
Abstract
Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease of the central nervous system with a poor prognosis and is primarily caused by JC virus (JCV) with a mutation called prototype. We encountered a case of PML with moderate progression and analyzed the mutational patterns of JCV in the cerebrospinal fluid (CSF). A 19-year-old Japanese woman with mild neurological symptoms was diagnosed with combined immunodeficiency following pneumocystis pneumonia. Brain magnetic resonance imaging scan showed multiple brain lesions, and real-time polymerase chain reaction testing detected JCV in the CSF, leading to the diagnosis of PML. The disease course of PML was stable after administration of mefloquine and mirtazapine with immunoglobulin replacement therapy. In the JCV genome cloned from the patient CSF, DNA sequences of the gene encoding the capsid protein (VP1) and the non-coding control region exhibited small mutations. However, they were quite similar to those of the archetype JCV, which persists asymptomatically in healthy individuals. These findings provide insight into the mutational characteristics of JCV in PML with mild symptoms and progression.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Progressive multifocal leukoencephalopathy (PML) is a demyelinating disease of the central nervous system caused by JC virus (JCV) in the context of immunodeficiency or immunosuppressive therapy (Cortese et al. 2021; Hadjadj et al. 2019). JCV establishes persistent asymptomatic infections in peripheral sites, such as the kidney and lymph nodes in humans (Cortese et al. 2021). The non-neuropathogenic, persistently infectious form of JCV, which can be detected in the urine of healthy individuals, is called an archetype and has a consistent nucleotide sequence within the viral genome (Yogo et al. 1990). In contrast, JCV isolates from the brain and cerebrospinal fluid (CSF) of PML patients commonly exhibit hypervariable mutations in the non-coding control region (NCCR) of the viral genome (Cortese et al. 2021; Gosert et al. 2010; Reid et al. 2011). Prototype JCV also often shows several types of mutations in the gene encoding a surface protein on the viral capsid (VP1) that affect its receptor specificity to host cells (McIlroy et al. 2019; Reid et al. 2011). Here, we report a case of atypical PML with mild progression and symptoms and analyzed the nucleotide sequences of the VP1 gene and NCCR of JCV in the CSF.
Methods
Patient
A 19-year-old female visited our hospital complaining of difficulty in speaking and using her right limbs for the past month. She had a history of herpes zoster in elementary school and pyelonephritis in high school. There was no family history of neurological disorders or immunodeficiency, except for her grandfather’s suspected amyotrophic lateral sclerosis. Neurologically, she had mild dysarthria, mild paralysis of her right upper and lower limbs, and ataxia of her left lower limb. Brain magnetic resonance imaging (MRI) scan showed multiple T2 and fluid-attenuated inversion recovery (FLAIR) hyperintense lesions without contrast enhancement in the cerebral white matter, brain stem, and cerebellum, and the cerebellar lesion was crescent-shaped (Fig. 1a–c). Her CSF was positive for oligoclonal bands. She was fulfilled the 2017 McDonald criteria and diagnosed with multiple sclerosis (MS) (Thompson et al. 2018). However, 2 months after the first visit, the patient developed fever and cough. We prescribed dimethyl fumarate, but she had never taken the medication because of suspicion of infection. One month later, she was admitted to our hospital for a thorough examination of fever and cough. Chest computed tomography scan showed diffuse ground-glass opacities and markedly high serum beta-d glucan levels (251.8 pg/mL) were detected. We made a clinical diagnosis of pneumocystis pneumonia and started sulfamethoxazole/trimethoprim therapy. She also had low immunoglobulin levels (IgG 137 mg/dL, IgA 30 mg/dL, and IgM 97 mg/dL) reduced response in the mitogen-induced lymphocyte proliferation test, and negative anti-HIV antibody. We diagnosed her with combined immunodeficiency and started immunoglobulin replacement therapy. A repeat brain MRI scan showed expansion of the lesions in the left centrum semiovale and left middle cerebellar peduncle compared with those at the initial visit, but these lesions remained without contrast enhancement (Fig. 1d). FLAIR lesions showed ring-shaped hyperintensities in diffusion-weighted imaging (data not shown). Spine MRI showed no intraspinal abnormal signals or abnormal enhancements suggestive of demyelinating diseases such as MS or neuromyelitis optica. CSF cell count and protein levels were within normal ranges. However, the CSF specimen was positive for JCV DNA, as determined by quantitative real-time polymerase chain reaction (PCR) testing (940 copies/mL). Based on the above findings, we re-diagnosed the patient with PML and believed that the first diagnosis of MS was wrong. After obtaining approval for off-label use from the institutional review board of our hospital, we started combination treatment with mefloquine and mirtazapine in addition to immunoglobulin replacement therapy at 6 months after the onset of symptoms. Follow-up MRI performed 1 month after the start of medication (7 months after the onset of disease) showed transient enlargement of the left centrum semiovale lesion and contrast enhancement (Fig. 1e), but these were not seen in the subsequent period (Fig. 1f). The amount of JCV DNA in the CSF decreased to a very low level of < 20% compared to that in the initial test (175 copies/mL) at 9 months after the start of treatment (15 months after onset). During the follow-up period, her activities of daily living were maintained, and there was no neurological deterioration (Fig. 1g). The patient and her family provided written informed consent for the JCV genome analysis.

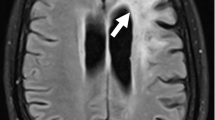
Brain MRI findings of the patient. a At the first visit, FLAIR MRI sequences revealed multiple hyperintense lesions in the cerebral white matter, b brain stem, and c cerebellum. The cerebellar lesion was crescent-shaped. d Three months later, the left centrum semiovale lesion had expanded. e Six months after the first visit, after 1 month and 3 months of concomitant administration with mefloquine and mirtazapine and immunoglobulin replacement therapy, respectively, the lesion temporarily enlarged. f Ten months after the first visit, the lesion had stopped expanding. g The clinical course of the patient is summarized in a timeline
Sequence analysis of the JCV genome
The study protocol was approved by the Ethical Committee for Biomedical Science of the National Institute of Infectious Diseases (approval number: 1247). Total DNA was extracted from CSF specimens using a QIAamp MinElute Virus Spin Kit (Qiagen, Valencia, CA, USA), and the copy number of JCV DNA in each sample was determined using a real-time PCR assay on the LightCycler 96 platform (Roche, Basel, Switzerland) as described previously (Nakamichi et al. 2019). When analyzing the mutation of the JCV genome in the CSF of the patient, it was difficult to amplify the entire nucleotide sequence using PCR because of the small number of viral copies. Therefore, the VP1 gene and NCCR were amplified, cloned into plasmids, and sequenced using the Sanger method. The complete fragment of the JCV VP1 gene in the CSF DNA was amplified using a pair of primers, VP1-F05 (5′-AAG ATC TGC TCC TCA ATG GAT G-3′) and VP1-R06 (5′-AGC TAA TGT TGG TAT GGG GAG AC-3′), and KOD One PCR Master Mix (Toyobo, Osaka, Japan) according to the manufacturer’s instructions. PCR products treated with 10 × A-attachment mix (Toyobo) were ligated to the pANT plasmid vector using the TA-Enhancer Cloning Kit (Nippon Gene, Tokyo, Japan) according to the manufacturer’s instructions. Competent Escherichia coli cells (ECOS competent E. coli DH5α; Nippon Gene) were transformed with the ligation mixture and plated on an LB agar medium containing ampicillin. After single colonies were picked up, the nucleotide sequences of the VP1 gene were determined in both directions using the Sanger method with universal primers (5′-TAA TAC GAC TCA CTA TAG GG-3′ and 5′-GGA AAC AGC TAT GAC CAT GA-3′) and additional primers designed for primer walking (5′-AGC AGT GGA GAG GAC TGT CC-3′ and 5′-GGA ACC CAA CAT TCA ACA GG-3′). The JCV NCCR in the CSF DNA was amplified using nested PCR with KOD One PCR Master Mix and two sets of primers reported previously (Nakamichi et al. 2013; Sugimoto et al. 1998). The NCCR fragment was ligated to the pANT vector and sequenced from both sides of the insert using universal primers, as in the analysis of the VP1 gene. The sequence data of the VP1 gene and NCCR were analyzed using the CLC Main Workbench Version 21.0.3 software (Qiagen). The nucleotide sequences were deposited in the DNA Data Bank of Japan and were assigned GenBank accession numbers LC627282 (VP1) and LC627283 (NCCR).
Results
To investigate the virological features of this case, the VP1 gene and NCCR were cloned from JCV in her CSF specimen at the initial testing time (designated here as Ks–286) and sequenced. In the VP1 gene, four clones were aligned, and all had identical sequences. The basic local alignment search tool (BLAST) search revealed that the nucleotide sequence of the Ks-286 VP1 gene had a high identity to JCV isolates 733 B (99.91%) and CY (99.81%), both of which were urine-derived archetypes belonging to type 7B. We then analyzed amino acid substitutions in VP1, which have been suggested to be associated with changes in prototype JCV in the affinity and specificity of cellular receptors (Gorelik et al. 2011; McIlroy et al. 2019; Sunyaev et al. 2009). In VP1 of Ks–286, while amino acids L55, K60, D66, S267, and S269 were not substituted and were identical to those of the archetype viruses, differences in E69D of Ks-286 were also seen in the archetype 733 B isolate (Fig. 2a). Nevertheless, a mutation at N265 was found in Ks–286, similar to the prototype JCV NIID11–68 isolate (Fig. 2a). Figure 2b illustrates the pattern of the NCCR sequences of Ks–286 and other JCVs. The nucleotide sequences of the 14 NCCR clones were aligned, and they all had the same sequence. Interestingly, Ks-286 presented the NCCR pattern similar to the archetype CY strain rather than the prototype, with only 13-bp and 9-bp deletions in the regions B, C, and F, respectively (Fig. 2b). These results suggest that the JCV detected, in this case, is very close to the archetype, although it has some prototypical features.

Mutational patterns of JCV in the CSF specimen. a Alignment of amino acid sequences of VP1 proteins encoded by JCV isolates. The numbers placed down the left side indicate the amino acid positions of VP1. The red background indicates the positions of amino acids located on the surface of VP1 that can be substituted in the prototype JCV. The yellow background illustrates the position where the amino acid difference of VP1 was observed in the JCV isolate of this case (Ks-286) and archetype JCV (733B). b Schematic diagram of the nucleotide sequences of NCCR. The NCCR sequences were compared to those of the archetype CY strain, and their patterns were illustrated using in silico analysis. The horizontal blue lines indicate DNA sequences identical to the archetype NCCR. The letters A to F are the regions assigned to the archetype NCCR. The numbers above or below the solid line and closed triangles represent the nucleotide positions of the archetype NCCR. The red lines, “Del,” and closed triangles indicate duplication, deletion, and single-base differences, respectively. The GenBank accession numbers of JCV sequences are as follows: AB038249 (CY), AY121912 (733 B), LC627282 (Ks-286, VP1), LC627283 (Ks-286, NCCR), AY536241 (SA296_02), AY536242 (SA28_03), D11364 (Mad11-Br), LC164353 (NIID11-68), AY536240 (SA84_00), AB038254 (Tky-1), AB038255 (Tky-2a), and J02226 (Mad-1)
Discussion
The details of the mechanisms that define the extent and progression of PML are not well understood, and an integrated clinical and virological approach is beneficial. We present a patient with mild PML as a background disease of combined immunodeficiency and the characteristics of JCV detected in the CSF. In a recent retrospective observational study of PML patients with underlying primary immunodeficiency, the median time from diagnosis to death was 8 months, and 5 out of 11 patients died within 6 months of diagnosis (Hadjadj et al. 2019). In our case, the patient was initiated on immunoglobulin replacement therapy and combination therapy of mefloquine and mirtazapine at 4 and 6 months after the onset of PML, respectively. During the follow-up period, no neurological deterioration was observed. The temporary enlargement of the lesion and the contrast-enhancing effect at 7 months after onset may indicate an inflammatory response and clearance of JCV. These were not observed in the later stages of the disease. Therefore, compared with previously reported cases, the disease progression, in this case, was slow, and the prognosis was favorable.
The mechanism underlying the mild conditions observed in this case is not well understood. However, considering the transient contrast-enhancing effect, it was suggested that local immune response to the virus had occurred. Another reason for the moderation of the disease was that JCV appearing, in this case, might have archetype-like characteristics based on the VP1 and NCCR sequences. We attempted to amplify and sequence the entire JCV genome, but this was difficult because of the low copy number of viral DNA in the CSF samples. Therefore, we cloned the VP1 gene and NCCR separately. The VP1 of JCV that emerged, in this case, was a perfect match to that of the archetype JCV 733B isolate, except for a substitution at the 265th position from asparagine to lysine among all 354 amino acid sequences. Other study groups have demonstrated that in the prototype JCV, amino acid substitutions from asparagine to aspartic acid, threonine, histidine, or serine can occur at position 265 on the surface of VP1 (Gorelik et al. 2011; McIlroy et al. 2019; Reid et al. 2011; Sunyaev et al. 2009). However, to the best of our knowledge, amino acid substitutions in lysine are rare. Thus, it is possible that VP1, which is not only very close to that of the archetype but also has atypical amino acid substitutions, may be related to the pathology in this case.
Another major finding, in this case, was the lack of complex mutations in the NCCR of the JCV genome. The CSF JCV from the patient can be considered an archetype-like virus in that the region D of the NCCR, which is frequently deleted in a prototype, was retained, and there were only a few deletions in other regions. In PML cases, most CSF JCV isolates are prototypes with variable mutations in the NCCR. However, there have been a few previous reports on the detection of archetypes or archetype-like viruses (Ferrante et al. 2003; Iannetta et al. 2013; Pfister et al. 2001; Seppälä et al. 2017). There have been cases, of long-term survival (Ferrante et al. 2003; Pfister et al. 2001) or short-term fatal outcomes (Iannetta et al. 2013; Seppälä et al. 2017) in patients, partly depending on the severity of the underlying disease. These lines of evidence suggest that complex mutations in the NCCR are not necessarily required for the development of PML itself and that some cases have a good prognosis when archetype or archetype-like viruses are detected. In the present case, it was assumed that the NCCR rearrangement did not proceed to owe to medication or that the virus did not increase its replication because of its archetype-like nature.
Conclusion
We found that archetype-like JCV was detected in a PML case with mild symptoms and progression. These observations serve as a basis for understanding the mutational mechanism of JCV and the pathogenesis of PML.
References
Cortese I, Reich DS, Nath A (2021) Progressive multifocal leukoencephalopathy and the spectrum of JC virus-related disease. Nat Rev Neurol 17:37–51. https://doi.org/10.1038/s41582-020-00427-y
Ferrante P, Delbue S, Pagani E, Mancuso R, Marzocchetti A, Borghi E, Maserati R, Bestetti A, Cinque P (2003) Analysis of JC virus genotype distribution and transcriptional control region rearrangements in human immunodeficiency virus-positive progressive multifocal leukoencephalopathy patients with and without highly active antiretroviral treatment. J Neurovirol 9(Suppl 1):42–46. https://doi.org/10.1080/13550280390195405
Gorelik L, Reid C, Testa M, Brickelmaier M, Bossolasco S, Pazzi A, Bestetti A, Carmillo P, Wilson E, McAuliffe M, Tonkin C, Carulli JP, Lugovskoy A, Lazzarin A, Sunyaev S, Simon K, Cinque P (2011) Progressive multifocal leukoencephalopathy (PML) development is associated with mutations in JC virus capsid protein VP1 that change its receptor specificity. J Infect Dis 204:103–114. https://doi.org/10.1093/infdis/jir198
Gosert R, Kardas P, Major EO, Hirsch HH (2010) Rearranged JC virus noncoding control regions found in progressive multifocal leukoencephalopathy patient samples increase virus early gene expression and replication rate. J Virol 84:10448–10456. https://doi.org/10.1128/JVI.00614-10
Hadjadj J, Guffroy A, Delavaud C, Taieb G, Meyts I, Fresard A, Streichenberger N, L’Honneur AS, Rozenberg F, D’Aveni M, Aguilar C, Rosain J, Picard C, Mahlaoui N, Lecuit M, Hermine O, Lortholary O, Suarez F (2019) Progressive multifocal leukoencephalopathy in primary immunodeficiencies. J Clin Immunol 39:55–64. https://doi.org/10.1007/s10875-018-0578-8
Iannetta M, Bellizzi A, Lo Menzo S, Anzivino E, D’Abramo A, Oliva A, D’Agostino C, d’Ettorre G, Pietropaolo V, Vullo V, Ciardi MR (2013) HIV-associated progressive multifocal leukoencephalopathy: longitudinal study of JC virus non-coding control region rearrangements and host immunity. J Neurovirol 19:274–279. https://doi.org/10.1007/s13365-013-0167-9
McIlroy D, Halary F, Bressollette-Bodin C (2019) Intra-patient viral evolution in polyomavirus-related diseases. Philos Trans R Soc Lond B Biol Sci 374:20180301. https://doi.org/10.1098/rstb.2018.0301
Nakamichi K, Kishida S, Tanaka K, Suganuma A, Sano Y, Sano H, Kanda T, Maeda N, Kira J, Itoh A, Kato N, Tomimoto H, Kurane I, Lim CK, Mizusawa H, Saijo M (2013) Sequential changes in the non-coding control region sequences of JC polyomaviruses from the cerebrospinal fluid of patients with progressive multifocal leukoencephalopathy. Arch Virol 158:639–650. https://doi.org/10.1007/s00705-012-1532-3
Nakamichi K, Kawamoto M, Ishii J, Saijo M (2019) Improving detection of JC virus by ultrafiltration of cerebrospinal fluid before polymerase chain reaction for the diagnosis of progressive multifocal leukoencephalopathy. BMC Neurol 19:252. https://doi.org/10.1186/s12883-019-1476-2
Pfister LA, Letvin NL, Koralnik IJ (2001) JC virus regulatory region tandem repeats in plasma and central nervous system isolates correlate with poor clinical outcome in patients with progressive multifocal leukoencephalopathy. J Virol 75:5672–5676. https://doi.org/10.1128/JVI.75.12.5672-5676.2001
Reid CE, Li H, Sur G, Carmillo P, Bushnell S, Tizard R, McAuliffe M, Tonkin C, Simon K, Goelz S, Cinque P, Gorelik L, Carulli JP (2011) Sequencing and analysis of JC virus DNA from natalizumab-treated PML patients. J Infect Dis 204:237–244. https://doi.org/10.1093/infdis/jir256
Seppälä H, Virtanen E, Saarela M, Laine P, Paulín L, Mannonen L, Auvinen P, Auvinen E (2017) Single-molecule sequencing revealing the presence of distinct JC polyomavirus populations in patients with progressive multifocal leukoencephalopathy. J Infect Dis 215:889–895. https://doi.org/10.1093/infdis/jiw399
Sugimoto C, Ito D, Tanaka K, Matsuda H, Saito H, Sakai H, Fujihara K, Itoyama Y, Yamada T, Kira J, Matsumoto R, Mori M, Nagashima K, Yogo Y (1998) Amplification of JC virus regulatory DNA sequences from cerebrospinal fluid: diagnostic value for progressive multifocal leukoencephalopathy. Arch Virol 143:249–262. https://doi.org/10.1007/s007050050284
Sunyaev SR, Lugovskoy A, Simon K, Gorelik L (2009) Adaptive mutations in the JC virus protein capsid are associated with progressive multifocal leukoencephalopathy (PML). PLoS Genet 5:e1000368. https://doi.org/10.1371/journal.pgen.1000368
Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, Correale J, Fazekas F, Filippi M, Freedman MS, Fujihara K, Galetta SL, Hartung HP, Kappos L, Lublin FD, Marrie RA, Miller AE, Miller DH, Montalban X, Mowry EM, Sorensen PS, Tintoré M, Traboulsee AL, Trojano M, Uitdehaag BMJ, Vukusic S, Waubant E, Weinshenker BG, Reingold SC, Cohen JA (2018) Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol 17:162–173. https://doi.org/10.1016/S1474-4422(17)30470-2
Yogo Y, Kitamura T, Sugimoto C, Ueki T, Aso Y, Hara K, Taguchi F (1990) Isolation of a possible archetypal JC virus DNA sequence from nonimmunocompromised individuals. J Virol 64:3139–3143. https://doi.org/10.1128/JVI.64.6.3139-3143.1990
Acknowledgements
The authors would like to thank the patient in this study.
Funding
This work was supported by the Research Committee of Prion Disease and Slow Virus Infection, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health and Labour Sciences Research Grants, The Ministry of Health, Labour and Welfare, Japan, and by JSPS KAKENHI (Grant Number 21K07450). We would like to thank Editage (www.editage.com) for English language editing.
Author information
Authors and Affiliations
Contributions
Kosuke Iwami, Masaaki Matsushima, Azusa Nagai, Shinichi Shirai, Sho Nakakubo, Ikuko Takahashi-Iwata, Masafumi Yamada, and Ichiro Yabe collected and interpreted the clinical data. Kazuo Nakamichi completed virological analyses. Kosuke Iwami and Kazuo Nakamichi wrote the manuscript and prepared the figures. Kosuke Iwami and Kazuo Nakamichi contributed equally to this work. All authors have critically revised and approved the manuscript.
Corresponding authors
Ethics declarations
Informed consent
The CSF was collected for clinical care, and written informed consent was obtained from the patient and her family for the use of the specimen for research purposes. The study was performed in accordance with the ethical standards of the Declaration of Helsinki after approval from the research institution.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Iwami, K., Nakamichi, K., Matsushima, M. et al. Progressive multifocal leukoencephalopathy with mild clinical conditions and detection of archetype-like JC virus in cerebrospinal fluid. J. Neurovirol. 27, 917–922 (2021). https://doi.org/10.1007/s13365-021-01017-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-021-01017-4