
Abstract
Neurotropic viruses can infiltrate the CNS by crossing the blood–brain barrier (BBB) through various mechanisms including paracellular, transcellular, and “Trojan horse” mechanisms during leukocyte diapedesis. These viruses belong to several families, including retroviruses; human immunodeficiency virus type 1 (HIV-1), flaviviruses; Japanese encephalitis (JEV); and herpesviruses; herpes simplex virus type 1 (HSV-1), Epstein-Barr virus (EBV), and mouse adenovirus 1 (MAV-1). For entering the brain, viral proteins act upon the tight junctions (TJs) between the brain microvascular endothelial cells (BMECs). For instance, HIV-1 proteins, such as glycoprotein 120, Nef, Vpr, and Tat, disrupt the BBB and generate a neurotoxic effect. Recombinant-Tat triggers amendments in the BBB by decreasing expression of the TJ proteins such as claudin-1, claudin-5, and zona occludens-1 (ZO-1). Thus, the breaching of BBB has been reported in myriad of neurological diseases including multiple sclerosis (MS). Neurotropic viruses also exhibit molecular mimicry with several myelin sheath proteins, i.e., antibodies against EBV nuclear antigen 1 (EBNA1) aa411–426 cross-react with MBP and EBNA1 aa385–420 was found to be associated with MS risk haplotype HLA-DRB1*150. Notably, myelin protein epitopes (PLP139-151, MOG35-55, and MBP87-99) are being used to generate model systems for MS such as experimental autoimmune encephalomyelitis (EAE) to understand the disease mechanism and therapeutics. Viruses like Theiler’s murine encephalomyelitis virus (TMEV) are also commonly used to generate EAE. Altogether, this review provide insights into the viruses’ association with BBB leakiness and MS along with possible mechanistic details which could potentially use for therapeutics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
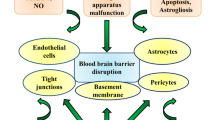
Numerous neurotropic viruses are commonly known to disrupt the blood–brain barrier (BBB) by triggering changes in the endothelial junctions through different entry mechanisms such as paracellular (between cells), transcellular (through cells), and “Trojan horse” (diapedesis of infected immune cells) (Chen and Li 2021; Spindler and Hsu 2012). These viruses include human T-cell leukemia virus (HTLV-1), human immunodeficiency virus-1 (HIV-1), West Nile virus (WNV), lymphocytic choriomeningitis virus (LCMV), mouse adenovirus type 1 (MAV-1), herpes simplex virus (HSV), and Epstein-Barr virus (EBV). WNV and its virus-like particles (VLPs) may pass through brain endothelial cell (EC) transcellular without affecting the integrity of the BBB while the paracellular entry of viruses triggers alteration in the tight junction (TJ) proteins, basal lamina, and cytoskeleton (Delorme-Axford and Coyne 2011). The functionality of BBB is governed by an intricate interaction of cells like endothelial, pericyte, and perivascular astrocytes (Takeshita and Ransohoff 2012). Pericytes are present on the abluminal side of the endothelial layer, have long cytoplasmic processes which make contact with numerous ECs, and thus, integrate signals along the vessel’s length and can also extend to more than one capillary (Bergers and Song 2005). During the disrupted BBB, the deposition of collagen, apoptotic changes in pericytes, and swelling of astrocytes have been observed (Fig. 1) (Claudio et al. 1995). Diminished BBB integrity showed continuous association with neurological diseases, namely, multiple sclerosis (MS), ischemic stroke, and traumatic brain injury (Profaci et al. 2020; Höftberger and Lassmann 2017). MS, being an immune-induced disease of the brain, is characterized by disruption of myelin sheath in white matter, loss of oligodendrocytes, changes in the BBB integrity, and leucocyte infiltration (Höftberger and Lassmann 2017). The leaky BBB has been found in MS lesions and revealed as the key step in its pathogenesis and development of inflammatory regions around the venules (Kolb et al. 2022). The leucocyte transmigration across the vasculature and leaky BBB aid in neuroinflammation as observed in MS (Bradl and Lassmann 2010; Stamatovic et al. 2008).

a Illustration of a healthy condition with no viral exposure and inflammatory molecules. b Representation of debilitated BBB due to viral, inflammatory and immune autoreactive insults. i Viruses activate different immune cells and trigger release of cytokines, which eventually target brain endothelial cells and exudate into the CNS. ii Several RNA and DNA viruses directly breach BBB transcellularly/paracellularly and enter into CNS, reactivating brain cells such as microglia and astrocytes. iii Autoantibodies primed against self-antigens (MBP, MOG, PLP, etc.) cross BBB follow the opsonization cascade and jeopardize the neuronal integrity by augmenting the cytokine storm. iv Several viruses follow the “Trojan horse” mechanism through diapedesis of immune cells. The disruption of BBB can be observed by the collagen deposition, pericyte apoptosis, and astrocyte swelling
Furthermore, the accumulation of perivascular fibrin provides an additional evidence for the chronic BBB anomaly in MS (Claudio et al. 1995). Several modulators like certain growth factors, cytokines/chemokines, matrix metalloproteinases (MMPs), various lipid mediators, and reactive oxygen species (ROS) affect this transcellular/paracellular BBB permeability of pathogens (Stamatovic et al. 2008). Likewise, chemokines also amend the binding affinity between the cell adhesion molecules (CAMs), triggering the infiltration of leukocytes and regenerative binding of T-cell with endothelium through intercellular adhesion molecule-1/lymphocyte function-associated antigen-1 (ICAM-1/LFA-1) and vascular cell adhesion molecule-1/alpha4beta1 integrin (VCAM-1/VLA-4) complexes (van Buul and Hordijk 2004). Then, the lymphocyte starts its migration process by adhering to the endothelial cell (Engelhardt and Wolburg 2004). Moreover, the increased permeability of BBB is also correlated with the clearance of therapeutic modalities against the neurotropic viruses in mice, suggesting an ineffective treatment against viral infections which eventually aid in MS (Fabis et al. 2007). The animal model commonly used for studying MS is experimental autoimmune encephalomyelitis (EAE), where autoimmunity is developed in the susceptible mice brain by administering the myelin and associated protein self-antigens (Robinson et al. 2014). Similarly, EAE can also be generated by using certain toxins like cuprizone and lysolecithin (Denic et al. 2011). Therefore, there are numerous studies available addressing these topics separately, and since these are overlapping topics, we aim to provide a summary of neurotropic virus-mediated BBB disruption and MS. Further, we have also mentioned the virus-associated mechanisms and highlight the significant viral as well as host protein molecules that can be a therapeutic target.
Viruses and unclasped blood–brain barrier
The BBB is held tightly by the TJs among the brain microvascular endothelial cells (BMECs). Pericytes and astrocytes cooperate with the BBB endothelium through several soluble factors along with neurons and form the neurovascular unit (NVU) (Sweeney et al. 2016). The disruption of BBB is a hallmark of central nervous system (CNS) infections that can instigate by host immune factors [tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β)] or viral factors (Erickson et al. 2012). Viruses may cross the BBB via numerous routes, including direct infection into BMECs, entry of infected peripheral leukocytes, or trans/paracellular viral migration through the endothelium (Chen and Li 2021). Neurotropic viruses such as retroviruses, rhabdoviruses, flaviviruses, picornaviruses, morbillivirus, bunyaviruses, alphaviruses, herpesviruses, and coronaviruses are known to invade the CNS (Ludlow et al. 2016) (Fig. 1). Viruses invade into the CNS directly by crossing the BBB or bypassing the BBB through non-hematogenous routes (Miner and Diamond 2016). The direct virus transit across the BBB includes the following: (i) a prominent level of viremia and inflammation-stimulating viruses spread over BMEC TJs; (2) direct virus entry into BMECs and transport of newly developed virions through basolateral membranes; (3) infected leukocytes migrate through the BBB to seed infectious virus in CNS which is known as “Trojan horse” pathway (Miner and Diamond 2016). Some viruses utilize non-hematogenous routes to get access into CNS. These mechanisms comprise the following: (i) reverse axonal transport of virions from the peripheral nervous system (PNS) to CNS and (ii) olfactory epithelium challenged by viruses triggers their transit to the CNS through the cribriform plate and olfactory bulb (Ludlow et al. 2016). It is highly plausible that certain viruses may use more than one pathway to enter the CNS. For instance, the Venezuelan equine encephalitis virus invades the CNS by cribriform plate followed by the delayed BBB opening that allows the second time viral neuro invasion straight through the BBB (Schäfer et al. 2011).
Involvement of retroviruses in blood–brain barrier permeability
Human immunodeficiency virus type 1
Among neurotropic viruses, the human retroviruses HIV-1, HTLV-1, usually invade through the hematogenous route (Miller et al. 2012). HIV-1 triggers acquired immunodeficiency syndrome (AIDS) and, due to the inflammatory status of AIDS-infected cells, lacks the crosstalk between the immune and endothelial cells. Inflammatory cytokines are known to increase the permeability of BBB (Miller et al. 2012) (Fig. 1), although the degree of BBB disruption differs among neurotropic viruses. For example, HIV-1 majorly enters the CNS by a “Trojan horse” mechanism and induces mild disruption, while the HSV leads to a higher disruption that can raise CSF albumin levels, 50 mg/dl which is three to six times higher than the normal levels (Erickson et al. 2012). The HIV-infected monocytes circumvent the BBB during normal patrolling of perivascular macrophages and eventually trigger the production of proinflammatory mediators, like chemokine ligand 2 (CCL2), which disrupt the BBB (Toborek et al. 2005). HIV-1 proteins, namely, trans-activator of transcription (Tat), glycoprotein 120 (gp120), negative factor (Nef), and HIV-1 viral protein R (Vpr), follow a mechanism of the cascade to disrupt the BBB and generate a neurotoxic effect (Maubert et al. 2017). Recombinant-Tat triggers downregulation in the expression of TJ proteins, e.g., zona occludens-2 (ZO-2), claudin-5, and claudin-1 in BBB cells (András et al. 2003). Likewise, the in-vivo challenge to recombinant-Tat triggered a redistribution of claudin-5 immunoreactivity (András et al. 2003). Besides, the Tat exposure in the brain hippocampus of mice (C57BL/6) resulted in the mitigation of ZO-1 mRNA levels and effectively reduced the ZO-1 continuity in brain microvessels (András et al. 2003). The above-stated changes are attributed to the induction of extracellular signal-regulated kinase 1/2 (ERK1/2), thus indicating that Tat-mediated oxidative stress is pivotally affecting BBB intactness via the ERK1/2 pathway (Pu et al. 2005). Moreover, Tat alters monocyte chemoattractant protein-1 (MCP-1) secretions from astrocyte/microglial cells and adhesion molecules in endothelial cells (Ajasin and Eugenin 2020). Intriguingly, an elevated risk of HIV-1-associated dementia (HAD) exhibited a correlation with the mutant MCP-1 allele (Ajasin and Eugenin 2020). In addition, BMECs express the HIV-1 co-receptors, i.e., C-X-C chemokine receptor type 4 (CXCR4) and C–C chemokine receptor type 5 (CCR5). Macrophage-derived gp120 (having CCR5 co-receptors) or lymphocytes (having CXCR4 co-receptors) were exposed to BMECs leading to reduce the BBB tightness and elevated monocyte migration across BBB (Kanmogne et al. 2007). In contrast, the BBB integrity was refurbished after dispensing gp120 (Kanmogne et al. 2007). According to an in-vitro study, antibodies against CCR5 and inhibitors for protein kinase C (PKC) and myosin light chain kinase blocked gp120-mediated surge of monocyte migration and eventually BBB permeability (Louboutin and Strayer 2012). Gp120 triggers the activation of three different isoforms of PKC in BMECs (PKC (pan)-bII, PKC-f/k, and PKC-a/bII). Besides, inhibitors against PKC targeting the ATP-binding and calcium release site block gp120-induced PKC activation and eventually increase the permeability of BBB (Kanmogne et al. 2007). Further, an in-vivo investigation also demonstrated the invasion of HIV-1 envelope protein into the murine brain (Banks et al. 2001). Depletion of capillary has shown that the radioactively labelled envelope glycoprotein+ (Env+) virus completely passes through the endothelial barrier of the brain to enter the CNS parenchyma while enveloping glycoprotein− (Env−) moiety has not shown any BBB crossing (Banks et al. 2001).
Human T-cell leukemia virus 1
HTLV-1 is also associated with numerous long-term neurological disorders called HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). HAM/TSP triggers BBB amendments such as the deposition of fibrinogen and immunoglobulin in the CNS parenchyma and the maneuver of lymphocytes carrying HTLV-1 through the BBB (Morris et al. 2019; Enose-Akahata et al. 2012). HTLV-1 tax protein epitope running from amino acid 346–353 unveiled cross-reactivity with a neuron-specific ribonucleoprotein (hnRNPA1); therefore, it develops autoantibodies, and such antibodies have also been detected in sera from HAM/TSP patients (Miner and Diamond 2016; Banks et al. 2001). The in-vitro and in-vivo studies have shown that HTLV-1 infected lymphocytes were able to change the structure of TJs, elevated paracellular permeability, and increased the transcellular migration due to IL-1α/TNF-α secretion (Futsch et al. 2018). In addition, feline immunodeficiency virus (FIV) exposure triggers impairments in the BBB and choroid plexus (Miller et al. 2012). Caprine arthritis encephalitis virus and Visna virus can also access the BBB by the “Trojan horse” mechanism (Miller et al. 2012) (Fig. 1).
An intricate interaction of flaviviruses with BBB
Japanese encephalitis virus
Several flaviviruses comprise neurotropic and neurovirulent characteristics, namely, Japanese encephalitis virus (JEV), dengue virus (DENV), zika virus (ZIKV), tick-borne encephalitis (TBEV), yellow fever virus (YFV), and WNV (Laureti et al. 2018). It is considered that these viruses enter the brain via a hematogenous route. JEV contributes to BBB disruption by decreasing the expression of claudin-5 and ZO-1. JEV is capable of infecting both BMECs and astrocytes (Liu et al. 2020). Interaction of BMECs with astrocytes is imperative for BBB permeability induced by JEV, which ultimately accelerates the secretion of inflammatory cytokines (Chang et al. 2015). These inflammatory species [i.e., IL-1β, IL-6, MCP-1, TNF-α, and inducible nitric oxide synthase (iNOS)] are released by microglia and create an environment that debilitates the endothelial barrier (Tohidpour et al. 2017) (Fig. 1). Subsequently, the activated microglia and astrocytes in the CNS of JEV-infected mice robustly produce cytokines: chemokine (C–C motif) ligand 5 (CCL5), C-X-C motif chemokine ligand 10 (CXCL10), chemokine (C–C motif) ligand 2 (CCL2), TNF-α, IL-6, and IFN-γ (Lannes et al. 2017). JEV RNA was detected in mice after 2 days post-infection (dpi) while BBB disintegration was visualized after 4 dpi (Li et al. 2015). However, the JEV-mediated breakdown of BBB suggests a bystander event rather than a direct virus infection in BBB endothelial cells (Chang et al. 2015).
West Nile virus
WNV infection triggers BMEC-intrinsic expression of inflammatory moieties (type I, type II IFNs, IFN-γ, IL-6, IL-1β, and TNF-α) at the BBB and eventually disrupts the cohesion among endothelial cells (Kumar et al. 2010) (Fig. 1). An in-vitro exposure of WNV to astrocytes results in the production of matrix metalloproteinases (MMP) that triggers the loss in TJ integrity (Roe et al. 2012). MMPs play a crucial role in WNV neuro invasion as established via a finding that revealed MMP9-/- mice showed resistance toward WNV (Wang et al. 2008). Exposure of human primary brain cortical astrocytes (HBCA) to WNV exhibited overexpression of MMP-1, -3, and -9 while a decrease of a tissue inhibitor of metalloproteinase-2 (TIMP-2) was observed. WNV supernatant when challenged to BMEC exhibited mitigation in the expression level of claudin and ZO-1 (Lazear et al. 2015). Contrarily, the reversed results were observed after the treatment of supernatants with inhibitors of MMP (Lazear et al. 2015). Therefore, it suggests that the production of MMPs induced by WNV may compromise BBB regardless of its infection to the endothelial cells. Notably, studies by Wang et al., and Morrey et al., showed an elevation in the BBB permeability upon exposure of WNV to mice (C57BL/6) (Wang et al. 2008; Morrey et al. 2008). Though it does not trigger any cytopathic effect, however, it alters claudin-1 and increases the level of E-selectin and vascular cell adhesion molecule 1 (VCAM-1) (Mustafá et al. 2019). Numerous pattern recognition receptors (PPRs) recognize the RNA of WNV and elicit innate immune responses. PPR-mediated induction of innate cytokines regulates the permeability of BBB and formation of TJs via proportionate activation of the Rac1 and RhoA (Daniels et al. 2014). It further regulates the transendothelial traffic of the virus. Attenuated type I interferon signalling or interferon induction [Ifnar (-/-) Irf7(-/-)] in mice showed increased BBB permeability and dysregulation of TJs upon WNV exposure (Daniels et al. 2014). Infection of WNV NY strain to mice (C57BL/6) results in upregulation of E-selectin, intercellular adhesion molecule (ICAM-1), and vascular cell adhesion molecule 1 (VCAM-1) in the brain. Knockout of ICAM-1 in mice have reduced the leukocyte infiltration via BBB, hence suggesting a decrease in the viral load of CNS (Hallahan and Virudachalam 1997). Several reports suggested that WNV follows a transcellular pathway to get entry into CNS without altering the BBB (Roe et al. 2012).
Zika virus
ZIKV has been detected in the brain of patients suffering from meningoencephalitis and microcephaly. Virus can invade the barrier of the brain by transcytosis (Fig. 1). The BBB model derived from a human-induced pluripotent stem cell (iPSC) revealed infection of ZIKV to infect brain endothelial cells (i-BECs) without creating a disturbance in the BBB barrier cohesiveness or permeability (Alimonti et al. 2018). Although no disintegration to the BBB was detected, post ZIKV infection and the newly generated virions were secreted from the BBB model on the abluminal side and infect the underlying neural progenitor cells (i-NPs) (Alimonti et al. 2018). Notably, i-BEC and i-NPs highly express the AXL (“anexelekto” in Greek means “uncontrolled”) which is a presumed ZIKV entry receptor (Alimonti et al. 2018).
Dengue virus
Association of DENV infection with neurological manifestations are still infrequent. In the case of severe dengue (SD), the leakage of plasma was observed in some patients (Li et al. 2017). The first report of DENV infection into brain cells was established using its serotype 2 with the primary cultures of human umbilical vein EC and rabbit cava vein (Calderón-Peláez et al. 2019). Endothelial cell surface-exposed intermediate filament, vimentin (rod domain), is being speculated to be entangled with domain III of its envelope protein (Yang et al. 2016). DENV infection induces Rho-associated coiled-coil-containing kinase (ROCK) reorganization and activation of vimentin which triggers the redistribution of endoplasmic reticulum for an effective viral cycle (replication and assembly) (Lei et al. 2013). In-vitro, by using primary culture and cell lines, DENV-infected endothelial cells were effectively low. DENV-4-infected mouse brain microvascular EC (MBEC) was proportionate to 7 and 12% at 24- and 48-h post-infection (hpi), respectively (Velandia-Romero et al. 2016), while the neuro-adapted D4MB-6 strain infected 50% of the cells at 48 hpi. In addition, activation of endothelial cells upon DENV infection is based on the secretion/expression of sICAM-1 and sVCAM-1 which was observed on the membrane of liver sinusoidal endothelial cells (LSEC) and HMEC-1 (Davies et al. 1993). Infection of D4MB-6 neuro-adapted strain to MBEC triggers expression of E-selectin, PECAM-1, VCAM-1, and ICAM-1 (Calderón-Peláez et al. 2019). Interestingly, the secretion of immune molecules by EC relies on different serotypes of DENV, e.g., DENV1 trigger the secretion of chemokine (C-X-C motif), ligand 1 (CXCL1), CCL2, CCL5, CCL20 IL-6, and TNF-a (Calderón-Peláez et al. 2019). Previous reports also suggested that the reaching of DENV4 in brain parenchyma is fatal, and its infection accelerates damage to neurons. Also, DENV4 was detected in serum, and the viral titer was higher in cerebrospinal fluid (CSF), suggesting a rapid virus entry through BBB (Calderón-Peláez et al. 2019). All the aforementioned molecules linked with enhanced endothelial permeability and amendments in the coagulator balance related to hemorrhages and dispersed intravascular coagulation were seen during SD (Calderón-Peláez et al. 2019).
Yellow fever virus
The neurovirulence of YFV has been usually evaluated using laboratory neuro-adapted strains and by intracerebral inoculation in mice. IFNA and/or IFNLR mice are more vulnerable to YFV infection. IFNAR-/- mouse showed high viremia and viral load in numerous organs like the kidney and spleen, but it remained same with time. IFNAR-dispensed mice not only showed reduced weight but also unveiled clinical manifestations (Erickson and Pfeiffer 2013). In contrast, IFNAR and IFNLR (ifnar-/-, ifnlr-/-) null mice succumb to YFV infection (Mustafá et al. 2019). In the aforementioned mice models, the permeability of BBB was detected after 5 dpi, while the presence of the virus was observed at 3 dpi (Erickson and Pfeiffer 2013). Thus, it poses the question of whether the disruption of BBB disruption is essential for the invasion of virus.
Tick-borne encephalitis
Yet, another virus, TBEV, is also known to manifest neurological disorders from mild meningitis to severe encephalomyelitis (Bogovic and Strle 2015). Various in-vitro analyses have estimated the virus cytopathic nature, efficient replication, and dissemination in neural cell lines and primary human neurons. Astrocytes are also prone to TBEV replication, though astrocytes are resistant to virus-mediated cytopathic effects. TBEV infects the primary astrocytes of rats persistently and no cellular toxicity was observed until 14 days (Ghita et al. 2021). TBEV infection affects cells in culture at a lower frequency, despite that the activation of astrocytes markedly showed upregulated response of glial fibrillary acidic protein (GFAP), an inflammatory cytokine, chemokines, and MMP-9 (Fares et al. 2020). Therefore, these results pointing toward the astrocyte exposed with TBEV affects the BBB permeability and ultimately the secretion of proinflammatory cytokines (type I IFN) and MMPs (Mustafá et al. 2019). Astrocytes from mice deficient with interferon-alpha/beta receptor (IFNAR) have nullified IFN response by using neutralizing antibodies and made mouse astrocytes more vulnerable to its infection (Lindqvist et al. 2016), whereas the infection with TBEV did not alter the expression and localization of TJ proteins along with adhesion molecules (Palus et al. 2017).
Association of BBB breaching with several other viruses
DNA viruses such as MAV-1 and HSV-1 have been reported to directly make amendments in the structure as well as in the function of BBB (Spindler and Hsu 2012). Adenoviruses enter and replicate after interacting with its receptor coxsackievirus and adenovirus receptor (CAR) which is a known TJ integral protein (Coyne et al. 2007). Nascently packed viruses come out from the basolateral surface and interact with CAR again and ultimately break the TJ to escape apically (Hou et al. 2016). HSV-1-mediated encephalitis obtains high morbidity which contributes to the aggravated immune response due to leukocyte infiltration (Michael et al. 2020). HSV-1 exposure to the murine model induces secretions of CXCL1 and CCL2 chemokines in the brain (Michael et al. 2020). Dispensing of CCL2 receptor in mice showed lower recruitment of monocytes, indefinite viral replication, and increased morbidity, whereas mice deficient with CXCL1 receptor have lower neutrophil recruitment, reduced BBB permeability, and morbidity without affecting the viral load (Michael et al. 2020). Astrocytes produce CXCL1 after inoculation with HSV-1 and CXCL1 is also crucial for neutrophil trans endothelial migration which corresponds to the breakdown of BBB. Thus, the CXCL1-CXCR2 axis depicts a plausible therapeutic target for HSV-1 mediated encephalitis (Michael et al. 2020). Moreover, EBV is also known to be involved with several neurological diseases but the mechanism of BBB crossing is not clear (Rani et al. 2023; Patra et al. 2023) (Fig. 1).
Besides, Reno virus are also known to insult TJ proteins for taking entry into the ependymal and neuronal cells by using junctional adhesion molecules (JAM) (Michalicová et al. 2017). Nonetheless, the hepatitis C virus (HCV) uses occluding-5 and claudin-1 as co-receptor for infection of the liver cells. Likewise, BMECs also express HCV receptors. Therefore, BBB permeability provides HCV with an extrahepatic infection target and contributes to neurological modalities (Fletcher et al. 2012). Also, the open reading frame 2 (ORF2) protein of hepatitis E virus (HEV) showed presence in the brain along with spinal cord sections of infected rabbits. After 48-h in-vitro inoculation of HBMVCs with HEV showed a decline in TJ proteins. Hence, these reports suggest that the BBB disruption might be a potential mechanism for invasion of HEV into CNS (Tian et al. 2019). Besides, infection with rabies virus (RV) is lethal for humans once it reaches the CNS. Several glycoproteins of RV regulate its pathogenesis by overseeing virus uptake rate and its replication and spread across synapses (Pulmanausahakul et al. 2008). External surface glycoprotein (G) of RV plays a key role in its uptake into neuronal cells (Dietzschold et al. 2005). An Asn194 → Lys194 mutation in RV G exhibited significant decrease in the internalization time (Dietzschold et al. 2008). Further, RV interacts at the neuromuscular junction with nicotinic acetylcholine receptors and travels within the motor and sensory axons to the CNS (Davis et al. 2015). Furthermore, the non-classical neurotropic viruses also show neuroinvasion in an age-dependent manner. For instance, chikungunya virus (CHIKV) is known to cause inflammatory arthritis and rarely involved with the neurological disease in adults, although in 2006 “La Reunion Island” CHIKV epidemic exhibited brain invasion (Soumahoro et al. 2011). Further, infection of CHIKV to neonates results various disease conditions like cerebral palsy, flaccid paralysis, seizure disorders, microcephaly, and even death (Mehta et al. 2018).
John Cunningham virus or human polyomavirus 2 (JCPyV/JCV) and DNA tumor virus infect ~ 80% human population in the early age (Safak and Khalili 2003; Assetta and Atwood 2017). JCPyV primarily infects oligodendrocytes which trigger demyelination of subcortical white matter in the CNS and subsequently stimulate the demyelinating disease like progressive multifocal leukoencephalopathy (PML) in the immunocompromised individuals (i.e., AIDS patients) (Elphick et al. 2004). The foci of JCPyV in the brain are related to blood vessels which can be corroborated with the hematogenous route as the primary site of infection, and eventually, it spread to numerous secondary sites such as kidney, lymphoid tissues, and brain (Chapagain and Nerurkar 2010). Notably, the detection of JCPyV genomic sequences in the brain autopsy samples obtained from immunocompromised patients suggests another site for its latency (Del Valle and Piña-Oviedo 2019). In the brain, this virus replicates into glial cells like oligodendrocytes and astrocytes and enter into these cells by using proteins like serotonin receptor 2A (5-HT2A-R) and β-arrestin (Mayberry et al. 2019). After maintaining the latency into B-lymphocytes, JCPyV can reach into the brain and potentially trigger PML (Harypursat et al. 2020). Reports also suggested the strong correlation of JCPyV entry into neural tissues directly by the choroid plexus (O’Hara et al. 2020). Intriguingly, the epithelial cells of primary choroid plexus express JCPyV receptors and exhibits its susceptibility for the viral infection. Upon entering to CNS, JCPyV may spread from cell to cell along the myelin sheath (Atkinson and Atwood 2020; Delbue et al. 2017). Moreover, coronaviruses, e.g., middle east respiratory syndrome-coronavirus (MERS-CoV) and severe acute respiratory syndrome coronavirus-1 (SARS-CoV-1), can potentially infect immune cells and may enter the brain. Thus, it also led to the speculation that SARS-CoV-2 may also enter into the CNS via infected immune cells (Morgello 2020).
Entanglement of neurotropic viruses with multiple sclerosis
The presence of several pathogens has been continuously overserved in MS clinical samples. Pathogens correlated with the generation and exacerbation of MS include viruses, such as herpesviruses, human endogenous retrovirus families, coronaviruses, and bacteria (Chlamydia pneumonia, Staphylococcus aureus) (Morgello 2020; Mentis et al. 2017). Nevertheless, to date, no single pathogenic agent is accepted as a causing agent. The involvement of viruses in MS has been speculated by the presence of anti-viral antibodies (CSF, brain tissue, and blood), viral DNA/RNA, and proteins of numerous viruses (Libbey et al. 2014a; Virtanen and Jacobson 2012). Certain common gastrointestinal, upper respiratory and urogenital tract infections also have some correlation with MS worsening (Marrodan et al. 2019). The contribution of different viruses in MS is hypothesized by different mechanisms such as direct viral toxicity, genetic/molecular mimicry, bystander activation, dual T-cell receptor, spreading of epitopes, and viral déjà vu (Fujinami et al. 2006; Donati 2020). MS pathogenesis has been contributed by many viruses, namely, EBV, HHV-6, Varicella-Zoster virus (VZV), Cytomegalovirus (CMV), JCPyV, human endogenous retroviruses (HERVs), HSV-1, and -2 (Virtanen and Jacobson 2012) (Fig. 1). Despite their belonging to different families, these viruses share the potential to alter the expression of host cell genes, dysregulation of the immune system, and disruption of myelin sheath (Fig. 1). Specifically, the ubiquitous prevalence of herpesviruses in adult populations can target neurons directly or indirectly and aid into tissue damage (Jakhmola et al. 2021). These infections remain asymptomatic and maintain lifelong latency in the immunocompetent hosts (Jha et al. 2016), while the reactivation of these viruses continues their replication cycle and results in cytotoxic effects in the virus-challenged cells (Jha et al. 2016).
Retroviruses and multiple sclerosis
The association of MS has been continuously reported in the upregulated reactivity to certain HERVs solely or in synchronization with herpes viruses (Cusick et al. 2013). Exogenous HIV involves in the activation of HERV via toll like receptor-4 (TLR-4) (Browne 2020). This connection was verified from brains of HIV patients through post-mortem and by checking the epitope cross-reactivity in T-cell responses toward HIV. An uneven ratio of CD8+ T-cell and CD4+ T-cell was also reported in HIV cases with demyelinating CNS (Stefanou et al. 2019) (Fig. 2). A study by Mameli et al. proposed that EBV activates HERV-W/MSRV/synctin-1 in astrocytes and peripheral blood-derived cells which hints a possible mechanism involved in MS (Mameli et al. 2012).

Antigen presenting cells such as dendritic cells presents unknown MS-related antigen/viral myelin sheath mimicking antigens to CD4+ T-cells, which in-turn become activated and eventually proliferate. These primed CD4+ T-cells initiate dendritic cells to activate CD8+ T-cells, and their clonal expansion into CD8+ regulatory (reg) T-cells and cytotoxic T lymphocytes (CD8+ CTL). CD8+ CTL can cause direct damage to the myelin layer and axons by releasing several cytokines and pro-apoptotic factors such as FasL. On the other hand, CD8+ regulatory cells reduce the inflammatory response by releasing IL-10. Activated CD4+ T-cells secret mainly pro-inflammatory cytokines and also activate B-cells, which produce myelin reactive antibodies. Cytokine interaction with BBB endothelial cells compromises the expression of tight junction proteins such as occludin and VCAM-1 and alters the activity of matrix metalloproteases. Furthermore, reactivated T-cells in the CNS increase the inflammatory responses. Brain cells, such as microglia and astrocytes, also become activated and release excessive inflammatory cytokines and excitotoxic substances, such as glutamate, nitric oxide (NO). Cytokines and chemokines (CCL2) expressed by microglia increases the infiltration of T-cells and macrophages, respectively. The complement factor, C5b-9, becomes activated, and cause directly damages the myelin sheath. These immune cascades lead to myelin sheath damage, destruction of oligodendrocytes, oligodendrocyte apoptosis, and axonal loss and promote neuronal hyperexcitability leading to the development of CNS neuropathies, such as MS
Besides, in the blood and/or brain and/or cerebrospinal fluid of MS, the complete virions, RNA, antibodies, gag, and env protein of HERV have been found, and the level of these moieties was associated with status/severity of disease (Morris et al. 2019). The single-nucleotide polymorphisms (SNPs) related to HERV susceptibility toward MS include rs662139 T/C of HERV-W env locus and rs391745 upstream to the HERV-Fc1 locus on the X chromosome, while SNPs in the tripartite motif-containing (TRIM) protein-encoding genes TRIM5 and TRIM22 showed negative correlation with MS (Morris et al. 2019).
Flaviviruses and multiple sclerosis
Among flavivirus, the non-structural protein, NS1 of ZIKV was detected in microglia by co-labelling with CD11b (Alves-Leon et al. 2019). Microglial role has already been investigated in several arbovirus infections such as dengue, affirming that it fabricates active immune defence during viral infection (Jhan et al. 2017). ZIKV have also showed its extensive presence in the acute disseminated encephalomyelitis (ADEM)-like syndrome and in the MRI lesions of MS (Alves-Leon et al. 2019). Moreover, antibodies against ZIKV was detected in the plasma (IgM) as well as in the brain biopsy samples (Stone et al. 2020). Besides, WNV is well known to trigger autoimmune diseases like CNS vasculitis with acute stroke and loss of vision. WNV is known to upregulate the both class major histocompatibility complex’s (MHC) in the PNS (i.e., Schwann cells) of Lewis’s rat (Leis and Stokic 2012). JEV infection into mice triggers demyelination. Reports has revealed the increased proliferation of myelin basic protein (MBP)-primed lymphocytes upon JEV exposure which possibly induce a cascade for deterioration of the axon-surrounding myelin (Tseng et al. 2011). Contrarily, MBP-specific antibodies were also found in some asymptomatic mice which suggests that MBP is not the sole target for myelin autoimmunity. Other myelin proteins (e.g., myelin oligodendrocyte glycoprotein (MOG) and proteolipid protein (PLP)] as well as non-myelin molecules also elicit antibodies in MS patients (Greer 2013).
Herpesviruses and their connection with MS
Alpha herpesviruses
It includes HSV-1, -2 and VZV where the reactivation of VZV was recognized in patients having difficulties in immunosuppressive therapies (i.e., Fingolimod) used for MS (Pfender et al. 2015). VZV DNA levels have also been correlated with the progression of MS specially in patients during the relapse and remission phase of MS (Mentis et al. 2017). Nonetheless, VZV-specific intrathecal IgG was found to be 35-fold higher in patients with MS compared to those with VZV reactivation (Otto et al. 2014). Peripheral lymphocytes derived from MS patients along with control showed that VZV antigens also induce and maintain the activity of HERVs (Mentis et al. 2017). Likewise, the complication faced during immunosuppressive therapy of MS includes HSV-mediated encephalitis which ended up with elevated levels of anti-HSV-1 and -2 antibodies. HSV-1 potentially triggers demyelinating encephalitis in in-vivo models like rat and mice (Duarte et al. 2019).
Beta-herpesviruses
CMV and HHV-6 are β-herpesviruses where the CMV infection is considered as an opportunistic reactivation in MS patients which ultimately leads to MS exacerbation (Sehrawat et al. 2018). Contrastingly, numerous studies revealed a negative correlation between the seropositivity of CMV with MS (Grut et al. 2021). Meta-analysis of 1341 MS patients and 2042 controls is however unable to draw an association between CMV infection and MS (Pakpoor et al. 2013). Indeed, numerous studies supported a link between MS pathogenesis and HHV-6. Neurotropism of HHV-6 has been observed as an accelerated prevalence of viral proteins and nucleic acid in CSF and MS relative to healthy individuals (Yao et al. 2009). Intriguingly, HHV-6 protein U-24 shares sequence homology with MBP and activates immune cross-reactive T-cells; however, it is pointing toward the molecular mimicry mechanism (Tejada-Simon et al. 2003). In addition, CD46 act as a receptor for HHV-6, and the absence of CD46 in rodents while widely expressed in the common marmoset makes them a potent model organism for MS and for studying the HHV-6 infection mechanism (Komaroff et al. 2020).
Gamma herpesviruses
EBV is known to be involved in numerous neurological modalities. The prevalence of EBV seropositivity in MS patients is approximately 99% (Meier et al. 2021). History of infectious mononucleosis and CSF-confined EBV-specific oligoclonal bands (OCB) is significantly high in patients suffering from MS (Castellazzi et al. 2014). A cohort study with a mean follow-up of 7 years on 147 clinically isolated syndrome (CIS) patients and 50 controls demonstrated immune responses against EBV, HHV-6, CMV, and measles (Lünemann et al. 2010). The aforementioned study also revealed an elevated immune response toward EBNA1. Thus, it suggests that the IgG titer of EBNA1 can use as a prognostic marker for the conversion and progression of the disease (Lünemann et al. 2010). The discrepant presence of EBV in MS lesions suggested that EBV-containing memory B-cells possibly lose the episomic EBV DNA during the replication process, although it hangs on to forbidden epitope recognition which is likely to activate a molecular mimicry mechanism (Laurence and Benito-León 2017). Numerous mechanisms have been proposed in favor of EBV involvement with MS, namely, immune cross-reactivity of the virus with myelin proteins, antibody, and complement system-dependent cytotoxicity and generation of auto-immune responses against alpha-crystallin (Füst 2011). The association of EBV with MS is well explained by a “two-hit hypothesis,” where the primary EBV infection would disrupt BBB and allow the activated immune cells to enter the CNS (Mohammed 2020). Besides, there are shreds of evidence suggesting an association of EBNA2 with neurological ailments, e.g., MS. EBNA2 can accelerate the expression of various host genes and signal recruitment of transcription activations factors such as immunoglobulin kappa J region (RBPJ) and the vitamin D receptor (VDR) (Ricigliano et al. 2015; Jog and James 2021). The above-stated factors are found to be playing a pivotal role in the pathology of MS.
Furthermore, an in-vitro study by Jha et al. has manifested that EBV effectively infects neuroblastoma (i.e., SH-SY5Y and Ntera2) as well as primary neuronal cells (Jha et al. 2015). Another study from our group have shown EBV infection to U-87 MG cells and suggested EBV aids to neuroinflammatory reactions by increasing proinflammatory cytokines (IL-6, and TNF-α) and also through peripheral blood mononuclear cell (PBMC) infiltration (Jakhmola and Jha 2021) (Fig. 2). Notably, the presentation of recombinant human MOG (rhMOG) by EBV-infected B-cells to CD8+ T-cells has been observed (‘T Hart et al. 2013). The higher expression of CD40 on B-cells from the relapsing patients of MS suggests increased antigen presentation by B-cells. Additionally, the EBV-infected B-cells were actively found in demyelinating lesions of relapsing–remitting multiple sclerosis (RRMS) patients who died of lethal relapse (Serafini et al. 2017). Also, the EBV (e.g., EBNA3A and EBNA3C) immortalized memory B-cells were undetectable through patrolling of T-cells by blocking B-cell differentiation into plasma cells by making a connection with the tumor suppressor genes (Styles et al. 2017). An increased titer of anti-EBV antibodies instigates the development of more contrast-enhancing lesions in magnetic resonance imagining (MRI) and higher disability scores (Höftberger and Lassmann 2017). The authors have also reported an opposite association between IgM against viral capsid antigen (VCA) and inflammatory activities (Höftberger and Lassmann 2017). Moreover, the presence of EBV DNA in the CSF of MS patients was also linked with elevated contrast-enhancing lesions (Virtanen and Jacobson 2012). Post-mortem analysis of white matter tissues of MS patients exhibited upregulation of inflammatory cytokine, i.e., IFN-α along with RNA positivity of EBV proteins was detected. In animal models, EBV-containing B-cells regulate the migration of lymphocytes via MS-related markers, namely, EBV-induced gene 2 (EBI2) and an orphan G-protein-coupled receptor. EBI2 activation by oxysterols (7-alpha-25-dihydroxycholesterol) regulates myelin development and effectively inhibits the secretion of pro-inflammatory cytokines, disruption of BBB, and microglial activation (Rutkowska et al. 2017). However, the infection of EBV stimulates the innate immune responses and further aggravates MS lesions (Guan et al. 2019).
Multiple sclerosis and other additional viruses
The peripheral infection with lymphocytic choriomeningitis virus (LCMV) developed in this virus-specific CD8+ T-cells invade the brain and induces neurological manifestation in mice (Matullo et al. 2011) (Fig. 2). Regardless of immune-mediated invasion, the virus enhances the replication of other viruses. For instance, the EBV-infected lymphoblastoid and Burkitt’s lymphoma cell lines showed enhanced torque-teno virus (TTV) replication which indicates the communication between two viruses (Borkosky et al. 2012). Likewise, the increased autoantibodies of measles were detected in the serum and CSF of MS patients which suggests dependency with the age and duration of the disease. Canine distemper virus triggers demyelinating leukoencephalitis which can be used to generate animal models for MS (Attig et al. 2019). Human coronaviruses (HCoV) are significantly known to cause infections in the upper respiratory tract and are also involved with the neurological disease’s initiation/aggravation. Furthermore, single T-cell analysis from MS patients showed HCoV-229E/MBP cross-reactive which implies a molecular mimicry mechanism (Bordeianu 1980).
Animal model use for understanding the pathogenesis of multiple sclerosis
Autoantigen-induced experimental autoimmune encephalomyelitis
Among these models, the most extensively studied is EAE, where the autoimmunity toward CNS is induced in the susceptible mice by immunizing them with myelin protein antigens (Constantinescu et al. 2011). Then, pertussis toxin (PT) and Freund’s adjuvant (CFA) are administered to generate the humoral immune response and develop transient symptoms of relapsing–remitting diseases, similar to MS (Procaccini et al. 2015). Demyelinating experiments have been performed on animal species like guinea pigs and monkeys, yet, rats and mice were proven to be good models to understand the relapsing–remitting and chronic progressive EAE through immunogenetic and acute monophasic (Constantinescu et al. 2011). Different genetic background mice (SJL/J, C57BL/6 and NOD) have been used as an EAE model by eliciting immune responses upon exposure to protein and peptide or can be by passive transfer of encephalitogenic T-cells (Table 1) (Miller and Karpus 2007). This immune response-generating immunogen can be derived from CNS proteins, namely, MBP, PLP, or MOG. An epitope of PLP139-151 induces relapsing–remitting disease in mice (SJL/J), while the chronic nature diseases are generated upon challenge to immunodominant MOG35-55 peptide concomitant administration of PT in C57BL6/J mice (Table 1). Autoimmune reactions are also induced by neurofascin (NF-155), contactin-2, NF-186, neurofilament-M, and S100 calcium-binding protein B (Midroni and Ashby 1989; Baumann and Pham-Dinh 2001; Derfuss et al. 2009). Relapsing–remitting EAE (RR-EAE) could also be developed by passive transfer of PLP-reactive T-cells, after then immunization of donor mice with PLP epitopes, subsequently, the isolation of peripheral lymphoid cells and subsequent transfer into naïve recipient (Table 1).
Virus-induced chronic demyelinating diseases
Numerous epidemiological studies have suggested that infections with several neurotropic viruses may elicit an immune-mediated response in the brain. These viral infections of the brain induce degradation of myelin sheath in mice; thus, it includes picornavirus, such as TMEV and a few coronavirus strains, i.e., MHV (Biswas et al. 2016). TMEV indicated as a neurotropic viral infection model for MS is divided into two subgroups, GDVII and TO, as per their potential to cause disease in the brain. GDVII subgroup strains, namely, GDVII and FA, can induce death in mice within 2 weeks indicating their high neurovirulence in mice (Table 1). TMEV TO subgroup strain includes BeAn8386 (BeAn) which commonly causes acute polio encephalomyelitis (Procaccini et al. 2015; Gautam et al. 1998) (Table 1). Notably, TMEV triggers this progressive inflammatory demyelinating only in mice and not in other different species, unlike EAE. Predominant infection of GDVII virus to neurons was observed during paucity in the recruitment of inflammatory mononuclear cell (MNC) (Oleszak et al. 2004). Contrary to infection of GDVII, CD3+ T-cells and parenchymal, perivascular, and subarachnoidal mononuclear cells (MNC) infiltrate in the gray matter of CNS during the acute phase of Daniels strain of TMEV (DA) infection (Tsunoda and Fujinami 2010). Additionally, MS and its animal model (EAE) are believed to be generated via severe inflammatory demyelination followed by axonal damage where lesions get generated from the outside (myelin) to the inside of the axon and depicted as outside-in model (Libbey et al. 2014b), whereas, the case of TMEV infection to neurons leads to an inside-out model of demyelination, and the disrupted axons spread was observed during the early phase of infection which eventually develops in the chronic phase (Sato et al. 2011). Therefore, these evidences pointing toward the degeneration of axons induced by the infiltration of macrophages and T-cells into the brain ultimately trigger myelin loss.
To get deeper knowledge about EBV involvement with MS, animal models are in use where mice infected with murine gamma herpesvirus shows EAE upon induction (Hassani and Khan 2022). Likewise, gamma-herpesvirus callitrichine herpesvirus 3 (CalHV3) naturally infects marmosets and CalHV3 immortalized B-cells like EBV (Fujiwara and Nakamura 2020). The CalHV3-infected B-cells behave as APCs for MHC class II and MHC class I to CD4+ and CD8+ T-cells, respectively. CalHV3 model shows a conversion from tolerogenic destructive processing of MOG peptide to productive processing and auto-aggressive T-cell activation (’t Hart 2020). Notably, the serum examination of MS patients unveiled that EB nuclear antigen 1 (EBNA1) 411–426 epitopes with antibodies cross-react with MBP (Jog et al. 2020). EBNA1 411-426 peptide inoculation in SJL/J and Balb/c mice develop signs of EAE (Jog et al. 2020). Besides, the L2 protein peptide from human papillomavirus (HPV) is overlooked by an MBP-specific T-cell clone from an MS patient and MBP-specific autoantibodies purified from MS brain tissue (Campos and Ozbun 2009). HPV-7 motif with the sequence “VHFFK” is similar to MBP aa87-99 suggesting molecular mimicry (Ruiz et al. 1999). This viral peptide induces papillomavirus-specific SJL mouse T-cells that cross-react with MBP aa87-99 which majorly is an encephalitogenic epitope for SJL mice (Falk et al. 2000). Histopathology of CNS and cytokine profiles of papillomavirus peptide-specific encephalitogenic T-cells showed similarity as induced by MBP (Ufret-Vincenty et al. 1998). Therefore, it signifies the aspects of self-antigen recognition and a cross-reactiveness of viral peptides by human, and murine MBP-specific T-cell receptors are conserved (Martinsen and Kursula 2022). A herpesvirus Saimiri (HVS) peptide (AAQRRPSRPFA) shares homology to MBP peptide 1–11 (ASQKRPSQRHG) and can set off a panel of MBP aa1-11-specific T-cell hybridomas and eventually leads EAE in mice (Table 1). These low doses of viral peptide-specific T-cell lines were highly encephalitogenic. The authors have also demonstrated the possibility of cross-recognition of these peptides by TCRs which can potentially lose the T-cell tolerance to self-antigens in autoimmune diseases employing cross-reactivity with unrelated peptides (Gautam et al. 1998).
The infection of CMV in mice with the expansion of CD4+CD28-/- T-cells has exhibited an aggravation of inflammation, myelin sheath disruption, and worsening of the EAE symptoms (Vanheusden et al. 2017). The intranasal exposure of HHV-6A and HHV-6B to non-human primates showed MS-like neuroinflammatory disease, EAE (Leibovitch et al. 2018). The viral antigens of HHV-6 in virus/EAE marmosets manifested remarkable elevation in the brain lesions (Constantinescu et al. 2011). Augmentation in the EAE was also observed upon exposure to the HSV-1 vector expressing IL-5 (Nygårdas et al. 2011). After being challenged with virus R8316 (IL-5), the authors have observed improvements in the disease score which counts on the cumulative disease index and the EAE mortality rate (Nygårdas et al. 2011). The above-mentioned vector of HSV-1 triggers an increase in the type-1 IFN response in CNS, especially IFN-β. Indeed, both type I and type II IFNs influenced the regulation and inflammation in MS along with neuromyelitis optica in EAE (Danastas et al. 2020). Further, the inoculation of 2D2 mice with influenza A virus also showed clinical and histological EAE in ∼ 29% of mice. Subsequently, the mice adapted influenza A virus showed that almost 90% of CD4+ T-cells in 2D2 mice express MOG aa35–55-specific-TCR and neurofilament-m (NF-M18–30); very few (4%) develop spontaneous classic EAE (Bettelli et al. 2003). Cumulatively, the influenza A virus can activate glia and has been correlated with increased relapse risk in MS patients (Blackmore et al. 2017). Nonetheless, another study used two different EAE rodent models to elucidate murine gammaherpesvirus-68 (+ MHV-68) ability in worsening of neurological symptoms (Flaño et al. 2002). SJL mice inoculated with UV inactivated + MHV-68 or intranasal + MHV-68 in due course immunization against PLP peptide aa139-151 (Peacock et al. 2003). The + MHV-68-mediated complications was due to remarkable viral replication within the CNS, and infectious-center assays signify no detectable virus in the brains and spinal cords of infected rodents undergoing EAE (Peacock et al. 2003).
Toxin-induced demyelination
Moreover, the toxin-generated animal models are mainly used to study the demyelination and remyelination processes. Among the most commonly used agent, cuprizone (bis-cyclohexanone-oxaldihydrazone) chelates copper and targets mature oligodendroglia cell death with demyelination, which have paramount effect on the astrocytes and microglial activation (Table 1) (Procaccini et al. 2015; Flaño et al. 2002). Deficiency of copper induces metabolic failure, and the exact reason it targets oligodendrocyte is still a question of debate (Emerson et al. 2001). Nonetheless, lysolecithin induces demyelination in the brain by subsiding the destruction to the adjacent cells (Procaccini et al. 2015). Injection of lysolecithin into the spinal cord of animals (cat, rabbit, rat, and mouse) induces demyelination in the focal areas by activating the phospholipase A2 (Table 1) (Wallace et al. 2003). Toxin induced degradation of myelin sheath because of detergents’ primary toxic effects instead of secondary effects on oligodendrocytes (Procaccini et al. 2015; Peacock et al. 2003).
Conclusion
Neurotropic viruses disrupt the homeostasis of the BBB and enters into the brain parenchyma through diverse number of routes where they trigger or aggravates the neurological diseases. Viruses act on the CNS by weakening the BBB integrity and exhibiting the molecular mimicry against host proteins which generates autoimmune responses and ultimately the MS. Animal models for MS (EAE) can aid in the detailed understanding of mechanism behind numerous devious strategies of viruses for their involvement in the brain diseases. Therefore, the neurotropic virus association with BBB leakiness and MS is crucial, and certain viral proteins can potentially be targeted therapeutically.
Abbreviations
- AIDS:
-
Acquired immunodeficiency syndrome
- BBB:
-
Blood-brain barrier
- BMECs:
-
Brain microvascular endothelial cells
- CMV:
-
Cytomegalovirus
- EBNA1:
-
EBV nuclear antigen 1
- EBV:
-
Epstein-Barr virus
- EAE:
-
Experimental autoimmune encephalomyelitis
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- CalHV3:
-
Gamma-herpesvirus callitrichine herpesvirus 3
- HCV:
-
Hepatitis C virus
- HSV-1:
-
Herpes simplex virus type 1
- HAD:
-
HIV-1-associated dementia
- HAD:
-
HIV-1-associated dementia
- HERVs:
-
Human endogenous retroviruses
- HHV-6 :
-
Human herpesvirus 6
- HIV-1:
-
Human immunodeficiency virus type 1
- HPV:
-
Human papillomavirus
- HTLV-1:
-
Human T-cell leukemia virus
- IL-1β:
-
Interleukin-1 beta
- JEV:
-
Japanese encephalitis
- JCV:
-
John Cunningham virus
- JAM:
-
Junctional adhesion molecules
- LCMV:
-
Lymphocytic choriomeningitis virus
- MMPs:
-
Matrix metalloproteinases
- MNC:
-
Mononuclear cell
- MAV-1:
-
Mouse adenovirus 1
- MS:
-
Multiple sclerosis
- MBP:
-
Myelin basic protein
- MOG:
-
Myelin oligodendrocyte glycoprotein
- MAG:
-
Myelin-associated glycoprotein
- NVU:
-
Neurovascular unit
- PT:
-
Pertussis toxin
- PLP:
-
Proteolipid protein
- TMEV:
-
Theiler’s murine encephalomyelitis virus
- TJs:
-
Tight junctions
- TTV:
-
Torque-teno virus
- TNF-α:
-
Tumor necrosis factor-alpha
- VZV:
-
Varicella-Zoster virus
- VLPs:
-
Virus-like particles
- WNV:
-
West Nile virus
References
Ajasin D, Eugenin EA (2020) HIV-1 Tat: role in bystander toxicity. Front Cell Infect Microbiol 10:61. https://doi.org/10.3389/fcimb.2020.00061
Alimonti JB, Ribecco-Lutkiewicz M, Sodja C et al (2018) Zika virus crosses an in vitro human blood brain barrier model. Fluids Barriers CNS 15:15. https://doi.org/10.1186/s12987-018-0100-y
Alves-Leon SV, Lima MDR, Nunes PCG et al (2019) Zika virus found in brain tissue of a multiple sclerosis patient undergoing an acute disseminated encephalomyelitis-like episode. Mult Scler 25:427–430. https://doi.org/10.1177/1352458518781992
András IE, Pu H, Deli MA et al (2003) HIV-1 Tat protein alters tight junction protein expression and distribution in cultured brain endothelial cells. J Neurosci Res 74:255–265. https://doi.org/10.1002/jnr.10762
Assetta B, Atwood WJ (2017) The biology of JC polyomavirus. Biol Chem 398:839–855. https://doi.org/10.1515/hsz-2016-0345
Atkinson AL, Atwood WJ (2020) Fifty years of JC polyomavirus: a brief overview and remaining questions. Viruses 12:969. https://doi.org/10.3390/v12090969
Attig F, Spitzbarth I, Kalkuhl A et al (2019) Reactive oxygen species are key mediators of demyelination in canine distemper leukoencephalitis but not in Theiler’s murine encephalomyelitis. IJMS 20:3217. https://doi.org/10.3390/ijms20133217
Banks WA, Freed EO, Wolf KM et al (2001) Transport of human immunodeficiency virus type 1 pseudoviruses across the blood-brain barrier: role of envelope proteins and adsorptive endocytosis. J Virol 75:4681–4691. https://doi.org/10.1128/JVI.75.10.4681-4691.2001
Baumann N, Pham-Dinh D (2001) Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev 81:871–927. https://doi.org/10.1152/physrev.2001.81.2.871
Bebo BF, Vandenbark AA, Offner H (1996) Male SJL mice do not relapse after induction of EAE with PLP 139–151. J Neurosci Res 45:680–689. https://doi.org/10.1002/(SICI)1097-4547(19960915)45:6%3c680::AID-JNR4%3e3.0.CO;2-4
Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 7:452–464. https://doi.org/10.1215/S1152851705000232
Bettelli E, Pagany M, Weiner HL et al (2003) Myelin oligodendrocyte glycoprotein-specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 197:1073–1081. https://doi.org/10.1084/jem.20021603
Biswas K, Chatterjee D, Addya S et al (2016) Demyelinating strain of mouse hepatitis virus infection bridging innate and adaptive immune response in the induction of demyelination. Clin Immunol 170:9–19. https://doi.org/10.1016/j.clim.2016.07.004
Bittner S, Afzali AM, Wiendl H, Meuth SG (2014) Myelin oligodendrocyte glycoprotein (MOG35–55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. JoVE. https://doi.org/10.3791/51275
Blackmore S, Hernandez J, Juda M et al (2017) Influenza infection triggers disease in a genetic model of experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1620415114
Bogovic P, Strle F (2015) Tick-borne encephalitis: a review of epidemiology, clinical characteristics, and management. World J Clin Cases 3:430–441. https://doi.org/10.12998/wjcc.v3.i5.430
Bordeianu CD (1980) Tenoplasty by marginal scleral division (author’s transl). J Fr Ophtalmol 3:671–674
Borkosky SS, Whitley C, Kopp-Schneider A et al (2012) Epstein-Barr virus stimulates torque teno virus replication: a possible relationship to multiple sclerosis. PLoS ONE 7:e32160. https://doi.org/10.1371/journal.pone.0032160
Bradl M, Lassmann H (2010) Oligodendrocytes: biology and pathology. Acta Neuropathol 119:37–53. https://doi.org/10.1007/s00401-009-0601-5
Browne EP (2020) The role of toll-like receptors in retroviral infection. Microorganisms 8:1787. https://doi.org/10.3390/microorganisms8111787
Calderón-Peláez M-A, Velandia-Romero ML, Bastidas-Legarda LY et al (2019) Dengue virus infection of blood-brain barrier cells: consequences of severe disease. Front Microbiol 10:1435. https://doi.org/10.3389/fmicb.2019.01435
Campos SK, Ozbun MA (2009) Two highly conserved cysteine residues in HPV16 L2 form an intramolecular disulfide bond and are critical for infectivity in human keratinocytes. PLoS ONE 4:e4463. https://doi.org/10.1371/journal.pone.0004463
Castellazzi M, Contini C, Tamborino C et al (2014) Epstein-Barr virus-specific intrathecal oligoclonal IgG production in relapsing-remitting multiple sclerosis is limited to a subset of patients and is composed of low-affinity antibodies. J Neuroinflammation 11:188. https://doi.org/10.1186/s12974-014-0188-1
Chang C-Y, Li J-R, Chen W-Y et al (2015) Disruption of in vitro endothelial barrier integrity by Japanese encephalitis virus-infected astrocytes: JEV disrupts endothelial barrier. Glia 63:1915–1932. https://doi.org/10.1002/glia.22857
Chapagain ML, Nerurkar VR (2010) Human polyomavirus JC (JCV) infection of human B lymphocytes: a possible mechanism for JCV transmigration across the blood-brain barrier. J Infect Dis 202:184–191. https://doi.org/10.1086/653823
Chen Z, Li G (2021) Immune response and blood-brain barrier dysfunction during viral neuroinvasion. Innate Immun 27:109–117. https://doi.org/10.1177/1753425920954281
Claudio L, Raine CS, Brosnan CF (1995) Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta Neuropathol 90:228–238. https://doi.org/10.1007/BF00296505
Constantinescu CS, Farooqi N, O’Brien K, Gran B (2011) Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 164:1079–1106. https://doi.org/10.1111/j.1476-5381.2011.01302.x
Coyne CB, Shen L, Turner JR, Bergelson JM (2007) Coxsackievirus entry across epithelial tight junctions requires occludin and the small GTPases Rab34 and Rab5. Cell Host Microbe 2:181–192. https://doi.org/10.1016/j.chom.2007.07.003
Cusick MF, Libbey JE, Fujinami RS (2013) Multiple sclerosis: autoimmunity and viruses. Curr Opin Rheumatol 25:496–501. https://doi.org/10.1097/BOR.0b013e328362004d
Danastas K, Miranda-Saksena M, Cunningham AL (2020) Herpes simplex virus type 1 interactions with the interferon system. Int J Mol Sci 21:5150. https://doi.org/10.3390/ijms21145150
Daniels BP, Holman DW, Cruz-Orengo L et al (2014) Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. mBio 5:e01476-e11414. https://doi.org/10.1128/mBio.01476-14
Davies MJ, Gordon JL, Gearing AJ et al (1993) The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol 171:223–229. https://doi.org/10.1002/path.1711710311
Davis BM, Rall GF, Schnell MJ (2015) Everything you always wanted to know about rabies virus (but were afraid to ask). Annu Rev Virol 2:451–471. https://doi.org/10.1146/annurev-virology-100114-055157
Del Valle L, Piña-Oviedo S (2019) Human polyomavirus JCPyV and its role in progressive multifocal leukoencephalopathy and oncogenesis. Front Oncol 9:711. https://doi.org/10.3389/fonc.2019.00711
Delbue S, Comar M, Ferrante P (2017) Review on the role of the human polyomavirus JC in the development of tumors. Infect Agents Cancer 12:10. https://doi.org/10.1186/s13027-017-0122-0
Delorme-Axford E, Coyne CB (2011) The actin cytoskeleton as a barrier to virus infection of polarized epithelial cells. Viruses 3:2462–2477. https://doi.org/10.3390/v3122462
Denic A, Johnson AJ, Bieber AJ et al (2011) The relevance of animal models in multiple sclerosis research. Pathophysiology 18:21–29. https://doi.org/10.1016/j.pathophys.2010.04.004
Derfuss T, Parikh K, Velhin S et al (2009) Contactin-2/TAG-1-directed autoimmunity is identified in multiple sclerosis patients and mediates gray matter pathology in animals. Proc Natl Acad Sci USA 106:8302–8307. https://doi.org/10.1073/pnas.0901496106
Dietzschold B, Li J, Faber M, Schnell M (2008) Concepts in the pathogenesis of rabies. Future Virol 3:481–490. https://doi.org/10.2217/17460794.3.5.481
Dietzschold B, Schnell M, Koprowski H (2005) Pathogenesis of rabies. Curr Top Microbiol Immunol 292:45–56. https://doi.org/10.1007/3-540-27485-5_3
Donati D (2020) Viral infections and multiple sclerosis. Drug Discov Today Dis Model 32:27–33. https://doi.org/10.1016/j.ddmod.2020.02.003
Duarte LF, Farías MA, Álvarez DM et al (2019) Herpes simplex virus type 1 infection of the central nervous system: insights into proposed interrelationships with neurodegenerative disorders. Front Cell Neurosci 13:46. https://doi.org/10.3389/fncel.2019.00046
Elphick GF, Querbes W, Jordan JA et al (2004) The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 306:1380–1383. https://doi.org/10.1126/science.1103492
Emerson MR, Biswas S, LeVine SM (2001) Cuprizone and piperonyl butoxide, proposed inhibitors of T-cell function, attenuate experimental allergic encephalomyelitis in SJL mice. J Neuroimmunol 119:205–213. https://doi.org/10.1016/S0165-5728(01)00394-0
Engelhardt B, Wolburg H (2004) Mini-review: transendothelial migration of leukocytes: through the front door or around the side of the house? Eur J Immunol 34:2955–2963. https://doi.org/10.1002/eji.200425327
Enose-Akahata Y, Abrams A, Johnson KR et al (2012) Quantitative differences in HTLV-I antibody responses: classification and relative risk assessment for asymptomatic carriers and ATL and HAM/TSP patients from Jamaica. Blood 119:2829–2836. https://doi.org/10.1182/blood-2011-11-390807
Erickson AK, Pfeiffer JK (2013) Dynamic viral dissemination in mice infected with yellow fever virus strain 17D. J Virol 87:12392–12397. https://doi.org/10.1128/JVI.02149-13
Erickson MA, Dohi K, Banks WA (2012) Neuroinflammation: a common pathway in CNS diseases as mediated at the blood-brain barrier. Neuroimmunomodulation 19:121–130. https://doi.org/10.1159/000330247
Fabis MJ, Scott GS, Kean RB et al (2007) Loss of blood–brain barrier integrity in the spinal cord is common to experimental allergic encephalomyelitis in knockout mouse models. Proc Natl Acad Sci USA 104:5656–5661. https://doi.org/10.1073/pnas.0701252104
Falk K, Rötzschke O, Santambrogio L et al (2000) Induction and suppression of an autoimmune disease by oligomerized T cell epitopes: enhanced in vivo potency of encephalitogenic peptides. J Exp Med 191:717–730. https://doi.org/10.1084/jem.191.4.717
Fares M, Cochet-Bernoin M, Gonzalez G et al (2020) Pathological modeling of TBEV infection reveals differential innate immune responses in human neurons and astrocytes that correlate with their susceptibility to infection. J Neuroinflammation 17:76. https://doi.org/10.1186/s12974-020-01756-x
Flaño E, Woodland DL, Blackman MA (2002) A mouse model for infectious mononucleosis. Immunol Res 25:201–218. https://doi.org/10.1385/IR:25:3:201
Fletcher NF, Wilson GK, Murray J et al (2012) Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology 142:634-643.e6. https://doi.org/10.1053/j.gastro.2011.11.028
Fujinami RS, von Herrath MG, Christen U, Whitton JL (2006) Molecular mimicry, bystander activation, or viral persistence: infections and autoimmune disease. Clin Microbiol Rev 19:80–94. https://doi.org/10.1128/CMR.19.1.80-94.2006
Fujiwara S, Nakamura H (2020) Animal models for gammaherpesvirus infections: recent development in the analysis of virus-induced pathogenesis. Pathogens 9:116. https://doi.org/10.3390/pathogens9020116
Füst G (2011) The role of the Epstein-Barr virus in the pathogenesis of some autoimmune disorders - similarities and differences. Eur J Microbiol Immunol (Bp) 1:267–278. https://doi.org/10.1556/EuJMI.1.2011.4.2
Futsch N, Prates G, Mahieux R et al (2018) Cytokine networks dysregulation during HTLV-1 infection and associated diseases. Viruses 10:691. https://doi.org/10.3390/v10120691
Gautam AM, Liblau R, Chelvanayagam G et al (1998) A viral peptide with limited homology to a self peptide can induce clinical signs of experimental autoimmune encephalomyelitis. J Immunol 161:60–64. https://doi.org/10.4049/jimmunol.161.1.60
Ghita L, Breitkopf V, Mulenge F et al (2021) Sequential MAVS and MyD88/TRIF signaling triggers anti-viral responses of tick-borne encephalitis virus-infected murine astrocytes. J Neurosci Res 99:2478–2492. https://doi.org/10.1002/jnr.24923
Glenn JD, Smith MD, Xue P et al (2017) CNS-targeted autoimmunity leads to increased influenza mortality in mice. J Exp Med 214:297–307. https://doi.org/10.1084/jem.20160517
Greer JM (2013) Autoimmune T-cell reactivity to myelin proteolipids and glycolipids in multiple sclerosis. Mult Scler Int 2013:151427. https://doi.org/10.1155/2013/151427
Grut V, Biström M, Salzer J et al (2021) Cytomegalovirus seropositivity is associated with reduced risk of multiple sclerosis—a presymptomatic case–control study. Eur J Neurol 28:3072–3079. https://doi.org/10.1111/ene.14961
Guan Y, Jakimovski D, Ramanathan M et al (2019) The role of Epstein-Barr virus in multiple sclerosis: from molecular pathophysiology to in vivo imaging. Neural Regen Res 14:373–386. https://doi.org/10.4103/1673-5374.245462
Hallahan DE, Virudachalam S (1997) Intercellular adhesion molecule 1 knockout abrogates radiation induced pulmonary inflammation. Proc Natl Acad Sci U S A 94:6432–6437. https://doi.org/10.1073/pnas.94.12.6432
Harypursat V, Zhou Y, Tang S, Chen Y (2020) JC Polyomavirus, progressive multifocal leukoencephalopathy and immune reconstitution inflammatory syndrome: a review. AIDS Res Ther 17:37. https://doi.org/10.1186/s12981-020-00293-0
Hassani A, Khan G (2022) What do animal models tell us about the role of EBV in the pathogenesis of multiple sclerosis? Front Immunol 13:1036155. https://doi.org/10.3389/fimmu.2022.1036155
Höftberger R, Lassmann H (2017) Inflammatory demyelinating diseases of the central nervous system. Handb Clin Neurol 145:263–283. https://doi.org/10.1016/B978-0-12-802395-2.00019-5
Hou J, Baker LA, Zhou L, Klein RS (2016) Viral interactions with the blood-brain barrier: old dog, new tricks. Tissue Barriers 4:e1142492. https://doi.org/10.1080/21688370.2016.1142492
Ignatius Arokia Doss PM, Roy A-P, Wang A et al (2015) The non-obese diabetic mouse strain as a model to study CD8+ T cell function in relapsing and progressive multiple sclerosis. Front Immunol. https://doi.org/10.3389/fimmu.2015.00541
Jakhmola S, Jha HC (2021) Glial cell response to Epstein-Barr virus infection: a plausible contribution to virus-associated inflammatory reactions in the brain. Virology 559:182–195. https://doi.org/10.1016/j.virol.2021.04.005
Jakhmola S, Upadhyay A, Jain K et al (2021) Herpesviruses and the hidden links to multiple sclerosis neuropathology. J Neuroimmunol 358:577636. https://doi.org/10.1016/j.jneuroim.2021.577636
Jha HC, Mehta D, Lu J et al (2015) Gammaherpesvirus infection of human neuronal cells. mBio 6:e01844-e1915. https://doi.org/10.1128/mBio.01844-15
Jha HC, Pei Y, Robertson ES (2016) Epstein-Barr virus: diseases linked to infection and transformation. Front Microbiol. https://doi.org/10.3389/fmicb.2016.01602
Jhan M-K, Tsai T-T, Chen C-L et al (2017) Dengue virus infection increases microglial cell migration. Sci Rep 7:91. https://doi.org/10.1038/s41598-017-00182-z
Jog NR, James JA (2021) Epstein Barr virus and autoimmune responses in systemic lupus erythematosus. Front Immunol 11:623944. https://doi.org/10.3389/fimmu.2020.623944
Jog NR, McClain MT, Heinlen LD et al (2020) Epstein Barr virus nuclear antigen 1 (EBNA-1) peptides recognized by adult multiple sclerosis patient sera induce neurologic symptoms in a murine model. J Autoimmun 106:102332. https://doi.org/10.1016/j.jaut.2019.102332
Kanmogne GD, Schall K, Leibhart J et al (2007) HIV-1 gp120 compromises blood-brain barrier integrity and enhances monocyte migration across blood-brain barrier: implication for viral neuropathogenesis. J Cereb Blood Flow Metab 27:123–134. https://doi.org/10.1038/sj.jcbfm.9600330
Kolb H, Al-Louzi O, Beck ES et al (2022) From pathology to MRI and back: clinically relevant biomarkers of multiple sclerosis lesions. NeuroImage Clin 36:103194. https://doi.org/10.1016/j.nicl.2022.103194
Komaroff AL, Pellett PE, Jacobson S (2020) Human herpesviruses 6A and 6B in brain diseases: association versus causation. Clin Microbiol Rev 34:e00143-e220. https://doi.org/10.1128/CMR.00143-20
Kumar M, Verma S, Nerurkar VR (2010) Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J Neuroinflammation 7:73. https://doi.org/10.1186/1742-2094-7-73
Lannes N, Summerfield A, Filgueira L (2017) Regulation of inflammation in Japanese encephalitis. J Neuroinflammation 14:158. https://doi.org/10.1186/s12974-017-0931-5
Laurence M, Benito-León J (2017) Epstein-Barr virus and multiple sclerosis: updating Pender’s hypothesis. Mult Scler Relat Disord 16:8–14. https://doi.org/10.1016/j.msard.2017.05.009
Laureti M, Narayanan D, Rodriguez-Andres J et al (2018) Flavivirus receptors: diversity, identity, and cell entry. Front Immunol 9:2180. https://doi.org/10.3389/fimmu.2018.02180
Lazear HM, Daniels BP, Pinto AK et al (2015) Interferon-λ restricts West Nile virus neuroinvasion by tightening the blood-brain barrier. Sci Transl Med 7:284ra59. https://doi.org/10.1126/scitranslmed.aaa4304
Lei S, Tian Y-P, Xiao W-D et al (2013) ROCK is involved in vimentin phosphorylation and rearrangement induced by dengue virus. Cell Biochem Biophys 67:1333–1342. https://doi.org/10.1007/s12013-013-9665-x
Leibovitch EC, Caruso B, Ha SK et al (2018) Herpesvirus trigger accelerates neuroinflammation in a nonhuman primate model of multiple sclerosis. Proc Natl Acad Sci U S A 115:11292–11297. https://doi.org/10.1073/pnas.1811974115
Leis AA, Stokic DS (2012) Neuromuscular manifestations of west nile virus infection. Front Neurol 3:37. https://doi.org/10.3389/fneur.2012.00037
Li F, Wang Y, Yu L et al (2015) Viral infection of the central nervous system and neuroinflammation precede blood-brain barrier disruption during Japanese encephalitis virus infection. J Virol 89:5602–5614. https://doi.org/10.1128/JVI.00143-15
Li G-H, Ning Z-J, Liu Y-M, Li X-H (2017) Neurological manifestations of dengue infection. Front Cell Infect Microbiol 7:449. https://doi.org/10.3389/fcimb.2017.00449
Libbey JE, Cusick MF, Fujinami RS (2014a) Role of pathogens in multiple sclerosis. Int Rev Immunol 33:266–283. https://doi.org/10.3109/08830185.2013.823422
Libbey JE, Lane TE, Fujinami RS (2014b) Axonal pathology and demyelination in viral models of multiple sclerosis. Discov Med 18:79–89
Lindqvist R, Mundt F, Gilthorpe JD et al (2016) Fast type I interferon response protects astrocytes from flavivirus infection and virus-induced cytopathic effects. J Neuroinflammation 13:277. https://doi.org/10.1186/s12974-016-0748-7
Liu Y-G, Chen Y, Wang X et al (2020) Ezrin is essential for the entry of Japanese encephalitis virus into the human brain microvascular endothelial cells. Emerg Microbes Infect 9:1330–1341. https://doi.org/10.1080/22221751.2020.1757388
Louboutin J-P, Strayer DS (2012) Blood-brain barrier abnormalities caused by HIV-1 gp120: mechanistic and therapeutic implications. ScientificWorldJournal 2012:482575. https://doi.org/10.1100/2012/482575
Ludlow M, Kortekaas J, Herden C et al (2016) Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol 131:159–184. https://doi.org/10.1007/s00401-015-1511-3
Lünemann JD, Tintoré M, Messmer B et al (2010) Elevated Epstein-Barr virus-encoded nuclear antigen-1 immune responses predict conversion to multiple sclerosis. Ann Neurol 67:159–169. https://doi.org/10.1002/ana.21886
Mameli G, Poddighe L, Mei A et al (2012) Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: inference for multiple sclerosis. PLoS ONE 7:e44991. https://doi.org/10.1371/journal.pone.0044991
Marrodan M, Alessandro L, Farez MF, Correale J (2019) The role of infections in multiple sclerosis. Mult Scler 25:891–901. https://doi.org/10.1177/1352458518823940
Martinsen V, Kursula P (2022) Multiple sclerosis and myelin basic protein: insights into protein disorder and disease. Amino Acids 54:99–109. https://doi.org/10.1007/s00726-021-03111-7
Matullo CM, O’Regan KJ, Curtis M, Rall GF (2011) CNS recruitment of CD8+ T lymphocytes specific for a peripheral virus infection triggers neuropathogenesis during polymicrobial challenge. PLoS Pathog 7:e1002462. https://doi.org/10.1371/journal.ppat.1002462
Maubert ME, Wigdahl B, Nonnemacher MR (2017) Opinion: inhibition of blood-brain barrier repair as a mechanism in HIV-1 disease. Front Neurosci 11:228. https://doi.org/10.3389/fnins.2017.00228
Mayberry CL, Soucy AN, Lajoie CR et al (2019) JC polyomavirus entry by clathrin-mediated endocytosis is driven by β-arrestin. J Virol 93:e01948-e2018. https://doi.org/10.1128/JVI.01948-18
McRae BL, Kennedy MK, Tan L-J et al (1992) Induction of active and adoptive relapsing experimental autoimmune encephalomyelitis (EAE) using an encephalitogenic epitope of proteolipid protein. J Neuroimmunol 38:229–240. https://doi.org/10.1016/0165-5728(92)90016-E
Mehta R, Gerardin P, de Brito CAA et al (2018) The neurological complications of chikungunya virus: a systematic review. Rev Med Virol 28:e1978. https://doi.org/10.1002/rmv.1978
Meier U-C, Cipian RC, Karimi A et al (2021) Cumulative roles for Epstein-Barr virus, human endogenous retroviruses, and human herpes virus-6 in driving an inflammatory cascade underlying MS pathogenesis. Front Immunol 12:757302. https://doi.org/10.3389/fimmu.2021.757302
Mentis A-FA, Dardiotis E, Grigoriadis N et al (2017) Viruses and endogenous retroviruses in multiple sclerosis: from correlation to causation. Acta Neurol Scand 136:606–616. https://doi.org/10.1111/ane.12775
Michael BD, Bricio-Moreno L, Sorensen EW et al (2020) Astrocyte- and neuron-derived CXCL1 drives neutrophil transmigration and blood-brain barrier permeability in viral encephalitis. Cell Rep 32:108150. https://doi.org/10.1016/j.celrep.2020.108150
Michalicová A, Bhide K, Bhide M, Kováč A (2017) How viruses infiltrate the central nervous system. Acta Virol 61:393–400. https://doi.org/10.4149/av_2017_401
Midroni G, Ashby P (1989) How synaptic noise may affect cross-correlations. J Neurosci Methods 27:1–12. https://doi.org/10.1016/0165-0270(89)90048-4
Miller F, Afonso PV, Gessain A, Ceccaldi P-E (2012) Blood-brain barrier and retroviral infections. Virulence 3:222–229. https://doi.org/10.4161/viru.19697
Miller SD, Karpus WJ (2007) Experimental autoimmune encephalomyelitis in the mouse. Curr Protoc Immunol Chapter 15:15.1.1-15.1.18. https://doi.org/10.1002/0471142735.im1501s77
Miner JJ, Diamond MS (2016) Mechanisms of restriction of viral neuroinvasion at the blood-brain barrier. Curr Opin Immunol 38:18–23. https://doi.org/10.1016/j.coi.2015.10.008
Mohammed EM (2020) Environmental influencers, microRNA, and multiple sclerosis. J Cent Nerv Syst Dis 12:117957351989495. https://doi.org/10.1177/1179573519894955
Morgello S (2020) Coronaviruses and the central nervous system. J Neurovirol 26:459–473. https://doi.org/10.1007/s13365-020-00868-7
Morrey JD, Olsen AL, Siddharthan V et al (2008) Increased blood-brain barrier permeability is not a primary determinant for lethality of West Nile virus infection in rodents. J Gen Virol 89:467–473. https://doi.org/10.1099/vir.0.83345-0
Morris G, Maes M, Murdjeva M, Puri BK (2019) Do human endogenous retroviruses contribute to multiple sclerosis, and if so, how? Mol Neurobiol 56:2590–2605. https://doi.org/10.1007/s12035-018-1255-x
Mustafá YM, Meuren LM, Coelho SVA, de Arruda LB (2019) Pathways exploited by flaviviruses to counteract the blood-brain barrier and invade the central nervous system. Front Microbiol 10:525. https://doi.org/10.3389/fmicb.2019.00525
Nygårdas M, Aspelin C, Paavilainen H et al (2011) Treatment of experimental autoimmune encephalomyelitis in SJL/J mice with a replicative HSV-1 vector expressing interleukin-5. Gene Ther 18:646–655. https://doi.org/10.1038/gt.2011.4
O’Hara BA, Morris-Love J, Gee GV et al (2020) JC virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog 16:e1008371. https://doi.org/10.1371/journal.ppat.1008371
Oleszak EL, Chang JR, Friedman H et al (2004) Theiler’s virus infection: a model for multiple sclerosis. Clin Microbiol Rev 17:174–207. https://doi.org/10.1128/CMR.17.1.174-207.2004
Otto C, Hofmann J, Finke C et al (2014) The fraction of varicella zoster virus-specific antibodies among all intrathecally-produced antibodies discriminates between patients with varicella zoster virus reactivation and multiple sclerosis. Fluids Barriers CNS 11:3. https://doi.org/10.1186/2045-8118-11-3
Pakpoor J, Pakpoor J, Disanto G et al (2013) Cytomegalovirus and multiple sclerosis risk. J Neurol 260:1658–1660. https://doi.org/10.1007/s00415-013-6912-4
Palus M, Vancova M, Sirmarova J et al (2017) Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology 507:110–122. https://doi.org/10.1016/j.virol.2017.04.012
Patra P, Rani A, Sharma N et al (2023) Unraveling the connection of Epstein-Barr virus and its glycoprotein M 146–157 peptide with neurological ailments. ACS Chem Neurosci 14:2450–2460. https://doi.org/10.1021/acschemneuro.3c00231
Peacock JW, Elsawa SF, Petty CC et al (2003) Exacerbation of experimental autoimmune encephalomyelitis in rodents infected with murine gammaherpesvirus-68. Eur J Immunol 33:1849–1858. https://doi.org/10.1002/eji.200323148
Pfender N, Jelcic I, Linnebank M et al (2015) Reactivation of herpesvirus under fingolimod: a case of severe herpes simplex encephalitis. Neurology 84:2377–2378. https://doi.org/10.1212/WNL.0000000000001659
Plemel JR, Michaels NJ, Weishaupt N et al (2018) Mechanisms of lysophosphatidylcholine-induced demyelination: a primary lipid disrupting myelinopathy. Glia 66:327–347. https://doi.org/10.1002/glia.23245
Procaccini C, De Rosa V, Pucino V et al (2015) Animal models of multiple sclerosis. Eur J Pharmacol 759:182–191. https://doi.org/10.1016/j.ejphar.2015.03.042
Profaci CP, Munji RN, Pulido RS, Daneman R (2020) The blood–brain barrier in health and disease: important unanswered questions. J Exp Med 217:e20190062. https://doi.org/10.1084/jem.20190062
Pu H, Tian J, Andras IE et al (2005) HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK1/2 activation. J Cereb Blood Flow Metab 25:1325–1335. https://doi.org/10.1038/sj.jcbfm.9600125
Pulmanausahakul R, Li J, Schnell MJ, Dietzschold B (2008) The glycoprotein and the matrix protein of rabies virus affect pathogenicity by regulating viral replication and facilitating cell-to-cell spread. J Virol 82:2330–2338. https://doi.org/10.1128/JVI.02327-07
Rani A, Tanwar M, Verma TP et al (2023) Understanding the role of membrane cholesterol upon Epstein Barr virus infection in astroglial cells. Front Immunol 14:1192032. https://doi.org/10.3389/fimmu.2023.1192032
Ricigliano VAG, Handel AE, Sandve GK et al (2015) EBNA2 binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin D receptor occupancy. PLoS ONE 10:e0119605. https://doi.org/10.1371/journal.pone.0119605
Robinson AP, Harp CT, Noronha A, Miller SD (2014) The experimental autoimmune encephalomyelitis (EAE) model of MS: utility for understanding disease pathophysiology and treatment. Handb Clin Neurol 122:173–189. https://doi.org/10.1016/B978-0-444-52001-2.00008-X
Roe K, Kumar M, Lum S et al (2012) West Nile virus-induced disruption of the blood–brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J Gen Virol 93:1193–1203. https://doi.org/10.1099/vir.0.040899-0
Ruiz PJ, Garren H, Hirschberg DL et al (1999) Microbial epitopes act as altered peptide ligands to prevent experimental autoimmune encephalomyelitis. J Exp Med 189:1275–1284. https://doi.org/10.1084/jem.189.8.1275
Rutkowska A, Sailer AW, Dev KK (2017) EBI2 receptor regulates myelin development and inhibits LPC-induced demyelination. J Neuroinflammation 14:250. https://doi.org/10.1186/s12974-017-1025-0
Safak M, Khalili K (2003) An overview: human polyomavirus JC virus and its associated disorders. J Neurovirol 9:3–9. https://doi.org/10.1080/13550280390195360
Sato F, Tanaka H, Hasanovic F, Tsunoda I (2011) Theiler’s virus infection: pathophysiology of demyelination and neurodegeneration. Pathophysiology 18:31–41. https://doi.org/10.1016/j.pathophys.2010.04.011
Schäfer A, Brooke CB, Whitmore AC, Johnston RE (2011) The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J Virol 85:10682–10690. https://doi.org/10.1128/JVI.05032-11
Sehrawat S, Kumar D, Rouse BT (2018) Herpesviruses: harmonious pathogens but relevant cofactors in other diseases? Front Cell Infect Microbiol 8:177. https://doi.org/10.3389/fcimb.2018.00177
Serafini B, Scorsi E, Rosicarelli B et al (2017) Massive intracerebral Epstein-Barr virus reactivation in lethal multiple sclerosis relapse after natalizumab withdrawal. J Neuroimmunol 307:14–17. https://doi.org/10.1016/j.jneuroim.2017.03.013
Soumahoro M-K, Boelle P-Y, Gaüzere B-A et al (2011) The Chikungunya epidemic on La Réunion Island in 2005–2006: a cost-of-illness study. PLoS Negl Trop Dis 5:e1197. https://doi.org/10.1371/journal.pntd.0001197
Spindler KR, Hsu T-H (2012) Viral disruption of the blood-brain barrier. Trends Microbiol 20:282–290. https://doi.org/10.1016/j.tim.2012.03.009
Stamatovic SM, Keep RF, Andjelkovic AV (2008) Brain endothelial cell-cell junctions: how to “open” the blood brain barrier. Curr Neuropharmacol 6:179–192. https://doi.org/10.2174/157015908785777210
Stefanou M-I, Krumbholz M, Ziemann U, Kowarik MC (2019) Human immunodeficiency virus and multiple sclerosis: a review of the literature. Neurol Res Pract 1:24. https://doi.org/10.1186/s42466-019-0030-4
Stone M, Bakkour S, Lanteri MC et al (2020) Zika virus RNA and IgM persistence in blood compartments and body fluids: a prospective observational study. Lancet Infect Dis 20:1446–1456. https://doi.org/10.1016/S1473-3099(19)30708-X
Styles CT, Bazot Q, Parker GA et al (2017) EBV epigenetically suppresses the B cell-to-plasma cell differentiation pathway while establishing long-term latency. PLoS Biol 15:e2001992. https://doi.org/10.1371/journal.pbio.2001992
Sweeney MD, Ayyadurai S, Zlokovic BV (2016) Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci 19:771–783. https://doi.org/10.1038/nn.4288
’t Hart BA (2020) A tolerogenic role of cathepsin G in a primate model of multiple sclerosis: abrogation by Epstein-Barr virus infection. Arch Immunol Ther Exp (warsz) 68:21. https://doi.org/10.1007/s00005-020-00587-1
‘T Hart BA, Jagessar SA, Haanstra K et al (2013) The primate EAE model points at EBV-infected B cells as a preferential therapy target in multiple sclerosis. Front Immunol. https://doi.org/10.3389/fimmu.2013.00145
Takeshita Y, Ransohoff RM (2012) Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immunol Rev 248:228–239. https://doi.org/10.1111/j.1600-065X.2012.01127.x
Tejada-Simon MV, Zang YCQ, Hong J et al (2003) Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann Neurol 53:189–197. https://doi.org/10.1002/ana.10425
Tian J, Shi R, Liu T et al (2019) Brain infection by hepatitis E virus probably via damage of the blood-brain barrier due to alterations of tight junction proteins. Front Cell Infect Microbiol 9:52. https://doi.org/10.3389/fcimb.2019.00052
Toborek M, Lee YW, Flora G et al (2005) Mechanisms of the blood–brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol 25:181–199. https://doi.org/10.1007/s10571-004-1383-x
Tohidpour A, Morgun AV, Boitsova EB et al (2017) Neuroinflammation and infection: molecular mechanisms associated with dysfunction of neurovascular unit. Front Cell Infect Microbiol 7:276. https://doi.org/10.3389/fcimb.2017.00276
Torkildsen O, Brunborg LA, Myhr K-M, Bø L (2008) The cuprizone model for demyelination. Acta Neurol Scand Suppl 188:72–76. https://doi.org/10.1111/j.1600-0404.2008.01036.x
Tseng Y-F, Wang C-C, Liao S-K et al (2011) Autoimmunity-related demyelination in infection by Japanese encephalitis virus. J Biomed Sci 18:20. https://doi.org/10.1186/1423-0127-18-20
Tsunoda I, Fujinami RS (2010) Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J Neuroimmune Pharmacol 5:355–369. https://doi.org/10.1007/s11481-009-9179-x
Ufret-Vincenty RL, Quigley L, Tresser N et al (1998) In vivo survival of viral antigen-specific T cells that induce experimental autoimmune encephalomyelitis. J Exp Med 188:1725–1738. https://doi.org/10.1084/jem.188.9.1725
van Buul JD, Hordijk PL (2004) Signaling in leukocyte transendothelial migration. Arterioscler Throm Vasc Biol 24:824–833. https://doi.org/10.1161/01.ATV.0000122854.76267.5c
Vanheusden M, Broux B, Welten SPM et al (2017) Cytomegalovirus infection exacerbates autoimmune mediated neuroinflammation. Sci Rep 7:663. https://doi.org/10.1038/s41598-017-00645-3
Velandia-Romero ML, Calderón-Peláez M-A, Castellanos JE (2016) In vitro infection with dengue virus induces changes in the structure and function of the mouse brain endothelium. PLoS ONE 11:e0157786. https://doi.org/10.1371/journal.pone.0157786
Virtanen JO, Jacobson S (2012) Viruses and multiple sclerosis. CNS Neurol Disord Drug Targets 11:528–544. https://doi.org/10.2174/187152712801661220
Wallace VCJ, Cottrell DF, Brophy PJ, Fleetwood-Walker SM (2003) Focal lysolecithin-induced demyelination of peripheral afferents results in neuropathic pain behavior that is attenuated by cannabinoids. J Neurosci 23:3221–3233. https://doi.org/10.1523/JNEUROSCI.23-08-03221.2003
Wang P, Dai J, Bai F et al (2008) Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J Virol 82:8978–8985. https://doi.org/10.1128/JVI.00314-08
Yang J, Zou L, Yang Y et al (2016) Superficial vimentin mediates DENV-2 infection of vascular endothelial cells. Sci Rep 6:38372. https://doi.org/10.1038/srep38372
Yao K, Honarmand S, Espinosa A et al (2009) Detection of human herpesvirus-6 in cerebrospinal fluid of patients with encephalitis. Ann Neurol 65:257–267. https://doi.org/10.1002/ana.21611
Acknowledgements
We appreciate our lab colleagues for their insightful discussions and advice. We gratefully acknowledge the Indian Institute of Technology Indore for providing facilities and support.
Funding
We thank Scheme for Promotion of Academic and Research Collaboration 2019 (grant no. 2018), Indo-German Science and Technology Centre (IGSTC) project no IGSTC/SING-2022/40/2021–22/332, and Center for Rural Development and Technology, IIT Indore grant no. IITI/CRDT/2022–23/05. Facilities received from the Department of Science and Technology (DST), Government of India, under the FIST Scheme (grant no. SR/FST/PSI-225/2016) are acknowledged. We extend our thanks to CSIR for providing stipend to Annu Rani.
Author information
Authors and Affiliations
Contributions
AR: conceptualization, data curation, original draft writing, and figure preparation; Dr. HCJ, Prof. SE, and Prof. SK: conceptualization, critical review, and editing. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rani, A., Ergün, S., Karnati, S. et al. Understanding the link between neurotropic viruses, BBB permeability, and MS pathogenesis. J. Neurovirol. 30, 22–38 (2024). https://doi.org/10.1007/s13365-023-01190-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-023-01190-8