
Abstract
Rabies virus is a neurotropic lyssavirus which is 100% fatal in its pathogenic form when reaching unprotected CNS tissues. Death can be prevented by mechanisms delivering appropriate immune effectors across the blood-brain barrier which normally remains intact during pathogenic rabies virus infection. One therapeutic approach is to superinfect CNS tissues with attenuated rabies virus which induces blood-brain barrier permeability and immune cell entry. Current thinking is that peripheral rabies immunization is sufficient to protect against a challenge with pathogenic rabies virus. While this is undoubtedly the case if the virus is confined to the periphery, what happens if the virus reaches the CNS is less well-understood. In the current study, we find that peripheral immunization does not fully protect mice long-term against an intranasal challenge with pathogenic rabies virus. Protection is significantly better in mice that have cleared attenuated virus from the CNS and is associated with a more robust CNS recall response evidently due to the presence in CNS tissues of elevated numbers of lymphocytes phenotypically resembling long-term resident immune cells. Adoptive transfer of cells from rabies-immune mice fails to protect against CNS challenge with pathogenic rabies virus further supporting the concept that long-term resident immune cell populations must be established in brain tissues to protect against a subsequent CNS challenge with pathogenic rabies virus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rabies virus (RABV) is a single-stranded RNA virus in the lyssavirus family. RABV spreads transaxonally from the site of infection into spinal and brain tissue, which limits immune recognition (Rupprecht 1996). Current methods of rabies post-exposure prophylaxis (PEP) include intramuscular inoculation with a killed vaccine strain of RABV and administration of RABV immunoglobulins (Rupprecht et al. 2009). This protocol results in the induction of a strong peripheral immune response that is effective at preventing rabies disease if given before the virus enters the central nervous system (CNS). However, once the virus reaches CNS tissues, the infection is nearly 100% fatal in a non-immune individual and current PEP regimens are rarely effective (Hemachudha et al. 2013; Hemachudha et al. 2002). The lack of efficacy against a CNS infection with pathogenic RABV in part arises from the inability of immune effectors to infiltrate into infected CNS tissues across the blood-brain barrier (BBB) (Roy et al. 2007), specialized neurovasculature that forms an anatomical and functional barrier between CNS tissues and blood. Immune cell and antibody entry into healthy CNS tissues is limited and occurs via specialized regions in the choroid plexus and subarachnoid space into the CSF (Wilson et al. 2010). The specialized nature and function of the neurovasculature known as the BBB generally impedes the entry into CNS tissues of potentially damaging circulating cells, antibodies, and other molecules without specific transport mechanisms. However, in response to certain infections of CNS tissues, including attenuated RABV, the tight neurovascular endothelial cell junctions that are in part responsible for the barrier function become altered such that CD4 T, CD8 T, and B cells can directly infiltrate the infected brain parenchyma (Phares et al. 2007; Fabis et al. 2008; Chai et al. 2014; Blanchette and Daneman 2015). The local production of IFNγ, likely through the activity of reactive oxygen species (ROS) (Phares et al., 2007; Spencer et al. 2016), causes alterations in the neurovasculature that facilitate immune cell extravasation through microvessel walls into the brain parenchyma (Wilson et al. 2010). Alterations include upregulation of the vascular adhesion molecule ICAM-1 and transient leakage of low molecular weight markers from the circulation into CNS tissues which accompanies the extravasation of lymphocytes into the perivascular space of CNS parenchymal capillaries and then deeper into the infected tissues (Phares et al. 2007; Fabis et al. 2008; Roy and Hooper 2008; Lebrun et al. 2015). This process is comparable to other neuroimmune responses where activated immune cells have been observed crossing the BBB in a step-wise manner with ICAM-1 expression by neurovasculature endothelial cells causing immune cells to arrest on their surface and then cross into CNS tissues (Ransohoff et al. 2003). CNS infection with wild-type RABV in a non-immune mouse does not trigger the loss of neurovascular integrity or cell infiltration into CNS tissues (Fabis et al. 2008; Roy et al. 2007; Phares et al. 2006).
The ability of RABV to enter the CNS transaxonally without disrupting BBB integrity allows the virus to hide from the immune system within the CNS (Fabis et al. 2008; Roy et al. 2007). The inability of immune effectors to enter CNS tissues in response to infection with pathogenic RABV likely contributes to the failure of PEP. Moreover, current PEP regimens use vaccines based on killed RABV which induce a type 2 immune response that elicits high serum titers of RABV virus neutralizing antibody but is deficient in clearing RABV from CNS tissues (Lebrun et al. 2015). These observations highlight the need to better understand how vaccination can protect CNS tissues.
RABV are unique tools to model the impact of different immunization regimens on CNS tissues. In non-immune mice, infection with pathogenic RABV invariably causes a lethal CNS infection regardless of the route of administration, with the BBB remaining intact even where a peripheral immune response to the virus develops (Roy and Hooper 2008). In contrast, a variety of attenuated RABV strains have been engineered in the laboratory that differ in their capacity to spread from the periphery to the CNS. If these strains reach the CNS, they are cleared because of immune infiltration into CNS tissues associated with BBB permeability (Fabis et al. 2008; Li et al. 2012). For example, SPBN-GAS-GAS-GAS (TriGAS) is an attenuated strain of RABV with three copies of a glycoprotein gene containing two attenuating mutations, Arg333 to Glu333 and Asn194 to Ser194, that has a severely reduced ability to spread due to its genomic structure and overproduction of attenuated glycoprotein (Faber et al. 2005; Schutsky et al. 2013; Faber et al. 2009b). The sequential transcription of mRNAs from the RABV monocistronic genome results in a transcription gradient with successful transcription diminishing as viral genomic length increases due to polymerase dissociation at each gene border. In addition, the glycoprotein of attenuated RABV is pro-apoptotic and evidently causes a cell stress response which temporarily shuts down viral replication resulting in delayed or inhibited spread (Faber et al. 2005; Medigeshi et al. 2007). Variants containing a single or double copy of the mutated glycoprotein, named GAS and BiGAS respectively, spread more efficiently due to their shorter genome length and decreased glycoprotein gene expression in comparison to TriGAS. Consequently, GAS and BiGAS more readily reach CNS tissues from a peripheral site of infection than does TriGAS. While the replication of TriGAS in CNS tissues induces an infiltrating immune response, its limited capacity to spread from cell to cell (Faber et al. 2005) suggests that if administered in the periphery, this variant may be able to induce therapeutic immunity to RABV antigens without spreading to the CNS. In this case, appropriate immune effectors may not reach CNS tissues.
Tissue resident immune cells demonstrated to participate in the clearance of secondary or challenge infections in the periphery can be identified by their expression of a variety of markers. These include CD138, a plasma cell marker, as well as CD27 and CD27L, which are markers of memory B cell development. Others are CD62L, also known as L-selectin, a key adhesion marker expressed on lymphocytes which increases their ability to migrate to and remain in inflamed tissue (Kaech et al. 2002) and CD127, the receptor for IL-7, which functions in homeostatic maintenance of long-lived populations (Bradley et al. 2005). Finally, CD69 is required for the early formation and maintenance of long-lived T cell populations (Schoenberger 2012). The aim of the current studies is to determine whether such cells become resident in CNS tissues because of the clearance of attenuated RABV from the CNS and whether their activity may be responsible for the subsequent clearance of a challenge of CNS tissues with pathogenic RABV.
To examine this possibility, mice were immunized by infection with TriGAS either in the gastrocnemius or masseter muscle, localizing the infection to the periphery or allowing it to reach the CNS respectively, and then challenged with pathogenic RABV in the CNS. The results suggest that establishment of long-lived resident immune effector cell populations in CNS tissues are necessary for long-term immune surveillance of the CNS and that this cannot be accomplished by a strictly peripheral immunization.
Materials and methods
Animal and study approval
C57BL/6 mice (JAX ™ Mouse Strain) were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in the Thomas Jefferson University Animal Facility. All procedures were conducted under Public Health Service Policy on Humane Care and Use of Laboratory Animals under protocols approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University (Animal Welfare Assurance Number A3085-01).
Viruses
SPBN-GAS (GAS) is a recombinant RABV with two mutations in the glycoprotein, Arg333 to Glu333 and Asn194 to Ser194, that attenuate the virus and prevent its reversion to a more pathogenic form. SPBN-GAS-GAS (Bi-GAS) contains two of these mutated glycoprotein genes and SPBN-GAS-GAS-GAS (TriGAS) contains three of these mutated glycoprotein genes as previously described (Faber et al. 2005). These viruses are all safe in immunocompetent mice, causing little to no signs of clinical RABV infection (Faber et al. 2009a, b). DRV4 is a pathogenic RABV, which was isolated from human brain tissue (Dietzschold et al. 2000; Li et al. 2012). DRV4 causes the death of non-immune mice within 8–12 days post infection (d.p.i.) regardless of site of infection. Signs of DRV4 infection are quantified by clinical sickness scores 0–5 with the following corresponding signs: 0: normal mouse; 1: atypical movement; 2: hunched back, abnormal gait; 3: shaking/trembling, labored/difficulty moving; 4: extreme difficulty in movement/no movement observed; and 5: unresponsive/moribund.
Infection of mice
Groups of C57BL/6 mice were immunized, under isoflurane (Vedco, St. Joseph, MO) anesthesia, intramuscularly (i.m.) (masseter or gastrocnemius) with 1 × 107 focus-forming units (FFU) of TriGAS in 100 μl PBS. Immunized and naïve mice were challenged intranasally (i.n.) with 1 × 105 FFU of DRV4 in 20 μl PBS and observed daily for signs of disease. All mice were humanely euthanized when unresponsive/moribund or after losing greater than 30% of starting body weight. Mice sacrificed for tissue analysis were anesthetized using a ketamine (Zoetis, Kalamazoo, MI): xylazine (Anased, Shenandoah, IA): PBS (3:2:1) mixture and perfused with 10 ml PBS and heparin, followed by 10 ml PBS.
Serum antibody titers
Serum antibody levels were measured by ELISA as previously described (Phares et al. 2006). Briefly, plates were coated with UV-inactivated ERA (Evelyn-Rokitnicki-Abelseth) virus, blocked with 1% BSA, and serum samples were added at varying dilutions. Mouse IgG-specific AKP-conjugated secondary antibody was used to detect bound antibody. Plates were developed using pNpp and read at 405 nm after 30 min.
Immunohistochemistry and co-localization quantification
Immunohistochemistry was performed as previously described (Lebrun et al. 2015). Briefly, brain tissues were embedded in OCT compound (Sakura Fintex, Torrance, CA) and blocks were cut using a Thermo Shandon cryostat (Pittsburgh, PA) into 15 μM sections. Sections were fixed and washed. After blocking, sections were incubated overnight in primary antibody (1:500) (Table 1) at 4 °C. Secondary antibody (1:1000) was added for 2 h at RT. Images were acquired with an upright Leica DM6000 microscope with the Leica Application Suite v4 program (Leica Microsystems, Heerbrugg, Switzerland). Brightness and contrast on pictures were adjusted using the GNU Image Manipulation Program.
qRT-PCR
Semi-quantitative and quantitative real-time PCR was performed as previously described (Phares et al. 2006). Briefly, brain tissues were homogenized, and total RNA was extracted using the RNeasyMini Kit (QIAGEN, Germantown, MD). cDNA was synthesized by reverse transcription using oligo (dt)15 primer (Promega Madison, WI). Quantitative real-time PCR was performed using iQ Supermix (Bio-Rad Laboratories, Hercules, CA), gene-specific primers and probes (Lebrun et al. 2015), and an iCycler iQ Real-Time PCR Detection System (Bio-Rad Laboratories). Synthetic cDNA standards specific for each gene were used to determine copy numbers. Semi-quantitative expression of CD69, CD27, CD27L, CD103, CD62L, and CD138 (Table 2) was determined using the SYBR Green reagent (Bio-Rad Laboratories) with the expression of these genes being analyzed using the DeltaDelta Ct method. All samples were normalized to their content of the housekeeping gene L13.
Statistical analysis
Statistical significant differences in gene expression and serum antibody titers between groups was assessed with one-way ANOVA followed by the Bonferroni multiple comparison test of all columns. Graphical design and statistical analysis was performed using GraphPad Prism 5.0 software (La Jolla, CA). Principle component analysis (PCA) to determine patterns in gene expression across samples was performed using MeV (Saeed et al. 2003).
Results
Peripheral immunization with inactivated or spread-deficient RABV fails to provide long-term protection against pathogenic RABV challenge directed at the CNS
Previous studies of the efficacy of RABV vaccination against CNS infection have generally used direct intracranial (i.c.) challenge with pathogenic RABV at the peak of the primary immune response (Seligman 1973). The objective here is to determine whether long-term protection can be conferred against pathogenic RABV, which persists long after the immunizing response has waned, without delivering the challenge via a needle stick which mechanically disrupts the BBB. To do so, we challenged at increasing intervals after vaccination using i.n. administration of pathogenic RABV. Our initial objective was to determine if a type 2 (Th2) immune response, which is the class of response normally elicited by administration of a killed RABV vaccine (Jackson 2011), can be effective against an i.n. challenge with DRV4, or can be skewed by superinfection by the administration of live-attenuated RABV to mice already infected with wild-type RABV toward a more type 1 (Th1)-biased response, which is known to be more effective in the clearance of a RABV CNS infection (Lebrun et al. 2015). Mice were immunized with the following protocols: (1) inoculation of UV-inactivated Bi-GAS into the gastrocnemius on day 0 then boosted with the same protocol on days 7 and 14 after the primary immunization; (2) inoculation of live BiGAS into the gastrocnemius on day 0, boosted on day 28 with live TriGAS in the gastrocnemius; and (3) inoculation of UV-inactivated BiGAS into the gastrocnemius on day 0, boosted on day 28 with live TriGAS in the gastrocnemius. At 60 days post initial immunization, all mice and non-immune controls were challenged with pathogenic DRV4 RABV i.n. No protection was seen in mice immunized with UV-inactivated BiGAS, reaching 100% mortality by 12 d.p.i. (Fig. 1a). As expected, these mice also showed a strong Th2 (IgG1) antibody bias (Fig. 1b). A boost with live TriGAS after initial immunization with inactivated virus only slightly improved the outcome with 30% of the mice surviving (Fig. 1a) despite a shift in bias from Th2 to mixed Th2/Th1 (IgG1 plus IgG2a) antibody production (Fig. 1b). Mice initially vaccinated with live BiGAS and boosted by live TriGAS were the best protected with 80% surviving (Fig. 1a). The RABV-specific antibody response in these animals was almost exclusively Th1 (IgG2a) (Fig. 1b).

Immunization with an inactivated RABV vaccine offers limited protection against a CNS challenge with pathogenic RABV which is associated with a predominantly Th2 RABV-specific antibody response. Groups of C57Bl/6 (n = 5) mice were immunized in the gastrocnemius with (1) three doses of UV-inactivated BiGAS (UV-GG); (2) a single dose of UV-inactivated BiGAS, boosted once with live TriGAS (GGG); or (3) a single dose of live BiGAS (GG), boosted once with live TriGAS. All mice and non-immune controls were challenged with DRV4 i.n. 60 days after the initial immunization as described in “Materials and methods” section. Mice were monitored for survival (a) and serum RABV-specific IgG1 and IgG2a levels prior to challenge and at sacrifice were assessed by ELISA (b)
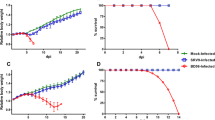
Antibodies produced by a type 2 response to RABV, as elicited by vaccination with a killed virus, prevent a subsequent infection with live virus from spreading to CNS tissues. Consequently, a secondary infection with live attenuated virus, despite inducing a type 1 immune response, may not result in the delivery of immune effectors into the CNS and the establishment of long-term protection against an i.n. challenge. To test this hypothesis, we sought to determine whether infection of the gastrocnemius versus masseter with the highly spread compromised TriGAS attenuated RABV, potentially causing differences in spread to CNS tissues, would result in differences in long-term protection against i.n DRV4 challenge. Mice were inoculated with TriGAS either in the masseter or gastrocnemius and challenged intranasally (i.n.) with DRV4 60 days post-immunization. While 90% of the masseter immunized mice survived, only 40% of those immunized via the gastrocnemius survived the challenge (Fig. 2). Despite the difference in outcome, serum RABV-specific IgG levels, commonly used as a marker of protection, were higher after TriGAS infection via the gastrocnemius (i.m.g.) by comparison with the masseter (i.m.m.) (Fig. 3a). However, analysis of virus spread confirmed that the inoculation of TriGAS into the masseter muscle results in the extensive infection of CNS tissue, as opposed to the minimal viral replication in the CNS detected when the virus was injected into the gastrocnemius (i.m.g.) (Fig. 3b). As shown in Fig. 3c, pathogenic DRV4 rapidly spreads through the CNS tissues of non-immune mice after infection (Fig. 3c). Spread of this virus is somewhat curtailed in mice immunized by TriGAS infection via the gastrocnemius, but insufficiently to prevent death in most of the animals. On the other hand, the prior clearance of TriGAS from CNS tissues prevents the spread of DRV4 with the majority of the mice surviving.

Long-term protection against a CNS challenge with pathogenic RABV is dependent upon the route of immunization. Groups of C57Bl/6 mice (n = 10) were immunized by infection with TriGAS either in the gastrocnemius (i.m.g.) or the masseter muscle (i.m.m.). Sixty days later, the immunized mice and non-immune controls were challenged i.n. with DRV4, then monitored for survival. The significance of differences in survival between the groups was assessed by the Kaplan Meier log rank test, *** p < .001

Long-term protection against a CNS challenge with pathogenic RABV is not associated with serum RABV-specific antibody titers but with spread of attenuated vaccine virus to CNS tissues. Serum RABV-specific IgG levels in non-immune and mice immunized by infection with TriGAS in either the masseter or gastrocnemius and then challenged with DRV4 were assessed by ELISA 8 days post challenge and are expressed as absorbance at 405 nm (a). Cortical and cerebellar tissues from groups of mice (n = 10) immunized by infection with TriGAS in the masseter versus gastrocnemius and unimmunized controls (b) or similarly immunized or left non-immune and then challenged with DRV4 i.n. (c) were analyzed by qRT-PCR for TriGAS and DRV4 mRNA levels respectively at 8 days after immunization or challenge as described in “Materials and methods” section. Mean ± SD copy numbers per 1000 copies of the housekeeping gene L13 are presented. The significance of differences between the groups was assessed by the Mann-Whitney u test, *p < .05
The inability of TriGAS to spread to CNS tissues from the gastrocnemius results in a reduced primary immune response in the brain as well as impaired long-term CNS immunity against DRV4
TriGAS infection induces a virus-clearing immune response when it reaches CNS tissues (Faber et al. 2009b). The data presented above suggests the failure of TriGAS infection in the gastrocnemius to establish long-term protection may be a consequence of its inability to spread to the CNS from this site thereby failing to trigger the infiltration of immune effectors into CNS tissues. To investigate this possibility, mice were immunized with TriGAS in the masseter or gastrocnemius, and then challenged 60 days later with DRV4 i.n. Cerebral cortex tissues were taken 8 days post challenge for analysis of immunologically relevant gene expression. Prior immunization by infection with TriGAS in the masseter results in higher cortical levels of mRNAs specific for CD4 (Fig. 4a), CD19 (Fig. 4b), CD8 (Fig. 4c), IFNγ (Fig. 4d), and antibody kappa light chain (Fig. 4e) than those detected in similar tissues from gastrocnemius immunized or non-immune mice.

DRV4 challenge i.n. causes greater elevations of IFNγ and antibody kappa light chain mRNAs in the CNS tissues of mice immunized by TriGAS infection in the masseter by comparison with the gastrocnemius. Cortical tissues from the mice described in panel c of Fig. 3 were assessed by qRT-PCR for CD4 (a) CD19 (b), CD8 (c), IFNγ (d), and antibody kappa light chain (e) mRNA levels 8 days post-challenge with DRV4. Data is presented as the mean ± SD copy numbers per 1000 copies of the housekeeping gene L13. The significance of differences between the groups was assessed by the Mann-Whitney u test, *p < .05; **p < .01; ***p < .001
To determine if DRV4 clearance from the CNS after masseter immunization also may result in the activity of resident, long-lived immune effector cell populations in brain tissues, the tissues described above were assessed for levels of mRNAs specific for long-term effector cell phenotype markers. At 8 days post DRV4 challenge, cortical tissues from mice immunized in the masseter muscle showed significantly higher expression levels of CD138 (Fig. 5a), CD62L (Fig. 5b), CD27 (Fig. 5c), CD27L (Fig. 5d), CD69 (Fig. 5e), and CD127 (Fig. 5f) mRNAs than similar tissues from their gastrocnemius immunized or non-immune counterparts, indicating that the responding cells bear these markers. PCA analysis of the expression levels of these long-lived resident immune phenotypic markers in the cerebral cortex tissues of the different mice shows clear clustering based on the immunization site (Fig. 5g). Naïve and i.m.g.-immunized mice cluster toward the center of the plot, indicating minimal expression, while i.m.m.-immunized mice skew toward the outside of the axes. These results were confirmed by IHC staining of infected cortical tissues for CD62L (Fig. 6a), CD138 (Fig. 6b), and CD27 (Fig. 6c). Mice originally immunized with TriGAS in the masseter consistently showed higher levels of expression for markers associated with long-lived resident immunity following pathogenic RABV challenge, while gastrocnemius-immunized mice more closely resembled naïve mice as they encounter RABV for the first time.

mRNAs encoding markers associated with long-lived immune effector cell populations are elevated to greater extents in the CNS tissues of DRV4-challenged mice immunized by infection with TriGAS in the masseter by comparison with gastrocnemius. Cortical tissues of mice immunized and challenged as described in “Materials and methods” section were assessed by qRT-PCR for changes in mRNA expression of CD138 (a), CD62L (b), CD27 (c), CD27L (d), CD69 (e), and CD127 (f) 8 days post-challenge with DRV4. Differences in mRNA expression levels between groups were also examined using principal component analysis (g). mRNA levels are expressed as relative change of expression between samples ± SD normalized to the housekeeping gene L13. The significance of differences between the groups was assessed by the Mann-Whitney u test, *p < .05; **p < .01; ***p < .001

Differences in expression of markers associated with long-lived cell populations can be visualized in the brain tissues of TriGAS immunized mice after challenge. Cortical tissues of mice immunized and challenged as described in “Materials and methods” section were harvested 8 days post challenge with DRV4 and stained for CD62L (red) and DAPI (blue) (a) CD138 (red) and DAPI (blue) (b), and CD27 (green) and DAPI (blue) (c). Scale bar indicates 100 μM. IHC images are representative of ten slices/brain of consecutive sections from three mice in two independent experiments
The adoptive transfer of lymphocytes from RABV-immune mice fails to protect naïve recipients against CNS challenge infection with pathogenic DRV4
Clearance from the CNS of an initial infection with attenuated RABV from CNS tissues is dependent on immune effector infiltration across the BBB (Roy et al. 2007), and we have shown that masseter immunization results in higher levels of lymphocyte infiltration into the CNS during immunization (Fig. 4). While our data suggests that immune cells that become resident in CNS tissues are responsible for long-term protection of CNS tissues, it remains possible that the process of RABV clearance from CNS tissues results in the generation of peripheral immune cell populations with the unique capacity to enter CNS tissues upon challenge. To test this hypothesis, mice were immunized with TriGAS i.m.m., their splenocytes were harvested 30 days later and administered i.p. to naïve recipients. The recipient mice were challenged 24 h afterwards with DRV4 in the masseter muscle with non-immune mice challenged as controls. While there appeared to be a small benefit to adoptive transfer with respect to survival, the difference in survival between non-immune animals and those that received cells from mice immunized via the masseter muscle with TriGAS was not statistically significant (Fig. 7a). Furthermore, weight loss (Fig. 7b) and clinical disease scores (Fig. 7c) were indistinguishable.

Immune cells primed in the periphery during immunization do not confer protection against CNS challenge. Spleen and lymph node cells from mice immunized by infection with TriGAS. i.m. in the masseter 14 days previously were adoptively transferred i.p. into naïve recipients as described in “Materials and methods” section. Twenty-four hours later, recipients of these cells and non-immune controls were challenged in the masseter muscle with DRV4. The mice were monitored for survival (a), percent original body weight (b), and clinical disease score (c). The difference in survival between the adoptively transferred and control groups was found to not be significant by the Kaplan-Meier log rank test
Discussion
The aim of the current investigation was to establish the basic parameters of an immunization protocol that can protect against a challenge with pathogenic RABV that is directed at CNS tissues. While rare in nature, this could occur through a bite to the face or inhalation of the virus. Current rabies PEP regimens are known to fail to prevent disease in a non-immune individual if administered after the virus has reached the CNS; however, little is known about the consequences of pathogenic RABV infection of the CNS at extended periods following vaccination. Our results show that clearance of attenuated RABV from the brain during immunization is likely to be required for long-term immune protection against a secondary infection of CNS tissues with pathogenic RABV. The key to this protection is the ability of an initial infection with attenuated RABV to induce the infiltration of immune effectors into CNS tissues.
Approaches used to attenuate RABV pathogenicity often impact the capacity of the viruses to spread from cell to cell (Li et al. 2012). Consequently, the inoculation of highly attenuated RABV into the gastrocnemius of immunocompetent mice rarely leads to appreciable infection of CNS tissues. After their primary response has subsided, mice vaccinated in this manner rarely survive when challenged with pathogenic wild-type RABV intranasally, a more direct route into CNS tissue for the virus than intramuscular challenge. In contrast, mice immunized by infection with spread-attenuated RABV in the masseter, which results in viral replication in CNS tissues, show a significantly higher survival rate from the same challenge. While long-term immune protection of CNS tissues has been established in these animals, adoptive transfer of their spleen cells fails to protect non-immune mice from a wild-type RABV challenge directed at the CNS. This confirms that intra-masseter priming alone does not confer RABV-specific immune cells the ability to infiltrate and function in CNS tissues. Therefore, we conclude that the generation of a RABV-specific immune response in CNS tissues resulting in the establishment of persisting local immune cells is required for long-term protection of CNS tissues from wild-type virus.
Immunization with live-attenuated RABV elicits a strong Th1 response in contrast to vaccination with UV-inactivated virus which induces a Th2 response known to be ineffective in clearing RABV infection from the brain (Lebrun et al. 2015). Since the induction of a Th1 response in brain tissue is most effective at establishing long-term immune protection of the CNS from RABV, the administration of a RABV vaccine based on killed virus would not be expected to confer long-term protection to pathogenic RABV that reaches CNS tissues. The data presented here support this concept.
Intranasal DRV4 challenge of mice that have been immunized by the clearance of attenuated TriGAS RABV from their CNS tissues results in elevated numbers of cells bearing markers associated with long-lived resident immune cell populations, and increased levels of the associated mRNAs, in brain tissues. This is not seen in mice that either had received TriGAS in their gastrocnemius or had been non-immune prior to challenge. At challenge, these putative memory cells are only seen in the CNS of masseter-immunized animals suggesting that their establishment in CNS tissues is necessary for long-term protection against a secondary infection of CNS tissues with pathogenic RABV.
At first glance, our findings may seem difficult to reconcile with the wealth of rabies vaccination data in the literature. Clearly, vaccines based on killed or live-attenuated RABV strains all elicit strong RABV-specific VNA responses that are highly effective at protecting against a peripheral challenge with a wide variety of pathogenic RABV variants. Antibody is undoubtedly the major effector in this scenario. The question we are addressing here is whether strictly peripheral immune memory can protect CNS tissues from pathogenic RABV that enters via a route that largely avoids peripheral defenses. During conventional rabies vaccine efficacy tests, mice are often challenged 14–28 days after immunization when the response to immunizing virus is still active and has not yet fully reached a memory state. Furthermore, the challenge route of choice is often intracranial, which necessitates the mechanical disruption of the BBB allowing peripheral immune cells to leak into the CNS. The needle stick also causes local inflammation that can further attract immune cells into the CNS. In such cases, the inadvertent delivery of peripheral immune effectors into CNS tissues may be sufficient for a therapeutic response. Infections with wild-type RABV are pathogenic at least in part because the BBB remains intact, preventing immune effectors from reaching the CNS. For proper assessment of long-term immune protection in CNS tissues, the challenge virus must therefore be delivered by a means that does not artificially compromise the BBB, to mice immunized well more than a month previously. Based on studies respecting this paradigm, we conclude that immune memory that is limited to the periphery offers little protection against a CNS infection with pathogenic RABV. The establishment of resident immune cells in CNS tissues during the immunization process is essential for long-term protection against an infection with wild-type RABV that reaches CNS tissues.
While providing insight into why conventional post-exposure regimens fail if initiated after the RABV has reached CNS tissues, the current findings are directed at providing a better understanding of the establishment of long-term immunity in CNS tissues in general. This applies to any situation following immune sensitization where a secondary immune response initiates in CNS tissues prior to, or in the absence of the loss of BBB integrity and a second wave of immune cell infiltration. Pertinent examples include not only the rare instances where an aerosol exposure to RABV occurs in an immunized individual but also persisting viral infections, CNS autoimmune conditions such as multiple sclerosis, and brain cancer immunotherapy.
References
Blanchette M, Daneman R (2015) Formation and maintenance of the BBB. Mech Dev 138:8–16
Bradley LM, Haynes L, Swain SL (2005) IL-7: maintaining T-cell memory and achieving homeostasis. Trends Immunol 26(3):172–176
Chai Q, He WQ, Zhou M, Lu H, Fu ZF (2014) Enhancement of blood-brain barrier permeability and reduction of tight junction protein expression are modulated by chemokines/cytokines induced by rabies virus infection. J Virol 88(9):4698–4710. https://doi.org/10.1128/JVI.03149-13
Dietzschold B, Morimoto K, Hooper DC, Smith JS, Rupprecht CE, Koprowski H (2000) Genotypic and phenotypic diversity of rabies virus variants involved in human rabies: implications for postexposure prophylaxis. J Hum Virol 3:50–57
Faber M, Dietzschold B, Li J (2009a) Immunogenicity and safety of recombinant rabies viruses used for oral vaccination of stray dogs and wildlife. Zoonoses Public Health 56(6–7):262–269. https://doi.org/10.1111/j.1863-2378.2008.01215.x
Faber M, Li J, Kean RB, Hooper DC, Alugupalli KR, Dietzschold B (2009b) Effective preexposure and postexposure prophylaxis of rabies with a highly attenuated recombinant rabies virus. Proc Natl Acad Sci U S A 106(27):11300–11305. https://doi.org/10.1073/pnas.0905640106
Faber M, Papaneri A et al (2005) A single amino acid change in rabies virus glycoprotein increases virus spread and enhances virus pathogenicity. J Virol 79(22):14141–14148. https://doi.org/10.1128/JVI.79.22.14141-14148.2005
Fabis MJ, Phares TW, Kean RB, Koprowski H, Hooper DC (2008) Blood-brain barrier changes and cell invasion differ between therapeutic immune clearance of neurotrophic virus and CNS autoimmunity. Proc Natl Acad Sci U S A 105(40):15511–15516. https://doi.org/10.1073/pnas.0807656105
Hemachudha T, Laothamatas J, Rupprecht CE (2002) Human rabies: a disease of complex neuropathogenetic mechanisms and diagnostic challenges. The Lancet Neurology 1(2):101–109. https://doi.org/10.1016/S1474-4422(02)00041-8
Hemachudha T, Ugolini G, Wacharapluesadee S, Sungkarat W, Shuangshoti S, Laothamatas J (2013) Human rabies: neuropathogenesis, diagnosis, and management. The Lancet Neurology 12(5):498–513. https://doi.org/10.1016/S1474-4422(13)70038-3
Jackson AC (2011) Research advances in rabies. Adv Virus Res 79:xvii-0-12-387040-7.00022-6. doi:https://doi.org/10.1016/B978-0-12-387040-7.00022-6 [doi].
Kaech SM, Wherry EJ, Ahmed R (2002) Effector and memory T-cell differentiation: implications for vaccine development. Nat Rev Immunol 2(4):251–262
Lebrun A, Portocarrero C, Kean RB et al (2015) T-bet is required for the rapid clearance of attenuated rabies virus from central nervous system tissue. JOI 195(9):4358–4368
Li J, Ertel A, Portocarrero C, Barkhouse DA, Dietzschold B, Hooper DC, Faber M (2012) Postexposure treatment with the live-attenuated rabies virus (RV) vaccine TriGAS triggers the clearance of wild-type RV from the central nervous system (CNS) through the rapid induction of genes relevant to adaptive immunity in CNS tissues. J Virol 86(6):3200–3210. https://doi.org/10.1128/JVI.06699-11
Medigeshi GR, Lancaster AM, Hirsch AJ, Briese T, Lipkin WI, DeFilippis V, Fruh K, Mason PW, Nikolich-Zugich J, Nelson JA (2007) West Nile virus infection activates the unfolded protein response, leading to CHOP induction and apoptosis. J Virol 81(20):10849–10860
Phares TW, Fabis MJ, Brimer CM, Kean RB, Hooper DC (2007) A Peroxynitrite-dependent pathway is responsible for blood-brain barrier permeability changes during a central nervous system inflammatory response: TNF-α is neither necessary nor sufficient. J Immunol 178(11):7334–7343. https://doi.org/10.4049/jimmunol.178.11.7334
Phares TW, Kean RB, Mikheeva T, Hooper DC (2006) Regional differences in blood-brain barrier permeability changes and inflammation in the apathogenic clearance of virus from the central nervous system. J Immunol 176(12):7666–7675
Ransohoff RM, KivisÃkk P, Kidd G (2003) Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 3:569–581
Roy A, Hooper DC (2008) Immune evasion by rabies viruses through the maintenance of blood-brain barrier integrity. J Neuro-Oncol 14(5):401–411. https://doi.org/10.1080/13550280802235924
Roy A, Phares TW, Koprowski H, Hooper DC (2007) Failure to open the blood-brain barrier and deliver immune effectors to central nervous system tissues leads to the lethal outcome of silver-haired bat rabies virus infection. J Virol 81(3):1110–1118. https://doi.org/10.1128/JVI.01964-06
Rupprecht CE (1996) Rhabdoviruses: rabies virus. In: Baron S (ed) Medical Microbiology, 4th edn. The University of Texas Medical Branch at Galveston, Galveston (TX).
Rupprecht CE, Briggs D, Brown CM, Franka R, Katz SL, Kerr HD, Lett S, Levis R, Meltzer MI, Schaffner W, Cieslak PR (2009) Evidence for a 4-dose vaccine schedule for human rabies post-exposure prophylaxis in previously non-vaccinated individuals. Vaccine 27(51):7141–7148. https://doi.org/10.1016/j.vaccine.2009.09.029
Saeed AI, Sharov V et al (2003) TM4: a free, open-source system for microarray data management and analysis. BioTechniques 32(2):374–378
Schoenberger SP (2012) CD69 guides CD4+ T cells to the seat of memory. Proc Natl Acad Sci 109(22):8358–8359
Schutsky K, Curtis D, Bongiorno EK et al (2013) Intramuscular inoculation of mice with the live-attenuated recombinant rabies virus TriGAS results in a transient infection of the draining lymph nodes and a robust, long-lasting protective immune response against rabies. J Virol 87(3):1834–1841. doi:https://doi.org/10.1128/JVI.02589-12
Seligman EB Jr (1973) The NIH test for potency. In Laboratory techniques in Rabies 3rd Edition, Eds MM Kaplan and H Koprowski WHO Geneva.
Spencer NG, Schilling T, Miralles F, Eder C (2016) Mechanisms underlying interferon-Î3-induced priming of microglial reactive oxygen species production. PLoS One 11(9):e0162497
Wilson EH, Weninger W, Hunter CA (2010) Trafficking of immune cells in the central nervous system. J Clin Invest 120(5):1368–1379. https://doi.org/10.1172/JCI41911
Funding
This work was supported by the National Institutes of Health Grants AI093369, U01 AI083045, and R01 AI093369-01 (to D.C.H.). Grant NCI 5 P30 CA056036 from the National Cancer Institute to the Sidney Kimmel Cancer Center provided support for the Laboratory Animal Facility Shared Resource used in the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Garcia, S.A., Lebrun, A., Kean, R.B. et al. Clearance of attenuated rabies virus from brain tissues is required for long-term protection against CNS challenge with a pathogenic variant. J. Neurovirol. 24, 606–615 (2018). https://doi.org/10.1007/s13365-018-0655-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-018-0655-z