieties, resulting in remarkable NLO responses [31]. Furthermore, the enlarged π-conformation with enormous absorption constraints of non-fullerene-based compounds has been improved by the association of electron-deficient terminal groups in the framework of the fullerene-free acceptors [28, 29]. The strong electron-withdrawing functional groups have been utilized to design fused-ring electron acceptors with a strong photovoltaic effect because of their enhanced electron-capturing capability [34]. Moreover, to design interesting NLO materials, a significant push–pull mechanism, higher asymmetric charge scattering, conjugation length and nature of electron-donating and electron-withdrawing groups are also important [3].
On account of these remarkable characteristics of π-conjugated non-fullerene chromophores, we have chosen NF-based chromophores in this research to discover their nonlinear optical outcomes from a hypothetical approach. To our understanding, NLO investigation on entitled chromophores (BDTD1–BDTD8) has not been reported so far. Hence in order to fulfil this research gap, density functional theory/ time-dependent density functional theory (DFT/TD-DFT)-based calculations have been carried out to discover NLO characteristics. Taking into account the significant characteristics of non-fullerene compounds, the synthesized compound (BDTX1) [35] has been selected as a parent compound. NF-based derivatives BDTD1–BDTD8 have been designed from modified reference compound (BDTR) via coupling of different strong electron-withdrawing acceptors, which comprise a phenyl ring fused with 1-one-3-dicyanovinylidenyl cyclopentane unit substituted with halogens on the phenyl ring or replacing phenyl ring by thiophene unit to fine-tune the electronic characteristics of NLO materials. This research work will proved to be an excellent addition in the development of NLO features as these novel NFA-based compounds might create an impactful NLO response in the future applications.
2 Computational Procedure
The quantum chemical calculations of non-fullerene benzodithiophene-based chromophore denoted as BDTR and its derivatives (BDTD1–BDTD8) having D–A configuration were investigated by employing the Gaussian 09 program [36]. For this purpose, M06 functional [37] with 6-311G(d,p) basis set [3, 35] of DFT/TD-DFT approaches was used. FMOs, TDMs, DOS, GRPs, absorption spectrum and hole-electron analyses were accomplished at fore-said functional via TD-DFT approach. NLO and NBO study was performed at M06/6-311G(d,p) functional of DFT. First of all, the structures were optimized to get true minima structures. The optimized geometries were then further employed to calculate the FMOs (energy difference between HOMO and LUMO), UV–Vis, transition density matrix (TDMs), DOS, NBOs and NLO insights at the aforementioned functional. The TDMs heat maps were developed to investigate charge dissociation patterns in the entitled molecules. The NBOs study was executed to evaluate the hyper-conjugative interactions as well as intramolecular charge transfer. The DOS computations were employed to regulate the energy states dispersion. UV–Vis absorption spectra were acquired to explore transitions of electrons. The values of the dipole moment (μ) [40], linear polarizability ⟨α⟩ [41], first-order hyperpolarizability (βtot) [42] and second-order hyperpolarizability (γtot) [43] are estimated by Eqs. (1–4).
where βx = βxxx+βxyy+βxzz, βy = βyxx+βyyy+βyzzandβz = βzxx+βzyy+βzzz.
Different softwares like GuassView 6.0 program [44], Avogadro [45], Chemcraft [46], PyMOlyze 2.0 [47] and Multiwfn 3.7 [48] were employed to obtain results from the input files.
3 Results and Discussion
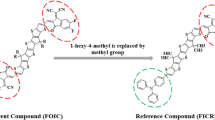
The current research is centred on quantum chemical analysis of non-fullerene-based NLO chromophores. The designed D-A natured molecules having donor and acceptor fragments show a strong push–pull mechanism with high polarity, improving their NLO characteristics. The architecture of parent compound (BDTX1) [35] is appeared to be A-D-A, and it is modified by altering long chain groups (C8H17,C6H13 and 3-methylhept-1-ene) with methyl (CH3) group to lower the computational cost and steric hindrance as replacement of bulky group with small groups does not show significant effect in NLO properties [49,50,51]. Consequently, the parent compound with A-D-A architecture is renamed from “BDTX1” to “BDTR” named as reference compound with D-A configuration, as illustrated in Fig. 1.

Modification of parent compound (BDTX1) to reference compound (BDTR)
A framework of compounds, namely BDTD1–BDTD8, was modified by substituting the weak electron-withdrawing group in the non-fullerene-based reference compound (BDTR), as illustrated in Figure S2. In BDTD1, 9-(4-(5''-(4,8-bis(4,5-dimethylthiophen-2-yl)-6-(3,4',5''-trimethyl-[2,2':5',2''-terthiophen]-5-yl)benzo[1,2-b:4,5-b']dithiophen-2-yl)-3',3''-dimethyl-[2,2':5',2''-terthiophen]-5-yl)phenyl)-9H-carbazole was utilized as donor, while 4-methoxy-2-methylene-3-oxobutanenitrile was utilized as acceptor. The acceptor part attached at one side of BDTD1 is substituted by adding up the new donor 9-phenyl-9H-carbazole. In the remaining designed compounds (BDTD2–BDTD8), the electron donor group was maintained, but the acceptor is altered with different well-known acceptor groups, demonstrated in Scheme 1. The enlisted acceptors with their IUPAC names and abbreviations are displayed in Figure S1; however, the IUPAC names of the entitled compounds are listed in Table S39 and data for Cartesian coordinates are presented in Tables S1–S9. The impact of a variety of acceptor units on absorption spectra, HOMO/LUMO energy gap, ICT, ⟨α⟩, βtot and γtot was examined. In the current research, we have used the parent molecule (E)-octyl 2-cyano-3-(5″-(6-(5″-(2-cyano-4-(octyloxy)-3-oxobut-1-en-1-yl)-4’-hexyl-3-methyl-[2,2’;5’,2″-terthiophene]-5-yl)-4,8-bis(4-hexyl-5-(2-vinylhexyl)thiophen-2-yl)benzo[1,2-b:4,5-b]dithiophen-2-yl)-3’-3″dihexyl-[2,2’:5’,2″-terthiophen]-5-yl) propanoate (BDTX1) [35] with an A-D-A configuration. This research might be an excellent accumulation in the field of NLO and also prompt the advance investigation in this area. The optimized tailored structures are shown in Figure S3, while the graphs of optimization frequency and tables of vibrational analysis of the compounds (BDTR) and (BDTD1–BDTD8) are illustrated in Figure S4 and Tables S10–S18, respectively.

The representation of the reference and the designed derivatives with different acceptors
3.1 Electronic Properties
The investigation of frontier molecular orbitals (FMOs) is extremely considerable to comprehend electronic and optical properties of species as well as to examine its chemical stability [52,53,54]. In FMOs exploration, frequently elaborated two significant molecular orbitals are HOMO and LUMO. Transition of electron out of HOMO towards LUMO is distinguished by the band gap (ΔE). The variables such as electrophilicity, hardness, softness, electronegativity, stability and reactivity are similarly regulated by ΔE [55, 56]. In addition, they can also calculate the mainly reactive part in π-conjugated structures and reveal the reaction types in resonating structures. The value of ΔE among HOMO and LUMO shows the charge-transmission capability of a compound. A small energy gap intimates a proficient charge transfer, increasing the NLO activity of chromophore. The energy gap outcomes for the examined chromophores are tabulated in Table 1. Energy difference and orbitals of HOMO-1/LUMO + 1 and HOMO-2/LUMO + 2 of the examined chromophores (BDTR–BDTD8) are illustrated in Figure S5 and Table S19.
From the data presented in Table 1, the theoretical value of ΔE possessed by BDTR is 2.594 eV comparable to the experimental ΔE of 1.65 eV. The design of the studied compounds includes donors and end-capped acceptors, which contributes to decrease in ΔE values in these compounds. This might be a consequence of modifying the A-D-A configuration into D-A, hence promoting proficient internal charge transfer, which reduces the energy band gap [57]. The ΔE value for BDTD1 decreases to 2.536 eV comparing with that of BDTR (2.594 eV) owing to strong push–pull configuration in combination with the enhanced resonance upon substituting one terminal acceptor with 9-phenyl-9H-carbazole [58] as donor and 4-methoxy-2-methylene-3-oxobutanenitrile as an acceptor unit. Moreover, the BDTD2 has lessen energy gap of 2.193 eV than BDTD1 because of the substitution of 4-methoxy-2-methylene-3-oxobutanenitrile with the 2-(6,7-dichloro-3-oxo-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophen-1-ylidene) malononitrile acceptor with two electron-withdrawing chloro (− Cl) groups. Correspondingly, the band gap of BDTD3 decreases to 2.057 eV because of the substitution of the − Cl with two nitro (− NO2) groups in 2-(2-methylene-6,7-dinitro-3-oxo-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophen-1-ylidene)malononitrile enhance the acceptor electron-accepting capability, forming an efficient pull–push structure. The value of ΔE in BDTD4 slightly increases to 2.197 eV owing to the substitution of two acetate (− COOCH3) groups in the acceptor part dimethyl1-(dicyanomethylene)-2-methylene-3-oxo-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophene-6,7-dicarboxylate. A minor decrease in ΔE (2.150 eV) is observed when the 2-(2-methylene-3-oxo-6,7-bis(trifluoromethyl)-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophen-1-ylidene) malononitrile acceptor moiety is introduced into BDTD5, where − COOCH3 groups are changed with − CF3 units, as − CF3 possess high electron-withdrawing effect and electronegativity as compared to − COOCH3. The introduction of the 1-(dicyanomethylene)-3-oxo-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophene-6,7-dicarbonitrile acceptor group in BDTD6 further decreases the energy band gap (2.075 eV), as –CF3 is substituted by cyano (− CN) units. This might be due to high electronegativity of − CN units in comparison with –CF3 group; hence, the enriched electron-withdrawing performance of 1-(dicyanomethylene)-3-oxo-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophene-6,7-dicarbonitrile results in improved push–pull conformation. Consequently, the large electronegativity of molecules has enhanced the transference of the electronic cloud towards acceptor owing to negative inductive effect. A small increase in ΔE (2.090 eV) value of BDTD7 contrasted to BDTD6 is because of acceptor moiety i.e. 1-(dicyanomethylene)-2-methylene-3-oxo-2,3-dihydro-1H-benzo[b]cyclopenta[d]thiophene-6,7-disulfonic acid, inBDTD7. A small increase in HOMO–LUMO energy gap (2.209 eV) in BDTD8 is due to 2-(6-methylene-7-oxo-6,7-dihydro-5H-cyclopenta[b]thieno[2,3-d]thiophen-5-ylidene) malononitrile used as an acceptor unit. This may be due to its diverse chemical structure, lower conjugation and least proficient push–pull configuration owing to the absence of electronegative units in comparison with other derivatives [59]. The existence of two nitro groups in BDTD3 reduces the ΔE value to 2.057 eV. The nitro groups linked with the acceptor region has a strong electron-accepting properties besides its electron-withdrawing potential is a result of a powerful negative inductive effect [60]. The perceived least ΔE value of BDTD3 between all the designed molecules is a result of the extensive conjugation occupied in the acceptor part containing nitro groups.
In a nutshell, the energy difference among HOMOs and LUMOs of the entitled chromophores reduces in the subsequent pattern: BDTR > BDTD1 > BDTD8 > BDTD4 > BDTD2 > BDTD5 > BDTD7 > BDTD6 > BDTD3. This sequence reveals that structural modulation by introducing a variety of electronegative functional groups at the acceptor region is an outstanding strategy to decrease ΔE and consequently improve the NLO outcomes. The ΔE values describe the effective ICT from donor to acceptor part and are favoured by π-linker that provides an insight about the correlated functional links of the NLO structures [54, 55]. The HOMOs and LUMOs of FMOs are employed to describe the electronic charge transfer, as indicated in Fig. 2. In BDTR, the HOMO charge density is generally located over the donors, though the LUMO is generally present on acceptor part. In the designed chromophores (BDTD1–BDTD8), the HOMO’s electronic cloud is mostly established across the donor. Conversely, overall electron density is located on the acceptor part. Literature describes that an indirect band gap can be considered, whenever the HOMO and LUMO contain topologically similar phase relationships in the interaction zones. Otherwise, the molecules can be considered with direct band gaps [63]. Moreover, our results indicate that the HOMO and the LUMO have not topologically similar phase relationships in the interaction regions (Fig. 2). Therefore, direct band gap is found in the entitled organic molecules.


HOMOs and LUMOs of the BDTR and BDTD1–BDTD8
This implicit that moieties which are electron-donating connect to electron-withdrawing groups by the assistance of π-linkers that promote charges transmission among the acceptor and donor molecules. Consequently, BDTD1–BDTD8 might appear to be effective NLO applicants.
3.2 Global Reactivity Parameters (GRPs)
To study global reactivity descriptors like electronegativity (X) [64], ionization potential (IP) [65], electron affinity (EA) [66], global softness (σ) [67], hardness (η) [68], chemical potential (μ) [69], and electrophilicity index (ω) [70], the energy values of designed compounds are estimated using the DFT approach conducive to investigate the reactivity as well as stability. Compounds with lower softness and hardness values are typically thought to produce excellent NLO outcomes. The studied compounds may offer fine NLO response because of their proficient global softness values [71]. The investigated compounds possess appropriate IP, EA, μ, and σ values, which is crucial indicator of efficient NLO response, obtained from the analysis of GRPs [72]. All of these aforesaid factors are computed via employing Koopmans's theorem [73] (Eqs. 5–11), and the results are composed and presented in Table S20.
The IP values (5.422–5.405 eV) of the designed compounds (BDTD1–BDTD8) are less than their reference chromophore (5.515 eV), which indicates prompt electron removal and less energy of polarization than BDTR.
The highest values of IP are exhibited by BDTD3 and BDTD4 at 5.444 eV, demonstrating improved electron donation capacity by the donor moiety towards the electron-withdrawing group. Whereas, BDTD8 exhibits the smallest value of IP (5.405 eV). Overall, it is discovered that IP increases in the following order: BDTD8 < BDTD2 < BDTD1 < BDTD6 < BDTD5 < BDTD7 < BDTD3 = BDTD4. More stability and chemical inertness are indicated by higher IP values. Similarly, the presence of strong acceptor moieties causes derivatives except BDTD1 to have higher EA values than BDTR, taking advantage of their higher tendency to receive electrons. Additionally, to recognize the compound’s stability chemical potential (μ) is considered, it is discovered that the μ of BDTD2–BDTD8 are substantially less (− 4.301 to − 4.416 eV) with greater softness (σ) values (0.486–0.453 eV) and that the softness of BDTD1 (0.394 eV) is somewhat higher than those of BDTR (0.386 eV). The NLO characteristics of such chromophores are improved by expressing the stronger chemical reactivity with greater polarizability. The amount of ΔNmax = − µ / ƞ represent the tendency of molecule for absorbing more electrical charge from its surroundings [72]. High and positive ω and ∆Nmax values indicate that the material is more likely to absorb an electron. On the other hand, if a compound has a low amount of these indices, it can act as an electron-donating group [74]. The charge transfer parameter (ΔNmax) and electrophilicity index (ω) for all the chromophores are in the range 3.252–4.291 eV and 6.804–9.478 eV, respectively. The high ΔNmax value of BDTD3 (4.291 eV) suggests that this compound has higher electron-absorbing tendency. Moreover, the hardness of a compound is openly linked with the ΔE, and a reverse interaction of ΔE is examined with the reactivity and global softness of the investigated chromophores. While a lower ΔE suggests a less stable, softer and highly reactive compound, a higher ΔE value indicates a more stable, harder and less reactive system. The maximum hardness is examined in reference chromophore BDTR (1.297 eV), which shows least reactivity and greater stability, while the minimum hardness is examined at 1.029 eV for BDTD3. The descending order of hardness in the tailored molecules is: BDTR > BDTD1 > BDTD8 > BDTD4 > BDTD2 > BDTD5 > BDTD7 > BDTD6 > BDTD3. The highest σ is considered to be 0.486 eV in BDTD3, which indicates its less stability and larger reactivity aspect, whereas the least value of 0.386 eV is examined in BDTR. The ascending order for σ is as follows: BDTR < BDTD1 < BDTD8 < BDTD4 = BDTD2 < BDTD5 < BDTD7 < BDTD6 < BDTD3. Consequently, all the investigated compounds have revealed large softness, which specifies more reactivity as well as polarizability resulting in exceptional NLO outcomes.
3.3 Density of States (DOS) Analysis
Density of states investigation is performed to further validate the inferences of FMOs charge distribution [75]. For the designed molecules, DOS investigation is performed at M06/6-311G(d,p) level of theory, and the results are represented in the form of graphs. In the designed chromophores, maximum charge density in the form of HOMO is distributed at donor with negative values of energy, while LUMO is distributed mostly at acceptor with positive values of energy [76]. Electronic charge distribution on both segments, i.e. donor and acceptor, as well as number of electronic states at specific energy are illustrated in graphs [77]. In DOS graph, energy in eV is plotted along x-axis and relative intensity along y-axis. In DOS pictographs, donor and acceptor are denoted by red and green coloured lines, accordingly. The peaks of graph vary from reference to designed compounds due to different strengths of donor and acceptor moieties and length of conjugation [78]. In the reference (BDTR) and designed compounds (BDTD1–BDTD8), donor displays electronic charge distribution as 96.7, 98.7, 99.1, 99.0, 99.3,99.0, 99.1, 99.0 and 99.2% to HOMO, which is relatively higher than that of LUMO at 62.4, 60.1, 27.2, 28.2, 26.6, 28.7, 28.2, 28.9 and 28.2%, respectively. This charge is significantly moves towards LUMO as we examined the charge distribution on acceptor part in BDTR and BDTD1–BDTD8; 3.3, 1.3, 0.9, 1.0, 0.7, 1.0, 0.9, 1.0 and 0.8% to HOMO, while 37.6, 39.9, 72.8, 71.8, 73.4, 71.3, 71.8, 71.1 and 71.8% to LUMO, respectively. This analysis significantly supported the FMOs counter surfaces charge distribution pattern (Fig. 2). In the pictographs of DOS analysis, the left side peaks charged values characterized by HOMOs, while right side charged values represents LUMOs beside the x-axis, with distance among them corresponds to the energy gap [79]. Figure 3 illustrates that for HOMO the charge is highly occupied on donor, whereas the LUMO relative density is much strong in acceptor. The comprehensive contribution pattern demonstrates that considerable charge transmission occurred out of donor towards acceptor. So this substantial charge distribution indicated the efficient NLO results from these fabricated chromophores.


Graphical representation of DOS for BDTR and BDTD1–BDTD8
3.4 UV–Vis Analysis
UV–Vis analysis gives further information regarding absorption spectra, oscillator frequency, transition energy and electronic states [80]. TD-DFT study was performed in chloroform and in gaseous phase via above-mentioned TD-DFT approach to analyse the electronic transitions of the reference (BDTR) and designed compounds (BDTD1–BDTD8). Significant results obtained in the form of excitation energy, oscillator strength (fos) and maximum absorbed wavelength (λmax) in solvent (chloroform) and gaseous phase are depicted in Tables S21 and S22 (Fig. 4). Maximum absorption values of wavelength are found in the chloroform solvent as protic solvents are predicted to increase the transitional characteristics of chromophores by increasing their bathochromic shifts and stabilizing the π-π* and n-π* transitions [81]. Hydrogen bonding and dipole interactions also take part in stabilizing the first singlet electron level in chromophores [82]. In gaseous and chloroform phase, absorption spectra of all the formulated compounds are located in visible region [83].

Absorption spectra of BDTR and BDTD1–BDTD8 in (a) chloroform solvent and b gaseous phase
The λmax in chloroform solvent existed in 592.942 to 717.211 nm range for studied chromophores. Reference compound (BDTR) shows the absorption spectra at 594.991 nm, while according to the experimental results the reference compound exhibits a maximum absorption peak at 530 nm, showing that strong intermolecular interaction is present in the solid film. By introducing various acceptor moieties in the designed compounds (BDTD2-BDTD8), λmax is observed at the different wavelengths. BDTD3 shows bathochromic shift in the absorption spectrum (717.211 nm) at excitation energy (1.729 eV) and oscillation strength of 0.880. BDTD3 has the maximum wavelength with minimum excitation energy than all the formulated derivatives owing to the presence of strong EWD groups, CN and NO2 in the acceptor moiety. Bathochromic shift is observed in BDTD3 due to excellent transfer of charges from D to A, less energy band gap is observed, and hence minimum energy is required for excitation from HOMO towards LUMO in chloroform. BDTD1 is the molecule with the lowest absorption wavelength of 592.942 nm, highest excitation energy of 2.091 eV in chloroform. The investigated compounds hold stronger absorption range as compared to the reference BDTR (594.991 nm) except BDTD1. The λmax of all the studied entities decreases as follows: BDTD3 > BDTD6 > BDTD7 > BDTD5 > BDTD2 > BDTD8 > BDTD4 > BDTR > BDTD1.
In gaseous phase, the range of wavelengths extends from 573.895 to 748.426 nm. Minimum λmax is observed in BDTD1 at 573.895 nm because of the presence of less electronegative-COOCH3 group resulting in less electron-withdrawing, reduced charge transfer and maximum energy required for excitation. Likewise, in chloroform, BDTD3 also secured the longest wavelength range of 748.426 nm in gaseous phase. Decreasing order of λmax in gaseous phase is observed in the order: BDTD3 > BDTD6 > BDTD7 > BDTD5 > BDTD2 > BDTD4 > BDTD8 > BDTR > BDTD1. All the developed chromophores exclusively BDTD3 hold lower excitation energy and is observed as an outstanding NLO compound with efficient charge transfer and broader absorption spectra.
3.5 Natural Bond Orbital (NBO) Analysis
NBO analysis illustrates the distribution, bonding, nature of intramolecular and intermolecular interactions among the associated systems [84]. The presence of non-covalent interactions, which cause shifting of electron density, binding and hyper-conjugation between the D and A moieties of the analysed systems, has been extensively investigated using NBOs analysis in recent years [85]. The NBOs investigation shows beneficial charge transfer values for each of the donor moieties, displaying the high donation capability of our designed compounds [85]. Meanwhile, the positive values of donors represent their ability to donate electrons, and all the acceptors have negative charge values, showing that all the entitled molecules are effective electron acceptors. Additionally, it is implied by the charge descriptions in terthiophenes that they may act as donor and help in effective electron donation towards acceptor [86]. The donor and acceptors are reported in the reference compound (BDTR) with values of − 0.569 and − 0.569, respectively, as indicated in Table S23. In the designed compounds (BDTD1–BDTD8), acceptor is substituted with a donor, which enhances their ability to donate protons with positive NBO charges. In case of donors, all compounds have positive values due to their electron donating ability. In tailored compounds, acceptor shows negatively charged values due to their electron-withdrawing characteristics. The second-order perturbation theory can be utilized to calculate the stabilization energy of molecules while performing NBOs analysis by using the succeeding equation:
The donor and acceptor parts are denoted by i, j subscripts, E(2) is the stabilization energy, qi, ɛi, ɛjand Fi,j characterize donor orbital occupation, diagonal and off-diagonal NBOs Fock matrix components [50], respectively.
For BDTR and BDTD1–BDTD8, the E(2) of various orbitals have been examined and are shown in Tables S24–S32. There are primarily six transitions types present in the formulated chromophores as; π → π*, σ → σ*, π → σ*, σ → π*, LP → π* and LP → σ*. The most prominent transitions that are detected in BDTR and BDTD1–BDTD8 are; π → π*, σ → σ*, LP → π* and LP → σ*. The least dominant transition is of σ → σ*, which is due to the excitation of electrons occupying a HOMO of a sigma bond towards LUMO of that bond. The highest dominant transition that is observed in the investigated compounds is π → π*, which provide the massive value of stabilization energies. In BDTR and BDTD1–BDTD8, the highest values of stabilization energy obtained through π → π* transitions are found to be 29.67, 28.76, 24.97, 25.01, 23.96, 24.12, 24.7, 24.42, 24.4 kcal mol−1 for π(C61-C63) → π*(C97-C100), π(C61-C63) → π *(C97-C100), π(C137-C138) → π*(C139-C140), π(C61-C63) → π*(C97-C132), π(C99-C101) → π*(C104-C106), π(C61-C63) → π*(C97-C132), π(C61-C63) → π*(C97-C132), π(C120-C122) → π*(C124-C126), π(C110-C111) → π*(C113-C116) transitions, respectively. On the other hand, the lowest values for this transition are 2.32, 6.02, 5.45, 4.76, 4.2, 7.99, 4.66, 8.07, and 5.17 kcal mol−1 exhibited by π(C99-O101) → π* (C97—C100), π(C21-C22) → π*(C1-C2), π(C1-C2) → π*(C21-C22), π(C5-C6) → π*(C15-C16), π(C131-O143) → π*(C130-C134), π(C133-C144) → π*(C97-C132), π(C15-C16) → π*(C5-C6), π(C133-C144) → π*(C97-C132), and π(C4-C5) → π*(C15-C16), respectively. σ → σ*, are the least dominant due to weak interactions, and among these transitions, the highest stabilization energy is provided by σ(C97-H98) → σ*(S60-C63), σ(C97-H98) → σ*(S60-C63), σ(C97-H98) → σ*(C132-C133), σ(C97-H98) → σ*(C132-C133), σ(C97-H98) → σ*(C132-C133), σ(C97-H98) → σ*(C132-C133), σ(C97-H98) → σ*(C132-C133), σ(C97-H98) → σ*(C132-C133) and σ(C97-H98) → σ*(C132-C133) which is 9.05, 8.94, 9.43, 9.17, 9.37, 9.34, 9.27, 9.23 and 9.39 kcal mol−1, respectively. The lowest values of stabilization for σ → σ* are 0.5, 0.5, 0.5, 0.5, 0.51, 0.5, 0.5, 0.5, 0.5 kcal mol−1 presented by σ(C92-H95) → σ*(C70—C92), σ (C4-S12) → σ*(C3-C9), σ (C1-S13) → σ*(C6-C7), σ(C136-S142) → σ*(C135-H141), σ (C160-H163) → σ*(C153-O155), σ(C1-S13) → σ*(C6-C7), σ(C1-S13) → σ*(C6-C7), σ(S153-O155) → σ*(C135-C140) and σ(C1-S13) → σ*(C6-C7), respectively. In BDTR and BDTD1–BDTD8, the higher stabilization energy values are important. These highest values of stabilization for LP → π* transitions are 28.85, 35.34, 35.4, 160.82, 48.56, 35.36, 35.38, 40.22, and 40.27 kcal mol−1, shown by LP2(O119) → π*(C99-O101), LP1(N114) → π* (C115—C117), LP1(N109) → π* (C110-C112), LP3(O151) → π* (N150-O152), LP2(O155) → π* (C153-O154), LP1(N109) → π*(C110-C112), LP1(N109) → π*(C110-C112), LP1(N109) → π*(C110-C111) and LP1(N109) → π*(C110-C111), respectively. Similarly, for LP → σ* the highest values of stabilization energy for LP2(O101) → σ*(C99-O119), LP2(O101) → σ*(C99—C100), LP2(O143) → σ*(C131-C132), LP2(O143) → σ*(C131-C132), LP2(O151) → σ*(C150-O152), LP1(N146) → σ*(C144-C145), LP2(O143) → σ*(C130-C131), LP3(O151) → σ* (S150-O157) and LP2(O141) → σ*(C131-C132) are 32.53, 21.72, 22.42, 33.73, 12.84, 22.83, 28.21 and 22.68 kcal mol−1. The outcomes illustrate that the π-electron bonding of carbon-to-carbon delocalized towards anti-bonding orbitals performs a considerable part in the ring strength. Furthermore, it improves the ICT characteristics that are crucial for the NLO response. The NBO analysis proved that all compounds under study (BDTR and BDTD1–BDTD8) exhibit substantial intramolecular charge transfer and extended hyperconjugation. The compounds are significantly stabilized according to NBO results. Data from NBO also clearly demonstrate charge transfer in compounds supporting possible NLO research.
3.6 Transition Density Matrix (TDMs)
The TDM study of fullerene-free acceptor-based three-dimensional molecules has been done to know nature and mode of excited state transition [87]. The extensive interactions and electron excitation among the donor and acceptor moieties and the hole–electron delocalization can be concluded by TDM calculations [88]. A TDMs exploration of non-fullerene-based chromophores (BDTD1–BDTD8) shows that the charge is effectively transferred from D towards A side. According to FMOs and DOS analyses, the charge density in HOMOs is primarily located on the donor part; however in LUMOs, it is primarily distributed over the acceptor, which results in a considerable variation in TDM pictographs. The electron density has been successfully transferred diagonally out of the donor to the acceptor part in all the formulated compounds, as shown by Fig. 5, enabling efficient charge transmission devoid of trapping. Charge localization from ground to excited state in TDMs investigation is characterized by pictograph containing band of colours. TDMs of studied chromophores are calculated at M06/6-311G(d,p) functional and basis set. The H-atoms have been omitted owing to their small contribution in valuable electron transfer phenomenon. The TDM pictographs of all the investigated chromophores reveal the mode of transitions, and the results are exhibited in Fig. 5.


TDMs pictographs of the entitled molecules (BDTR and BDTD1–BDTD8)
The TDMs diagram shows that in BDTR, charge is transferred transversely, which is found majorly on the π-spacer and donor parts. In the investigated compounds, slight charge consistency is observed on the donor and the major part on the A group. A significant diagonal charge transfer is attained in modified structures BDTD1–BDTD8. The donor (terthiophene) works as a connection and facilitates the movement of electrons from the D towards the NFAs. The diagonal charge is proficiently relocated from the D to A area, which transfers the charge without trapping showing charge coherence. It is also investigated that the coupling of electrons in all the modified chromophores (BDTD1–BDTD8) may be inferior to that of BDTR. However, this might be predicted as very significant and straightforward excitation dissociation in the excited state.
3.7 Hole-Electron Analysis
A hole–electron excitation occurs when an electron in excitation from the hole region moves to the electron region [89,90,91]. This technique is extremely efficient as well as broadly employed to determine the localization of electron density in a compound [92]. Therefore, to comprehend the movement of charge in the formulated chromophores, we performed the hole–electron analysis with the help of Multiwfn 3.7 [48]. In all the explored compounds, the highest transition of electrons is observed in acceptor part on C(67) in BDTR, C(61) in BDTD1 and on C(83) in BDTD2–BDTD8. Figure S6 shows that the carbon atoms in the strongly electron-withdrawing acceptor region, which are further connected with the cyano (-CN) groups, exhibit intense electron intensity. This might occurs owing to the presence of highly electron-withdrawing character of cyano groups over the acceptor unit. The presence of HOMO on the donor ends of all the molecules under investigation suggest that charge would be transported from the donor towards the acceptor ends by means of the hole transport materials, and the presence of LUMO on the acceptor core of all the molecules under study is strong evidence for this transference. The reference BDTR and the designed derivatives BDTD1–BDTD8 appear to be electron-type materials, because the electron intensity in the electronic band is higher than the hole intensity in the hole band. This research clearly specified that the end-capped acceptor modification is a proficient approach for designing high-performance NLO featured chromophores.
3.8 Exciton Binding Energy (E b) Analysis
A considerable and capable feature to approximate optoelectronic characteristics is the Eb analysis that assists in determining the excitation dissociation potential [93]. The Eb values of compounds have been investigated with the M06/6-311G(d,p)level of DFT. To explore the hole–electron interface, Eb calculations for all the compounds are examined. It is a significant parameter for the evaluation of columbic force interaction and exciton dissociation capacity, as lower the Eb, less will be the columbic forces among hole and electron. Lower Columbic interactions and smaller binding energies result in a more intense exciton dissociation in excited state. Greater charge mobility is observed if Eb is lower [94]. Eb of investigated compounds (BDTD1–BDTD8) is calculated by using Eq. 13.
In Eq. 13, EL-H is the energy band gap in between HOMO and LUMO and Eopt (eV) is the first singlet excitation energy [95]. The estimated binding energies of BDTR and entitled compounds are elucidated in Table 2.
The binding energy of BDTR is considered to be 0.510 eV. The Eb values of BDTD1–BDTD8 are 0.445, -0.303, 0.328, -0.328, − 0.353, 0.323, 0.333 and -0.302 eV, correspondingly. The ascending order of Eb is following: BDTD5 < BDTD4 < BDTD2 < BDTD8 < BDTD6 < BDTD3 < BDTD7 < BDTD1 < BDTR. All the designed derivatives (BDTD1–BDTD8) give smaller Eb values in contrast to the reference chromophore (BDTR). These results of Eb values for tailored chromophores are in accordance with the results of TDMs. In a nutshell, these NF-based tailored structures have the smallest binding energies, which maintain the compounds high polarizability, which makes them suitable substances to utilize in the NLO applications.
3.9 Nonlinear Optical Properties
In quantum and classical optics, the basic procedure for optical frequency conversion is second-order hyperpolarizability, which is a considerable subdivision of science that started with the innovation of second harmonic generation (SHG) [96]. Conjugated organic compounds in general have useful applications in a variety of technical fields of optical limiting, optical switching, optical memory devices, optical communication, signal processing, photodynamic therapy, etc., because of their inherent nonlinear optical (NLO) behaviour and durability [97, 98]. As a consequence, various modelling efforts are made for different materials, including inorganic and organic compounds. Organic complexes are thought to be best options in NLO compounds because of their simple reaction chemistry, reasonable advancement and structural modulations, all of which enable us to alter them chemically for a distinctive NLO response [99]. In order to develop a new proton–donor–acceptor system, ICT among electron donor and acceptor groups can be used to reduce the band gap and regulate the transitions by using different donor or acceptor moieties with higher first hyper-polarizability values [100]. The degree of NLO efficiency is accomplished by the strength of the electron-withdrawing and electron-donating groups, the arrangement in which they are assembled [101]. The electrical individualities of the whole chromophores are associated with the ⟨α⟩, βtot and γtot outcomes, which are accountable for optical activity [102]. The µtot is a three-dimensional vector that indicates the molecular charge distribution. As a consequence, it can be implemented as an identifier to demonstrate how the positive and negative charge centres influence charge transfer through a molecule [101]. To study the change in the π- linker, the ⟨α⟩, beside x, y, and z directions, the βtot and γtot values of the designed molecules (BDTD1–BDTD8) are illustrated in Table 3. The results for all tensors are displayed in Tables S33–S38.
Urea is considered as a reference substance for evaluation of the preliminary dipole moment and hyperpolarizability values of BDTD1–BDTD8, which are observed as: 6.789, 6.100, 12.00, 6.964, 7.747,12.90, 11.13 and 3.107 D, accordingly. It is illustrated that the value of µtot of all the proposed chromophores is larger comparative to urea (1.373 D) [103]. The µtot of BDTD6 is the highest (12.90 D) amongst all the entitled molecules. The values of dipole moments are presented in general decreasing order as follows: BDTD6 > BDTD3 > BDTD7 > BDTR > BDTD5 > BDTD4 > BDTD1 > BDTD2 > BDTD8. The outcomes of Table S25 have exhibited that linear polarizability tensor along z-axis is more prominent in BDTD4, while x-axis is foremost in BDTD3, and y-axis is prominent in BDTD6 among all polarizability tensors and its involvement towards linear polarizability is much prominent than others. The literature studies reveal that the energy gap between LUMO and HOMO manipulates the polarizability of a compound. Greater ⟨α⟩ value is indicated by chromophores with least energy gap value. The descending order of ⟨α⟩ values of the tailored chromophores are found to be BDTD7 > BDTD3 > BDTD6 > BDTD4 > BDTD5 > BDTD2 > BDTD8 > BDTD1 > BDTR. For the designed molecule BDTD7, it is demonstrated that the maximum polarizability is the average polarizability. An unexpected addition in its value is observed, i.e., (3.261 × 10–22 esu). This is due to the existence of two -SO3H functional units that has enhanced the charge density close to the acceptor part and increase the electron-accepting capability. At the aforementioned functional and basis set, the βtot is calculated along with its constituent tensors, and the resulting values are listed in Table S26. Among all the designed compounds, BDTD8 exhibit the maximum (9.682 × 10–28 esu) βtot amplitude along x-axis while along y-axis BDTD7 exhibit the maximum value (9.241 × 10–29 esu). Similarly along z-axis, BDTD1 exhibit the maximum value (5.102 × 10–32 esu). βtot value and molecular geometry are efficiently interlinked with each other. The βtot normally enhances with an increment in the strength of the functional groups such as F, Cl and CN groups connected to the acceptor moiety, which influence the nonlinearity of a molecule. Additionally, as the replacements occur, the involvement of the conjugated system is extensive and βtot is further prevailing. For all customized structures, the βtot values are presented in the decreasing order: BDTD3 > BDTD7 > BDTD6 > BDTD5 > BDTD2 > BDTD4 > BDTD1 > BDTD8 > BDTR.
Furthermore, BDTR has the lowest value of βtot, i.e. (5.726 × 10–28 esu) in comparison with the other studied molecules. Among all the derivatives (BDTD1–BDTD8), the enhanced NLO response is observed in BDTD3 in contrast to the other designed molecules owing to the integration of a nitro group, which increase the electron-accepting affinity of compounds from the donor molecules. The –NO2 group is more electrons accepting, therefore, successfully led to improved NLO dimensions for the molecule BDTD3. A better electron delocalization means decrease of a HOMO/LUMO energy gap and an increase in βtot values in the entitled chromophores. The results found in Table S27 for second hyperpolarizability showed that γz tensor is more prevailing in BDTD5 with the value 2.802 × 10–34 esu, while γx and γy tensors are prominent in BDTD3 with the values 3.937 × 10–32 esu and 3.406 × 10–33 esu. Highest γtot value of 4.302 × 10–32 esu is also indicated by BDTD3, while smallest γtot value of 2.487 × 10–32 esu is exhibited in BDTD1. The decreasing order of all the developed molecules is as follows: BDTD3 > BDTD6 > BDTD7 > BDTD5 > BDTD2 > BDTR > BDTD8 > BDTD4 > BDTD1. The modern study presents perceptions into the attractive NLO data of the aforesaid π-conjugated push–pull organic molecules for current NLO purposes. Additionally, owing to their sophisticated NLO results, the tailored structures might be employed as targets for advance investigation.
3.10 Natural Population Analysis (NPA)
For the quantum chemical exploration of any molecule, the atomic charges play a significant part. The distribution of charges on an atom strongly influences the dipole moment, chemical reactivity, electrostatic interactions between atoms and compounds, as well as a number of other properties of the chemical system. The valuable estimation of atomic charge of the designed chromophores is significant for the better interpretation of the conjugated system [104, 105]. The charges of BDTR and BDTD1–BDTD8 are computed at M06/6-311G(d,p) functional and are demonstrated in Figure S7. The NBO-based charges indicate substantial influence on the molecular configuration and bonding capacity. Its main objective is to exhibit the reorganization of charges over the tailored compounds (BDTD1–BDTD8) in order to conclude their reactive sites. In NBO procedures, sulphur and hydrogen atoms possess net positive charge. On the other hand, carbon hold negative and positive charges but mainly negative charge owing to steric effects. Nitrogen is found in all the investigated molecules (BDTD1–BDTD8) and has highest electronegativity as compared to all other atoms in all these compounds. Fluorine present in BDTD5 and chlorine in BDTD2 are electronegative hetero-atoms and also have negative charges but less than nitrogen. NPA is examined as a superior technique in elucidating the differences in atomic electro-negativities in compound. Extensively, this approach has anticipated reactive behaviour in all the designed compounds, i.e. BDTD1–BDTD8.
4 Conclusion
The major objective of this investigation was to design and evaluate the NLO properties of benzodithiophene-based fullerene-free organic chromophores (BDTD1–BDTD8) having D-A configuration. All such chromophores were developed via A-D-A-type compound (BDTR) through the modification of structures with different acceptor groups. The impact of diverse acceptors was significant for nonlinear amplitudes. The UV–Vis results predicted highest λmax value in both chloroform and gaseous phases of 717.211 and 748.426 nm, respectively, in BDTD3. FMO results exposed that all of the structures have a lower energy band gap in the range of 2.536–2.057 eV as compared to BDTR (2.594 eV) along with higher bathochromic absorption spectra. The Eb was illustrated to be in ascending order as: BDTD5 < BDTD4 < BDTD2 < BDTD8 < BDTD6 < BDTD3 < BDTD7 < BDTD1 < BDTR. Our designed compounds were found with lesser Eb values as compared to BDTR. An efficient charge transference is noted from donor towards acceptor in HOMO/LUMO via DOS and TDM analyses. The values of ⟨α⟩, βtot and γtot were greater for the designed derivatives in contrast to the reference (BDTR). Among the tailored structures, the molecule (BDTD7) showed the highest value of ⟨α⟩ (3.261 × 10−22esu), while the compound (BDTD3) displayed a large βtot and γtot values of 2.324 × 10−27and 4.302 × 10–32esu, respectively. Among all the designed compounds, BDTD3 proved to be the best derivative owing to its nonlinearity-based results in all activities. It can be concluded that this work might assist in the advancement of non-fullerene-based organic chromophores for the betterment of optical devices and proved to be the valuable materials in the field of NLO.
Data Availability
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Sivasankari, B.; Roopan, S.M.: L-Malic acid-doped Guanidinium Carbonate crystal: a New NLO material and its photoluminescence study. Optik 226, 165909 (2021)
Xu, H.-L.; Sun, S.-L.; Muhammad, S.; Su, Z.-M.: Three-propeller-blade-shaped electride: remarkable alkali-metal-doped effect on the first hyperpolarizability. Theoret. Chem. Acc. 128, 241–248 (2011)
Khalid, M.; Ali, A.; Jawaria, R.; Asghar, M.A.; Asim, S.; Khan, M.U.; Hussain, R.; Ur Rehman, M.F.; Ennis, C.J.; Akram, M.S.: First principles study of electronic and nonlinear optical properties of A-D–π–A and D–A–D–π–A configured compounds containing novel quinoline–carbazole derivatives. RSC Advances. 10, 22273–22283 (2020)
Saeed, A.; Muhammad, S.; Rehman, S.; Bibi, S.; Al-Sehemi, A.G.; Khalid, M.: Exploring the impact of central core modifications among several push-pull configurations to enhance nonlinear optical response. J. Mol. Graph. Model. 100, 107665 (2020)
Khan, M.U.; Khalid, M.; Ibrahim, M.; Braga, A.A.C.; Safdar, M.; Al-Saadi, A.A.; Janjua, M.R.S.A.: First theoretical framework of triphenylamine–dicyanovinylene-based nonlinear optical dyes: structural modification of π-linkers. J. Phys. Chem. C. 122, 4009–4018 (2018)
Kosar, N.; Ayub, K.; Mahmood, T.: Surface functionalization of twisted graphene C32H15 and C104H52 derivatives with alkalis and superalkalis for NLO response; a DFT study. J. Mol. Graph. Model. 102, 107794 (2021)
Zhong, R.-L.; Xu, H.-L.; Muhammad, S.; Zhang, J.; Su, Z.-M.: The stability and nonlinear optical properties: encapsulation of an excess electron compound LiCN⋯ Li within boron nitride nanotubes. J. Mater. Chem. 22, 2196–2202 (2012)
Vasumathi, S.; Jeyakumar, H.J.; Selvarajan, P.: Spectral, NLO, thermal, hardness and SEM studies of phosphate doped bis-urea oxalic acid crystals for laser applications. Chin. J. Phys. 73, 1–12 (2021)
Ahsin, A.; Ayub, K.: Remarkable electronic and NLO properties of bimetallic superalkali clusters: a DFT study. J. Nanostruct. Chem. 2, 1–17 (2021)
Khan, B.; Khalid, M.; Shah, M.R.; Tahir, M.N.; Asif, H.M.; Aliabad, H.A.R.; Hussain, A.: Synthetic, spectroscopic, SC-XRD and nonlinear optical analysis of potent hydrazide derivatives: a comparative experimental and DFT/TD-DFT exploration. J. Mol. Struct. 1200, 127140 (2020)
Akram, M.; Adeel, M.; Khalid, M.; Tahir, M.N.; Khan, M.U.; Asghar, M.A.; Ullah, M.A.; Iqbal, M.: A combined experimental and computational study of 3-bromo-5-(2, 5-difluorophenyl) pyridine and 3, 5-bis (naphthalen-1-yl) pyridine: Insight into the synthesis, spectroscopic, single crystal XRD, electronic, nonlinear optical and biological properties. J. Mol. Struct. 1160, 129–141 (2018)
Ivanov, I.P.; Li, X.; Dolan, P.R.; Gu, M.: Nonlinear absorption properties of the charge states of nitrogen-vacancy centers in nanodiamonds. Opt. Lett. 38, 1358–1360 (2013)
Zhang, B.; Shi, G.; Yang, Z.; Zhang, F.; Pan, S.: Fluorooxoborates: beryllium-free deep-ultraviolet nonlinear optical materials without layered growth. Angew. Chem. Int. Ed. 56, 3916–3919 (2017)
Li, M.; Li, Y.; Zhang, H.; Wang, S.; Ao, Y.; Cui, Z.: Molecular engineering of organic chromophores and polymers for enhanced bulk second-order optical nonlinearity. J. Mater. Chem. C. 5, 4111–4122 (2017)
Zhao, Y.; Li, H.; Shao, Z.; Xu, W.; Meng, X.; Song, Y.; Hou, H.: Investigation of regulating third-order nonlinear optical property by coordination interaction. Inorg. Chem. 58, 4792–4801 (2019)
Issa, Y.M.; Abdel-Latif, S.A.; El-Ansary, A.L.; Hassib, H.B.: The synthesis, spectroscopic characterization, DFT/TD-DFT/PCM calculations of the molecular structure and NBO of the novel charge-transfer complexes of pyrazine Schiff base derivatives with aromatic nitro compounds. New J. Chem. 45, 1482–1499 (2021)
Abdel-Kader, N.S.; Abdel-Latif, S.A.; El-Ansary, A.L.; Sayed, A.G.: Spectroscopic studies, density functional theory calculations, non-linear optical properties, biological activity of 1-hydroxy-4-((4-(N-(pyrimidin-2-yl) sulfamoyl) phenyl) diazenyl)-2-naphthoic acid and its chelates with Nickel (II), Copper (II), Zinc (II) and Palladium (II) metal ions. J. Mol. Struct. 1223, 129203 (2021)
Janjua, M.R.S.A.: Nonlinear optical response of a series of small molecules: quantum modification of π-spacer and acceptor. J. Iran. Chem. Soc. 14, 2041–2054 (2017)
Coe, B.J.; Foxon, S.P.; Harper, E.C.; Raftery, J.; Shaw, R.; Swanson, C.A.; Asselberghs, I.; Clays, K.; Brunschwig, B.S.; Fitch, A.G.: Nonlinear optical and related properties of iron (II) pentacyanide complexes with quaternary nitrogen electron acceptor units. Inorg. Chem. 48, 1370–1379 (2009)
Janjua, M.R.S.A.; Jamil, S.; Mahmood, A.; Zafar, A.; Haroon, M.; Bhatti, H.N.: Solvent-dependent non-linear optical properties of 5, 5′-disubstituted-2, 2′-bipyridine complexes of ruthenium (II): a quantum chemical perspective. Aust. J. Chem. 68, 1502–1507 (2015)
Janjua, M.R.S.A.; Yamani, Z.H.; Jamil, S.; Mahmood, A.; Ahmad, I.; Haroon, M.; Tahir, M.H.; Yang, Z.; Pan, S.: First principle study of electronic and non-linear optical (NLO) properties of triphenylamine dyes: interactive design computation of new NLO compounds. Aust. J. Chem. 69, 467–472 (2015)
Lacroix, P.G.; Malfant, I.; Lepetit, C.: Second-order nonlinear optics in coordination chemistry: an open door towards multi-functional materials and molecular switches. Coord. Chem. Rev. 308, 381–394 (2016)
Kato, S.; Diederich, F.: Non-planar push–pull chromophores. Chem. Commun. 46, 1994–2006 (2010)
Roncali, J.: Molecular bulk heterojunctions: an emerging approach to organic solar cells. Acc. Chem. Res. 42, 1719–1730 (2009)
Mahmood, A.; Abdullah, M.I.; Nazar, M.F.: Quantum chemical designing of novel organic non-linear optical compounds. Bull. Korean Chem. Soc. 35, 1391–1396 (2014)
Wadsworth, A.; Moser, M.; Marks, A.; Little, M.S.; Gasparini, N.; Brabec, C.J.; Baran, D.; McCulloch, I.: Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells. Chem. Soc. Rev. 48, 1596–1625 (2019)
Liu, W.; Xu, X.; Yuan, J.; Leclerc, M.; Zou, Y.; Li, Y.: Low-bandgap non-fullerene acceptors enabling high-performance organic solar cells. ACS Energy Lett. 6, 598–608 (2021)
Lin, Y.; Zhan, X.: Non-fullerene acceptors for organic photovoltaics: an emerging horizon. Mater. Horiz. 1, 470–488 (2014)
Zhao, W.; Li, S.; Yao, H.; Zhang, S.; Zhang, Y.; Yang, B.; Hou, J.: Molecular optimization enables over 13% efficiency in organic solar cells. J. Am. Chem. Soc. 139, 7148–7151 (2017)
Liang, N.; Jiang, W.; Hou, J.; Wang, Z.: New developments in non-fullerene small molecule acceptors for polymer solar cells. Mater. Chem. Front. 1, 1291–1303 (2017)
Saeed, M.U.; Iqbal, J.; Mehmood, R.F.; Akram, S.J.; El-Badry, Y.A.; Noor, S.; Khera, R.A.: End-capped modification of Y-Shaped dithienothiophen[3,2-b]-pyrrolobenzothiadiazole (TPBT) based non-fullerene acceptors for high performance organic solar cells by using DFT approach. Surf. Interfaces. 30, 101875 (2022)
Wang, L.; An, Q.; Yan, L.; Bai, H.-R.; Jiang, M.; Mahmood, A.; Yang, C.; Zhi, H.; Wang, J.-L.: Non-fullerene acceptors with hetero-dihalogenated terminals induce significant difference in single crystallography and enable binary organic solar cells with 17.5% efficiency. Energy Environ. Sci. 15, 320–333 (2022)
Ge, G.-Y.; Li, J.-T.; Wang, J.-R.; Xiong, M.; Dong, X.; Li, Z.-J.; Li, J.-L.; Cao, X.-Y.; Lei, T.; Wang, J.-L.: Unveiling the interplay among end group, molecular packing, doping level, and charge transport in N-doped small-molecule organic semiconductors. Adv. Func. Mater. 32, 2108289 (2022)
Yao, C.; Yang, Y.; Li, L.; Bo, M.; Peng, C.; Huang, Z.; Wang, J.: Replacing the cyano (–C [triple bond, length as m-dash] N) group to design environmentally friendly fused-ring electron acceptors. Phys. Chem. Chem. Phys. 23, 18085–18092 (2021)
Yu, Q.; Xu, J.; Fu, J.; Xu, T.; Yan, X.; Chen, S.; Chen, H.; Sun, K.; Kan, Z.; Lu, S.: Crystallinity dictates the selection of fullerene or non-fullerene acceptors in a small molecule organic solar cell. Dyes Pigm. 187, 109085 (2021)
Frisch, M.J.; Clemente, F.R.: Gaussian 09, revision a. 01, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, V. Barone, B. Mennucci, GA Petersson, H. Nakatsuji, M. Caricato, X. Li, HP Hratchian, AF Izmaylov, J. Bloino, G. Zhe. 20–44 (2009)
Bryantsev, V.S.; Diallo, M.S.; Van Duin, A.C.; Goddard, W.A., III.: Evaluation of B3LYP, X3LYP, and M06-class density functionals for predicting the binding energies of neutral, protonated, and deprotonated water clusters. J. Chem. Theory Comput. 5, 1016–1026 (2009)
Bibi, A.; Muhammad, S.; UrRehman, S.; Bibi, S.; Bashir, S.; Ayub, K.; Adnan, M.; Khalid, M.: Chemically modified quinoidal oligothiophenes for enhanced linear and third-order nonlinear optical properties. ACS Omega 6, 24602 (2021)
Khalid, M.; Khan, M.U.; Hussain, R.; Irshad, S.; Ali, B.; Braga, A.A.C.; Imran, M.; Hussain, A.: Exploration of second and third order nonlinear optical properties for theoretical framework of organic D–π–D–π–A type compounds. Opt. Quant. Electron. 53, 1–19 (2021)
Hudson, J.J.; Sauer, B.E.; Tarbutt, M.R.; Hinds, E.A.: Measurement of the electron electric dipole moment using YbF molecules. Phys. Rev. Lett. 89, 023003 (2002)
Alparone, A.: Linear and nonlinear optical properties of nucleic acid bases. Chem. Phys. 410, 90–98 (2013)
Oudar, J.-L.; Chemla, D.S.: Hyperpolarizabilities of the nitroanilines and their relations to the excited state dipole moment. J. Chem. Phys. 66, 2664–2668 (1977)
Naik, V.S.; Patil, P.S.; Wong, Q.A.; Quah, C.K.; Gummagol, N.B.; Jayanna, H.S.: Molecular structure, linear optical, second and third-order nonlinear optical properties of two non-centrosymmetric thiophene-chalcone derivatives. J. Mol. Struct. 1222, 128901 (2020)
Dennington, R.; Keith, T.; Millam, J.: Semichem Inc. Shawnee Mission KS, GaussView, Version. 5, (2009)
Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R.: Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 4, 1–17 (2012)
Zhurko, G.A.: Chemcraft: http://www.chemcraftprog. com. Received: October. 22, (2014)
O’boyle, N.M.; Tenderholt, A.L.; Langner, K.M.: Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845 (2008)
Lu, T.; Chen, F.: Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012)
Afzal, Z.; Hussain, R.; Khan, M.U.; Khalid, M.; Iqbal, J.; Alvi, M.U.; Adnan, M.; Ahmed, M.; Mehboob, M.Y.; Hussain, M.: Designing indenothiophene-based acceptor materials with efficient photovoltaic parameters for fullerene-free organic solar cells. J. Mol. Model. 26, 1–17 (2020)
Hussain, R.; Khan, M.U.; Mehboob, M.Y.; Khalid, M.; Iqbal, J.; Ayub, K.; Adnan, M.; Ahmed, M.; Atiq, K.; Mahmood, K.: Enhancement in photovoltaic properties of N, N-diethylaniline based donor materials by bridging core modifications for efficient solar cells. ChemistrySelect 5, 5022–5034 (2020)
Khan, M.U.; Hussain, R.; Mehboob, M.Y.; Khalid, M.; Ehsan, M.A.; Rehman, A.; Janjua, M.R.S.A.: First theoretical framework of Z-shaped acceptor materials with fused-chrysene core for high performance organic solar cells. Spectrochimica Acta Part A. 245, 118938 (2021)
Haroon, M.; Al-Saadi, A.A.; Janjua, M.R.S.A.: Insights into end-capped modifications effect on the photovoltaic and optoelectronic properties of S-shaped fullerene-free acceptor molecules: a density functional theory computational study for organic solar cells. J. Phys. Org. Chem. 35, e4314 (2022)
Janjua, M.R.S.A.: Prediction and understanding: quantum chemical framework of transition metals enclosed in a B12N12 inorganic nanocluster for adsorption and removal of DDT from the environment. Inorg. Chem. 60, 10837–10847 (2021)
Janjua, M.R.S.A.: How does bridging core modification alter the photovoltaic characteristics of triphenylamine-based hole transport materials? Theoretical understanding and prediction. Chem. A Eur. J. 27, 4197–4210 (2021)
Adeel, M.; Khalid, M.; Ullah, M.A.; Muhammad, S.; Khan, M.U.; Tahir, M.N.; Khan, I.; Asghar, M.; Mughal, K.S. (2010) Exploration of CH⋯F & CF⋯H mediated supramolecular arrangements into fluorinated terphenyls and theoretical prediction of their third-order nonlinear optical response. RSC Adv. 11, 7766–7778.
Khalid, M.; Ali, A.; Abid, S.; Tahir, M.N.; Khan, M.U.; Ashfaq, M.; Imran, M.; Ahmad, A.: Facile ultrasound-based synthesis, SC-XRD, DFT exploration of the substituted acyl-hydrazones: an experimental and theoretical slant towards supramolecular chemistry. ChemistrySelect 5, 14844–14856 (2020)
Jiao, C.; Guo, Z.; Sun, B.; Meng, L.; Wan, X.; Zhang, M.; Zhang, H.; Li, C.; Chen, Y.: An acceptor–donor–acceptor type non-fullerene acceptor with an asymmetric backbone for high performance organic solar cells. J. Mater. Chem. C. 8, 6293–6298 (2020)
Cai, K.; Wu, H.; Hua, T.; Liao, C.; Tang, H.; Wang, L.; Cao, D.: Molecular engineering of the fused azacycle donors in the D-A-π-A metal-free organic dyes for efficient dye-sensitized solar cells. Dyes Pigm. 197, 109922 (2022)
Haroon, M.; Janjua, M.R.S.A.: Prediction of NLO response of substituted organoimido hexamolybedate: First theoretical framework based on p-anisidine adduct [Mo6O18 (p-MeOC6H4N)] 2. Mater. Today Commun. 26, 101880 (2021)
Jezuita, A.; Ejsmont, K.; Szatylowicz, H.: Substituent effects of nitro group in cyclic compounds. Struct. Chem. 32, 179–203 (2021)
Pham, P.-T.; Xia, Y.; Frisbie, C.D.; Bader, M.M.: Single crystal field effect transistor of a Y-shaped ladder-type oligomer. J. Phys. Chem. C. 112, 7968–7971 (2008)
Cai, X.; Burand, M.W.; Newman, C.R.; da Silva Filho, D.A.; Pappenfus, T.M.; Bader, M.M.; Brédas, J.-L.; Mann, K.R.; Frisbie, C.D.: N-and P-channel transport behavior in thin film transistors based on tricyanovinyl-capped oligothiophenes. J. Phys. Chem. B 110, 14590–14597 (2006)
Seo, D.-K.; Hoffmann, R.: Direct and indirect band gap types in one-dimensional conjugated or stacked organic materials. Theoret. Chem. Acc. 102, 23–32 (1999)
Pearson, R.G.: Absolute electronegativity and absolute hardness of Lewis acids and bases. J. Am. Chem. Soc. 107, 6801–6806 (1985)
Parr, R.G.; Yang, W.: Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 106, 4049–4050 (1984)
Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E.: Electronegativity: the density functional viewpoint. J. Chem. Phys. 68, 3801–3807 (1978)
Parthasarathi, R.; Padmanabhan, J.; Elango, M.; Subramanian, V.; Chattaraj, P.K.: Intermolecular reactivity through the generalized philicity concept. Chem. Phys. Lett. 394, 225–230 (2004)
Parr, R.G.; Pearson, R.G.: Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516 (1983)
Politzer, P.; Truhlar, D.G.: Introduction: the role of the electrostatic potential in chemistry. Chem. Appl. 5, 1–6 (1981)
Chattaraj, P.K.; Roy, D.R.: Update 1 of: electrophilicity index. Chem. Rev. 107, PR46–PR74 (2007)
Amiri, S.S.; Makarem, S.; Ahmar, H.; Ashenagar, S.: Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1, 3, 4-oxadiazol-2-yl) thio) benzene-1, 2-diol. J. Mol. Struct. 1119, 18–24 (2016)
Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K.: Electrophilicity-based charge transfer descriptor. J. Phys. Chem. A 111, 1358–1361 (2007)
Koopmans, T.: The classification of wave functions and eigen-values to the single electrons of an atom. Physica. 1, 104–113 (1934)
Mirkamali, E.S.; Ahmadi, R.; Kalateh, K.; Zarei, G.: 251. Adsorption of Melphalan anticancer drug on the surface of carbon nanotube: a comprehensive DFT study. Int. J. 9, 11 (2020)
Arshad, M.N.; Khalid, M.; Asad, M.; Braga, A.A.; Asiri, A.M.; Alotaibi, M.M.: Influence of peripheral modification of electron acceptors in nonfullerene (O-IDTBR1)-based derivatives on nonlinear optical response: DFT/TDDFT study. ACS Omega 7, 11631–11642 (2022)
Azeem, U.; Khera, R.A.; Naveed, A.; Imran, M.; Assiri, M.A.; Khalid, M.; Iqbal, J.: Tuning of a A-A–D–A–A-type small molecule with benzodithiophene as a central core with efficient photovoltaic properties for organic solar cells. ACS Omega 6, 28923–28935 (2021)
Khan, M.U.; Mehboob, M.Y.; Hussain, R.; Afzal, Z.; Khalid, M.; Adnan, M.: Designing spirobifullerene core based three-dimensional cross shape acceptor materials with promising photovoltaic properties for high-efficiency organic solar cells. Int. J. Quantum Chem. 120, e26377 (2020)
Mahmood, A.; Abdullah, M.I.; Khan, S.U.-D.: Enhancement of nonlinear optical (NLO) properties of indigo through modification of auxiliary donor, donor and acceptor. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 139, 425–430 (2015)
Lu, X.: DC-C.: Fractal geometry and architecture design: case study review. Chaotic Model. Simul. (CMSIM). 311, 322 (2012)
Khalid, M.; Lodhi, H.M.; Khan, M.U.; Imran, M.: Structural parameter-modulated nonlinear optical amplitude of acceptor–π–D–π–donor-configured pyrene derivatives: a DFT approach. RSC Adv. 11, 14237–14250 (2021)
Mahmood, A.; Khan, S.U.-D.; Rana, U.A.; Tahir, M.H.: Red shifting of absorption maxima of phenothiazine based dyes by incorporating electron-deficient thiadiazole derivatives as π-spacer. Arab. J. Chem. 12, 1447–1453 (2019)
Adeoye, M.D.; Adeogun, A.I.; Adewuyi, S.; Ahmed, S.A.; Odozi, N.W.; Obi-Egbeedi, N.O.: Effect of solvents on the electronic absorption spectra of 9, 14 dibenzo (a, c) phenazine and tribenzo (a, c, i) phenazine. Sci. Res. Essays. 4, 107–111 (2009)
Uzun, S.; Esen, Z.; Koç, E.; Usta, N.C.; Ceylan, M.: Experimental and density functional theory (MEP, FMO, NLO, Fukui functions) and antibacterial activity studies on 2-amino-4-(4-nitrophenyl)-5, 6-dihydrobenzo [h] quinoline-3-carbonitrile. J. Mol. Struct. 1178, 450–457 (2019)
Khalid, M.; Arshad, M.N.; Murtaza, S.; Shafiq, I.; Haroon, M.; Asiri, A.M.; de AlcântaraMorais, S.F.; Braga, A.A.: Enriching NLO efficacy via designing non-fullerene molecules with the modification of acceptor moieties into ICIF2F: an emerging theoretical approach. RSC Adv. 12, 13412–13427 (2022)
Khalid, M.; Khan, M.U.; Shafiq, I.; Hussain, R.; Ali, A.; Imran, M.; Braga, A.A.; Fayyaz ur Rehman, M.; Akram, M.S.: Structural modulation of π-conjugated linkers in D–π–A dyes based on triphenylamine dicyanovinylene framework to explore the NLO properties. R. Soc. Open Sci. 8, 210570 (2021)
Won, Y.S.; Yang, Y.S.; Kim, J.H.; Ryu, J.-H.; Kim, K.K.; Park, S.S.: Organic photosensitizers based on terthiophene with alkyl chain and double acceptors for application in dye-sensitized solar cells. Energy Fuels 24, 3676–3681 (2010)
Ans, M.; Iqbal, J.; Ayub, K.; Ali, E.; Eliasson, B.: Spirobifluorene based small molecules as an alternative to traditional fullerene acceptors for organic solar cells. Mater. Sci. Semicond. Process. 94, 97–106 (2019)
Ans, M.; Iqbal, J.; Ahmad, Z.; Muhammad, S.; Hussain, R.; Eliasson, B.; Ayub, K.: Designing three-dimensional (3D) non-fullerene small molecule acceptors with efficient photovoltaic parameters. ChemistrySelect 3, 12797–12804 (2018)
Liu, Z.; Lu, T.; Chen, Q.: An sp-hybridized all-carboatomic ring, cyclo [18] carbon: Electronic structure, electronic spectrum, and optical nonlinearity. Carbon 165, 461–467 (2020)
Liu, Z.; Lu, T.: Optical properties of novel conjugated nanohoops: Revealing the effects of topology and size. J. Phys. Chem. C. 124, 7353–7360 (2020)
Yuan, J.; Yuan, Y.; Tian, X.; Wang, H.; Liu, Y.; Feng, R.: Photoswitchable boronic acid derived salicylidenehydrazone enabled by photochromic spirooxazine and fulgide moieties: multiple responses of optical absorption, fluorescence emission, and quadratic nonlinear optics. J. Phys. Chem. C. 123, 29838–29855 (2019)
Hassan, T.; Hussain, R.; Khan, M.U.; Habiba, U.; Irshad, Z.; Adnan, M.; Lim, J.: Development of non-fused acceptor materials with 3D-Interpenetrated structure for stable and efficient organic solar cells. Mater. Sci. Semicond. Process. 151, 107010 (2022)
Köse, M.E.: Evaluation of acceptor strength in thiophene coupled donor–acceptor chromophores for optimal design of organic photovoltaic materials. J. Phys. Chem. A 116, 12503–12509 (2012)
Kim, B.-G.; Zhen, C.-G.; Jeong, E.J.; Kieffer, J.; Kim, J.: Organic dye design tools for efficient photocurrent generation in dye-sensitized solar cells: exciton binding energy and electron acceptors. Adv. Func. Mater. 22, 1606–1612 (2012)
Ans, M.; Iqbal, J.; Eliasson, B.; Ayub, K.: Opto-electronic properties of non-fullerene fused-undecacyclic electron acceptors for organic solar cells. Comput. Mater. Sci. 159, 150–159 (2019)
Shkir, M.; Muhammad, S.; AlFaify, S.; Chaudhry, A.R.; Al-Sehemi, A.G.: Shedding light on molecular structure, spectroscopic, nonlinear optical and dielectric properties of bis (thiourea) silver (I) nitrate single crystal: a dual approach. Arab. J. Chem. 12, 4612–4626 (2019)
Ali, A.; Khalid, M.; Rehman, M.F.U.; Haq, S.; Ali, A.; Tahir, M.N.; Ashfaq, M.; Rasool, F.; Braga, A.A.C.: Efficient synthesis, SC-XRD, and theoretical studies of O-Benzenesulfonylated pyrimidines: role of noncovalent interaction influence in their supramolecular network. ACS Omega 5, 15115–15128 (2020)
Yang, C.-C.; Zheng, X.-L.; Tian, W.Q.; Li, W.-Q.; Yang, L.: Tuning the edge states in X-type carbon based molecules for applications in nonlinear optics. Phys. Chem. Chem. Phys. 24, 7713–7722 (2022)
Khalid, M.; Khan, M.U.; Shafiq, I.; Hussain, R.; Mahmood, K.; Hussain, A.; Jawaria, R.; Hussain, A.; Imran, M.; Assiri, M.A.: NLO potential exploration for D–π–A heterocyclic organic compounds by incorporation of various π-linkers and acceptor units. Arab. J. Chem. 14, 103295 (2021)
Ashfaq, M.; Ali, A.; Tahir, M.N.; Khalid, M.; Assiri, M.A.; Imran, M.; Munawar, K.S.; Habiba, U.: Synthetic approach to achieve halo imine units: solid-state assembly, DFT based electronic and non linear optical behavior. Chem. Phys. Lett. 803, 139843 (2022)
Buvaneswari, M.; Santhakumari, R.; Usha, C.; Jayasree, R.; Sagadevan, S.: Synthesis, growth, structural, spectroscopic, optical, thermal, DFT, HOMO–LUMO, MEP, NBO analysis and thermodynamic properties of vanillin isonicotinic hydrazide single crystal. J. Mol. Struct. 1243, 130856 (2021)
Mahmood, A.; Khan, S.U.-D.; Rana, U.A.; Janjua, M.R.S.A.; Tahir, M.H.; Nazar, M.F.; Song, Y.: Effect of thiophene rings on UV/visible spectra and non-linear optical (NLO) properties of triphenylamine based dyes: a quantum chemical perspective. J. Phys. Org. Chem. 28, 418–422 (2015)
Adant, C.; Dupuis, M.; Bredas, J.L.: Ab initio study of the nonlinear optical properties of urea: electron correlation and dispersion effects. Int. J. Quantum Chem. 56, 497–507 (1995)
Demircioğlu, Z.; Kaştaş, G.; Kaştaş, Ç.A.; Frank, R.: Spectroscopic, XRD, Hirshfeld surface and DFT approach (chemical activity, ECT, NBO, FFA, NLO, MEP, NPA& MPA) of (E)-4-bromo-2-[(4-bromophenylimino) methyl]-6-ethoxyphenol. J. Mol. Struct. 1191, 129–137 (2019)
Demircioğlu, Z.; Kaştaş, Ç.A.; Büyükgüngör, O.: X-ray structural, spectroscopic and computational approach (NBO, MEP, NLO, NPA, fukui function analyses) of (E)-2-((4-bromophenylimino) methyl)-3-methoxyphenol. Mol. Cryst. Liq. Cryst. 656, 169–184 (2017)
Acknowledgements
Dr. Muhammad Khalid gratefully acknowledges the financial support of HEC Pakistan (project no. 20-14703/NRPU/R&D/HEC/2021). Authors are thankful for cooperation and collaboration of A.A.C.B from IQ-USP, Brazil, especially for his continuous support and providing computational laboratory facilities. A.A.C.B. (grant 2015/01491-3) is highly thankful to Fundação de Amparo à Pesquisa do Estado de São Paulo for the cooperation and financial assistance. The authors thank the Researchers Supporting Project number (RSP2023R29), King Saud University, Riyadh, Saudi Arabia.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Khalid, M., Maqsood, R., Shafiq, I. et al. Theoretical Approach towards Benzodithiophene-Based Chromophores with Extended Acceptors for Prediction of Efficient Nonlinear Optical Behaviour. Arab J Sci Eng 49, 339–359 (2024). https://doi.org/10.1007/s13369-023-08136-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13369-023-08136-6