
Abstract
HIV-1-infected mononuclear phagocytes release soluble factors that affect the homeostasis in tissue. HIV-1 can prompt metabolic encephalopathy with the addition of neuronal dysfunction and apoptosis. Recently, we reported that HIV-1 enhances the expression and secretion of bioactive cathepsin B in monocyte-derived macrophages, ultimately contributing to neuronal apoptosis. In this research, we asked if microglia respond to HIV infection similarly by modifying the expression, secretion, and neurotoxic potential of cathepsin B and determined the in vivo relevance of these findings. HIV-1ADA-infected human primary microglia and CHME-5 microglia cell line were assessed for expression and activity of cathepsin B, its inhibitors, cystatins B and C, and the neurotoxicity associated with these changes. Human primary neurons were exposed to supernatants from HIV-infected and uninfected microglia in the presence of cathepsin B inhibitors and apoptosis was assessed by TUNEL. Microglial expression of cathepsin B was validated in brain tissue from HIV encephalitis (HIVE) patients. HIV-infected microglia secreted significantly greater levels of cathepsin B, cystatin B, and cystatin C compared to uninfected cells. Increased apoptosis was observed in neurons exposed to supernatants from HIV-1 infected microglia at day 12 post-infection. The cathepsin B inhibitor CA-074 and cathepsin B antibody prevented neuronal apoptosis. Increased microglia-derived cathepsin B, cystatin B, and cystatin C and caspase-3+ neurons were detected in HIVE brains compared to controls. Our results suggest that HIV-1-induced cathepsin B production in microglia contributes to neuronal apoptosis and may be an important factor in neuronal death associated with HIVE.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Microglia are the brain macrophages that participate in the protection and support of neurons. (Rock et al. 2004). One of the most important functions of microglia is to supply primary defense of the central nervous system (CNS) against infectious diseases (Rock et al. 2004) and to play a pivotal role of innate immunity in the CNS (Nguyen et al. 2002). Microglia exert their protective functions by working as scavengers (Voyvodic 1996), maintaining homeostasis (Tambuyzer et al. 2009), and recruiting immune cells from the periphery to the CNS (Alexaki et al. 2008). However, the protective phenotype can evolve into a destructive one under chronic disease conditions such as HIV-1 infection and its neurological complications by producing inflammatory cytokines and chemokines that can damage bystander cells including neurons (Ghorpade et al. 1998a).
The hallmark for HIV-associated neurocognitive disorders (HAND) is the release of soluble viral and cellular neurotoxins from infected and/or activated microglia and perivascular macrophages that can damage bystander cells. In a recent study, we used monocyte-derived macrophages (MDM) differentiated from blood monocytes from healthy donors as a model of perivascular macrophage to explore the role of cathepsin B on neuronal apoptosis in the brain during HIV infection (Rodriguez-Franco et al. 2012). Cathepsin B is a cysteine protease of lysosomal origin involved in various cellular processes (Honey and Rudensky 2003) including; apoptosis (Kingham and Pocock 2001), inflammation, and neurodegeneration (Nixon et al. 2000). In the previous study, we reported that HIV-infected MDM secreted high levels of bioactive cathepsin B that were associated with increased neuronal apoptosis (Rodriguez-Franco et al. 2012). We also demonstrated that following chronic HIV infection, cathepsin B does not interact with the lysosome membrane. Absence of interactions between cathepsin B and both of its inhibitors, cystatins B and C, were observed in HIV-1-infected MDM compared to uninfected controls (Rodriguez-Franco et al. 2012). The increased expression of cathepsin B was also observed in peripheral blood monocytes from the Hispanic women cohort with HAND (Cantres-Rosario et al. 2013), from post-mortem brain tissue from a patient with HAND (Rodriguez-Franco et al. 2012), and from MDM and brain tissue of patients exposed to cocaine (Zenón et al. 2014). However, the contribution of cathepsin B from HIV-infected microglia remained to be determined.
In the present study, we asked if HIV-1 triggers similar responses in resident microglia. We compared the expression of cathepsin B and cystatins B and C in HIV-1-infected and uninfected human primary microglia. Results demonstrated that HIV infection in vitro affected cathepsin B expression and secretion in human primary microglia and the microglia cell line CHME-5. The results revealed an induced secretion of cathepsin B in HIV-infected microglia that prompted neuronal apoptosis. The results with CHME-5 cells were comparable to primary microglia albeit at lower levels. To assess the relevance of these findings, we determined expression patterns of these enzymes in post-mortem brain tissue from HIV encephalitis (HIVE) and control cases. We found that cathepsin B expression and cystatin C were increased in HIVE cases and was associated with microglial cells and perivascular macrophages. Expression of cystatin B increased in microglial cells with HIV infection and in HIV+ subsyndromic cases. Taken together, our findings support an important role for cathepsin B and cystatins B and C in microglial response to HIV infection of the CNS.
Materials and methods
Human primary neuron and microglia isolation and culture
Human primary neurons and microglia were provided by Temple University’s Comprehensive NeuroAIDS Center. Fetal brain tissue (gestational age 16–18 weeks) was obtained from elective abortion procedures performed in full compliance with National Institutes of Health and Temple University ethical guidelines. The tissue was washed with cold Hanks balanced salt solution (HBSS) and meninges and blood vessels were removed as previously described [Huang et al 2011; Ghorpade et al. 1998a, b]. For microglia, brain tissue was supplemented with Ca2+ and Mg2+ and then digested with 0.25 % trypsin (Sigma) for 30 min at 37 °C. Trypsin was neutralized with fetal bovine serum (FBS), and the tissue was further dissociated to obtain single-cell suspensions. The cells were cultured in DMEM (Sigma), supplemented with a mixture containing 10 % heat-inactivated FBS, 1,000 U of purified recombinant human macrophage colony-stimulating factor (MCSF) per milliliter, penicillin and streptomycin (50 mg/ml), and 100 mg/ml of neomycin. The mixed culture was maintained under 5 % CO2 for 7 days, and the medium was fully replaced to remove any cell debris. The microglia cells released with further incubation were collected and purified by preferential adhesion. Microglia were seeded as an adherent monolayer at a density of 3 × 106 cells/well in a 6-well plate and floating cells were removed after 4 h. For neurons, tissue in HBSS was digested with papain (Sigma-Aldrich, St. Louis, MO) for 30 min at 37 °C. Tissue was then dissociated to obtain single-cell suspensions by repeated pipetting with small bore fire polished glass Pasteur pipettes. Cells were plated in poly-D lysine-coated dishes at a density of ~1.8 × 106 cells/60 mm dish in neurobasal media with B27 supplement, horse serum, and gentamicin (Invitrogen). After 2 h of attachment, media was replaced with fresh complete neurobasal media. Twenty-four hours later, cultures were replaced with neurobasal media without horse serum. Four days later, one fourth of the media was removed and replaced with neurobasal media supplemented with fluorodeoxyuridine (FDU) and uridine. Following FDU treatment, neurons were cultured in neurobasal medium with Glutamax and B27 supplement. One half volume of media was replaced every 48 h. Purity of cell type-specific cultures was assessed by immunolabeling for cell type-specific markers, including IBA-1 for microglia, GFAP for astrocyte and MAP2, neurofilament, and synaptophysin for neurons.
CHME-5 microglia
The microglia cell line CHME-5 was kindly provided by Dr. Randal Davis from Oklahoma State University. It was originally obtained from human fetal microglia (Peudenier et al. 1991) transfected with the T-antigen of SV-40 virus (Janabi et al. 1995). Cells were cultured to 70 % confluence in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma Chemical Company, St. Louis, MO) supplemented with 5 % inactivated fetal bovine serum (FBS), 1 % l-glutamine, and 1 % pen/strep. All cells were maintained at 37 °C in a 5 % CO2 incubator.
Detection of HIV-1 receptors CD4 and CCR5 and intracellular cystatin B in CHME-5 cells by flow cytometry
CHME-5 microglia were grown as previously described until confluence for surface labeling of CD4 and CCR5 receptors. At approximately 75 % confluence, cells were detached from the flask by repeated pipetting with fresh media or PBS against the cell monolayer. Detached cells were transferred to 5-ml polystyrene tubes (BD Falcon, Franklin Lakes, NJ) at a concentration of 5 × 105 cells/ml. The cell suspension was then centrifuged for 3 min at 4 °C and 1200 rpm to obtain the cell pellet. Cells were washed with 1 ml of wash buffer (3 % FBS/PBS) and labeled with CD4-FITC (1:100; Becton Dickinson) and CCR5-FITC (1:100; R & D Systems) antibodies or left in the wash buffer alone (unlabeled control) for 30 min at 4 °C. For detection of intracellular cystatin B levels, CHME-5 were permeabilized using the BD Cytofix/Cytoperm kit (BD Biosciences, San Jose, CA), incubated with anti-cystatin B (Sigma) antibody (1:1000) for 1 h at 4 °C. After incubation with antibodies, cells were washed twice with 1 ml of wash buffer and centrifuged. Finally, cells were fixed in 1 ml of 1 % paraformaldehyde. Fluorescence was read within 1 h using a FACSort (BD Immunocytometry Systems, Franklin Lakes, NJ). Flow cytometry data acquisition and multivariate analysis was performed using the Cell Quest software (BD Biosciences, CA). Cells were gated in forward/side scatter dot plots and FITC emission measured in the FL1 (band pass filter 525 nm) channel. Data on scatter parameters and histograms were acquired in log mode. Ten thousand events were evaluated for each sample and the median peak channel obtained from the histograms was used to determine levels of CD4, CCR5, or cystatin B.
HIV-1 infection of primary human microglia and CHME-5 cells
HIV-1ADA was obtained from Dr. Howard Gendelman (University of Nebraska) following propagation and titration in MDM from HIV-1-seronegative donors after as previously described (Gendelman et al. 1990). Primary microglia and CHME-5 were inoculated with HIV-ADA as described in Rodriguez-Franco et al. 2012. Cultures were followed for 12 days post-infection and media collected at 3, 6, and 12 days post-infection (dpi) and stored at −80 °C for p24 antigen assay, protein expression, and activity. Cells were lysed for protein studies were prepared according the previous published work of Rodriguez-Franco et al 2012.
Western blot analysis
Cell lysates containing 30 μg of total protein were lyophilized at 4 °C and rehydrated in 10 μl of sample buffer (4.75 μl of Laemmli sample buffer, 0.25 μl of β-mercaptoethanol [BioRad] and 5.00 μl of ultrapure water (HPLC), and heated at 70 °C for 10 min. Samples were loaded into 4–20 % Tris-HCl 15-well Ready Gels (BioRad) and positive controls for cystatin B, cystatin C, and cathepsin B (isolated human protein from liver, Bio Vision, New Minas, N Nova Scotia, Canada). Proteins were separated at 150 V for 1 h according to the NuPAGE® Protocol (Life Technologies, Grand Island, NY). After electrophoresis, proteins were transferred to a PVDF membrane using the semi-dry transfer method on a BioRad Trans Blot Turbo Transfer System for 7 min at 1.3 A–25 V per gel. After transfer, membranes were blocked with non-fat dry milk in 1× T-TBS for 1 h at room temperature with shaking. Membranes were incubated overnight at 4 °C with primary antibodies including mouse anti-human cathepsin B (1:500, Sigma-Aldrich), mouse anti-human cystatin B (1:2000, Sigma), mouse anti-human cystatin C (1:500, R&D Systems), and rabbit GAPDH (1:500, BIOSS) followed by secondary anti-mouse Ig G-HRP (1:10,000, Sigma), anti-rabbit Ig G-HRP (1:10,000, Sigma), or anti-mouse IgG-HRP (1:10,000, Sigma). All incubations with primary antibodies were conducted overnight at 4 °C; all incubations with secondary antibodies were conducted for 1 h at room temperature. Following incubations with primary and secondary antibodies, membranes were washed with 1× T-TBS at room temperature, five times for 5–10 min. Chemiluminescence (Super Signal West Femto Detection Kit; Pierce, Rockford, IL) was used for signal detection at BioRad Chemi Doc XR. The density of protein bands was determined using the Image Lab software (BioRad) and normalized against monoclonal rabbit anti-GAPDH (Sigma).
Secretion of cathepsin B and its inhibitors
The secretion of cathepsin B and its inhibitors was determined from uninfected and HIV-1-infected microglial supernatants collected at 3, 6, 9, and 12 dpi. Sandwich ELISA was used for the quantification of expression of secreted cathepsin B (R & D), cystatin B (R & D), and cystatin C (Bio Vendor) according to the manufacturer’s instructions.
Cathepsin B activity assay
The bioactivity of secreted cathepsin B was determined using the cathepsin B Activity Assay kit (Bio Vision, New Mines, Nova Scotia) according to manufacturer’s instructions AFC substrate was quantified using fluorometry as described (Rodriguez-Franco et al. 2012). Microglial supernatants collected from cell cultures at 3, 6, 9, and 12 dpi were assayed in duplicates. Signals were detected using a fluorescence plate reader equipped with 400-nm excitation filter and 505-nm emission filters VersaFluor TM Fluorometer (BioRad).
Determination of neuronal apoptosis from microglia condition media
Supernatants from primary microglia and CHME-5 cells were collected at days 3, 6, and 12 dpi, and fresh serum-free media was added for 24 h prior to collection (conditioned media). To determine neuronal apoptosis induced from primary microglia supernatants, we followed the method described in Rodriguez-Franco et al. 2012. Human primary neurons were cultured until confluence and incubated with conditioned media derived from HIV-infected primary microglia at 12 dpi and treated with cathepsin B inhibitor (50 μM) or cathepsin B monoclonal antibody (50 ng/ml). Apoptosis was measured using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay at 12 days post-infection. Confocal images were obtained on a Zeiss confocal microscope Axiovert 200 M with a LSM 510 under an excitation wavelength of 488 nm, 20× magnification. Quantitative analysis of staining ratio of apoptotic (green)/non-apoptotic (blue) nuclei was performed using Image-based Tool for Counting Nuclei (ITNC) from Image J software (Rasband, 1997–2009) to determine the percentage of apoptotic neurons.
Neuronal apoptosis was assessed using SK-N-SH cell line obtained from human metastatic neuroblastoma tissue (Biedler et al. 1973; Biedler et al. 1978; Nishida et al. 2008; Baratchi et al. 2011). The SK-N-SH cells were cultured until ~85 % confluence and incubated with conditioned media from uninfected and HIV-infected microglia at 1:4 dilution in serum-free DMEM. The CHME-5-conditioned medium with or without cathepsin B inhibitor (50 μM, Sigma) or cathepsin B monoclonal antibody (50 ng/ml, Sigma) was added to some wells. Cathepsin B inhibitor and antibody were titrated previous to selection of final concentration for the assay. After incubation, conditioned media was removed, and neuronal apoptosis was measured by TUNEL as described previously (Rodriguez-Franco et al. 2012).
Determination of cathepsin B, cystatin B, and cystatin C from microglia in post-mortem brain tissue with HIV encephalitis
Human brain tissue was obtained from the National NeuroAIDS Tissue Consortium (NNTC) and the California NeuroAIDS Tissue Consortium (CNTN) (San Diego, CA), in accordance with Human Subjects Protections and the Institutional Review Board (Temple and UPR). Serial sections of formalin-fixed, paraffin-embedded frontal cortex tissues from HIVE patients or normal controls were placed on electromagnetically charged glass slides. Sections were deparaffinized at 60 °C for 40 min, placed in xylene, and rehydrated through descending grades of ethanol up to water. After non-enzymatic antigen retrieval in 0.01 M sodium citrate buffer (pH 6.0) for 30 min at 97 °C in a vacuum oven, slides were washed with 1× PBS and placed in a blocking solution (5 % normal goat or horse serum; Vector Laboratories) for 2 h. Primary antibodies utilized included IBA (1:100; Wako, Richmond, VA), cathepsin-B (1:100; Sigma-Aldrich), cystatin C (1:100; R&D Systems), cystatin B (1:500, Sigma-Aldrich), and caspase-3 (1:15, Millipore, Danvers, MA). Sections were incubated with primary antibody overnight in a dark, humidified chamber at room temperature, rinsed 3× with PBS, and incubated with fluorescein isothiocyanate (FITC) (1:200; Vector Laboratories) or Rhodamine Red (1:200; Vector Laboratories)-conjugated secondary antibodies for 1 h at room temperature in the dark. Sections were again washed 3× with PBS, cover-slipped with an aqueous-based mounting medium containing DAPI for nuclear labeling (Vectashield; Vector Laboratories), visualized with a Nikon ultraviolet inverted microscope, and processed with deconvolution software (Slidebook 4.0; Intelligent Imaging, Denver, CO). Deconvolution was performed using SlideBook4 software, allowing acquisition of multiple 0.25-μm thick digital sections and 3-D reconstruction of the image. For cystatin B and IBA immunofluorescence, sections were labeled with mouse anti-human cystatin B (1:100, Sigma-Aldrich), IBA and DAPI. Conjugated goat anti-mouse Alexa-488 and goat anti-rabbit Alexa-546 secondary antibodies (Life Technologies) were used at 1:200 dilution. Images were acquired using a Nikon Eclipse E400 fluorescence microscope equipped with a SPOT Insight QE camera and analyzed using SPOT 5.1 software. Mean flluoresence intensity for each field (3 fields per case) was measured using Imaris x64 6.2 software and analyzed with Kruskal Wallis with Dunns post-test in GraphPad®.
Statistical analysis
Data from each experiment were tested for normal distribution using the Shapiro-Wilk test of normality. For those data that were normally distributed, descriptive statistics included mean and standard deviation. Differences between HIV-positive and HIV-negative samples over days of infection were assessed using analysis of variance (ANOVA). For those data that were not normally distributed, descriptive statistics included median and interquartile range (25th and 75th percentile of the distribution). Differences between groups and over days of infection were assessed using median one-way or two-way analysis of variance one-way ANOVA, as needed. Statistical significance was considered at p < 0.05 for all comparisons. All statistics were performed using SAS, version 9.2 (The SAS Institute, Cary, NC) or Prism GraphPad 5.0 software (GraphPad software, Inc., CA).
Results
HIVADA replication is productive in human primary microglia and CHME-5 cells
Human primary microglia were inoculated with HIVADA and productivity of infection determined from cell supernatants by ELISA. Levels of HIV-1 infection were indicated by increased HIV-1 p24 antigen over the time of infection with a mean significant increase of ~5000 pg/ml at 12 days post-infection (dpi) (Fig. 1).

HIV-1ADA replicates productively in primary microglia. a–c In vitro HIV-infected primary microglia from three different donors (triangles) shows productive infection at 12 dpi. HIV replication was measured in cell supernatants by p24 antigen ELISA. d Mean of three experiments
To determine the susceptibility of CHME-5 microglia to HIV-1 infection, we first determined the expression of the CD4 receptor and CCR5 co-receptor by flow cytometry, as both receptors are used for HIV-1 entry. Results showed that CHME-5 microglia cells expressed the CD4 and CCR5 receptors with a mean fluorescence intensity (MFI) of 20 and 60, respectively (Supplementary Figure 1A-C). These results are similar to human primary fetal microglia cells (Lecointe et al. 2002). After HIV-1 infection, CHME-5 microglia showed an increase in p24 antigen levels after 3 dpi (p < 0.05), reaching 500 pg/ml at 12 dpi (Supplementary Figure 1D). The levels of HIV p24 were 10× lower than human primary microglia but showed an increase in HIV-1 replication over time in culture. These data demonstrate that both human primary microglia and CHME-5 cells were productively infected by HIV-1 albeit at lower levels than MDM (Lee et al. 1993; Ioannidis et al. 1995).
Intracellular expression of cathepsin B and its inhibitors in HIV-infected microglia
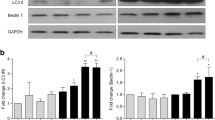
Cathepsin B and its natural inhibitors cystatin B and cystatin C expression was determined in HIV-infected human primary microglia at 3, 6, and 12 dpi by Western blot analyses. Intracellular levels of cathepsin B remained at similar levels in uninfected and HIV-infected cells (Fig. 2a, b), while cystatin B increased at 12 dpi in HIV-infected cells (Fig. 2c). Cystatin C did not show differences in expression between HIV-infected and uninfected cells (Fig. 2d). Following densitometry analysis, no significant differences were observed between uninfected and HIV-infected primary human microglia for cathepsin B or its inhibitors (Fig. 2a–d).

Intracellular expression of cathepsin B, cystatin B, and cystatin C in human primary microglia. a Western blot analyses were performed with cell lysates from uninfected or HIV-1-infected human primary microglia using mouse monoclonal anti-cystatin B, anti-cystatin C, anti-cathepsin B, and anti-β-actin antibodies. Cathepsin B (25 and 35 kDa), cystatin B (11 kDa), and cystatin C (14 kDa) are observed in all samples from 3, 6, 9, and 12 days post-infection, as well as the loading control GAPDH. Densitometry analyses were performed for all the proteins at the four time points after normalization with GAPDH, indicating there were no differences in the expression of b cystatin B, c cathepsin B, or d cystatin C. The figure is representative of three biological replicates. A two-way ANOVA was used as statistical analysis under 95 % confidence interval
Protein expression levels of cathepsin B, cystatin B, and cystatin C were monitored in CHME-5 cells following HIV-1 infection. Cathepsin B levels increased in HIV-1 infected CHME-5 cells at 3 and 6 dpi; however, these levels decreased at 12 dpi (Supplementary Figure 2A-C). Cystatin B did not show significant differences between uninfected and HIV-infected cells (Supplementary Figure 2B and D). However, intracellular levels of cystatin C were increased in HIV-infected over uninfected CHME-5 microglia at 12 dpi (Supplementary Figure 2E).
Since Western blot analyses revealed little or no expression of cystatin B in uninfected and HIV-1-infected CHME-5 cells (Supplementary Figure 1A, B, and D), we conducted flow cytometry analyses to confirm intracellular expression of this protein. Analysis confirmed the presence of very low intracellular levels of cystatin B in uninfected (Supplementary Figure 3A) and HIV-infected CHME-5 cells (Supplementary Figure 3B). No differences were observed in mean fluorescence intensities for cystatin B between uninfected and HIV-infected CHME-5 cells (Supplementary Figure 3C).
Increased levels of cathepsin B secretion in HIV-infected primary microglia and CHME-5 cells
The concentration of secreted cathepsin B from human primary microglia of three different donors was measured. Data shows that at 3 dpi the levels of cathepsin B in uninfected controls were 8 ng/ml. However, at 6 and 12 dpi the levels of secreted cathepsin B in HIV-infected microglia supernatants increased to 25 and 26 ng/ml, respectively. These data suggest that HIV-1 replication significantly increased the levels of cathepsin B secretion in primary microglia at 6 and 12 dpi (Fig. 3a). Cathepsin B inhibitors, cystatins B and C were measured from HIV-1-infected and uninfected primary microglia supernatants collected at 3,6, and 12 dpi. Results showed a similar levels of cystatin B over time in culture (Fig. 3b). Cystatin B levels reached 8 ng/ml in uninfected and HIV-infected cells (Fig. 3b). Cystatin C increased at 3 dpi in HIV infected cells from 20 to 60 ng/ml but remained at similar levels at and 12 dpi (Fig. 3c). No differences were observed in the ratio of cathepsin B to cystatins B or C, respectively (Fig. 3d, e).

Secretion and activity of cathepsin B and its inhibitors in primary microglia. a–f HIV-infected or uninfected supernatants from 3 different donors were collected at 3, 6, and 12 dpi, and a levels of cathepsin B secretion was measured by ELISA. A significant increase in cathepsin B secretion was observed in HIV-infected microglial supernatants at 6 and 12 dpi compared with uninfected controls (*p < 0.05). b No differences were observed in the levels of cystatin B. c Significantly increased levels of cystatin C (**p < 0.01) were observed at 3 dpi in HIV-infected microglia compared with uninfected controls. d, e No differences were observed in the cathepsin B/cystatin B or the cathepsin B/cystatin C ratios. f Supernatants of 12 dpi show no significant differences in cathepsin B activity between HIV-infected and uninfected controls
Extracellular secretion of cathepsin B and its inhibitors were also measured in CHME-5 cells. Cathepsin B increased in HIV-infected cells at 6 dpi, and although it decreased over time in culture, it remained significantly increased over uninfected controls (Supplementary Figure 4). However, the levels of cathepsin B in CHME-5 cells were 40-fold lower (reaching 0.5 ng/ml) than the levels in primary microglia that reached 25 ng/ml (Fig. 3a). Cystatin C levels were similar in uninfected and HIV-infected CHME-5 cells (Supplementary Figure 4). The levels of cystatin C in HIV-infected and uninfected CHME-5 were 0.25 ng/ml, which is approximately 200-fold lower than in primary microglia (Fig. 3c). The levels of extracellular cystatin B in CHME-5 were undetectable in both uninfected and HIV-infected cultures (data not shown). However, the levels of cystatin B secretion between HIV-infected and uninfected primary microglia although not significant reached 8 ng/ml (Fig. 3b) which is 8-fold higher than CHME-5 cells.
Taken together, these results indicate that both primary microglia and CHME-5 cells showed increased cathepsin B in response to HIV infection but the levels of its secretion and of the inhibitors were different.
Bioactive cathepsin B is secreted from human primary microglia and CHME-5 cells upon HIV-1 infection
The activity of secreted cathepsin B was measured in culture supernatants from HIV-infected and uninfected primary microglia. No differences were detected between HIV-infected and uninfected cells at all days post-infection (Fig. 3f), but cathepsin B activity is significantly higher at 12 dpi for both uninfected and HIV-positive microglia compared to 3 and 6 dpi. Activity of cathepsin B in CHME-5 cells was similar in uninfected and HIV-infected CHME-5 cells (Supplementary Figure 4C). When compared to primary microglia, the levels of cathepsin B activity as 25-fold higher in CHME-5 than human primary microglia.
Taken together, the results of cathepsin B in HIV-infected and uninfected primary microglia and CHME-5 cells indicate that HIV-1 infection induces increased levels of cathepsin B in both types of cells during HIV infection. While the levels of cathepsin B in primary cells are much higher than CHME-5, the level of activity in CHME-5 was higher suggesting that the combination of concentration and activity in both types of cells is elevated during HIV infection.
Human primary microglia and CHME-5-conditioned media induce neuronal apoptosis
To further determine the potential contribution of human primary microglia-derived cathepsin B to neuronal apoptosis, primary neurons were treated with supernatant from primary microglia infected with HIV-1ADA 6 and 12 dpi. Neuronal apoptosis was assessed using the TUNEL assay, and images were analyzed by fluorescence microscopy. The results show a significant increase of neuronal apoptosis at 12 dpi (Fig. 4). However, the apoptosis can be ameliorated with primary microglia-conditioned medium treated with cathepsin B inhibitor (CA-074) or pretreated medium with anti-cathepsin B antibody. The percentage of cathepsin B-mediated neuronal apoptosis induced by HIV-infected microglial supernatant was determined by quantitative analysis of apoptotic (green)/non-apoptotic (blue) nuclei using Image-based Tool for Counting Nuclei (ITNC) from Image J software (NIH). A significant increase in the percentage of apoptotic neurons was observed at 12 dpi (p < 0.001) compared to neurons treated with uninfected primary microglia-conditioned media. However, inhibition of cathepsin B by CA-074 or anti-cathepsin B significantly decreased the percentage of apoptotic neurons at 12 dpi (p < 0.001) (Fig. 4).

HIV-infected primary microglia secrete cathepsin B that induces neurotoxicity. Human primary neurons were incubated with conditioned media from HIV-infected primary microglia until 12 dpi with or without cathepsin B inhibitor (50 μM) or with cathepsin B monoclonal antibody (50 ng/ml). Apoptosis was measured using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay at 12 dpi. Apoptotic cells are shown in green. Primary neurons exposed to conditioned media from a uninfected microglia or from b HIV-infected microglia HIV+ supernatant show significantly more apoptosis+neurons (green) compared to uninfected supernatant. c Inhibition of cathepsin B with the specific inhibitor, CA-074, decreased the neuronal apoptosis. d Little or no apoptosis was observed in neurons treated with microglia-conditioned medium pretreated with cathepsin B antibody. e Positive control shows neuronal apoptosis as compared with f negative control of culture media. Confocal images were obtained on a Zeiss confocal microscope Axiovert 200M with a LSM 510 under an excitation wavelength of 488 nm, 20× magnification. g Quantitative analysis of staining ratio of apoptotic (green)/non-apoptotic (blue) nuclei was performed using Image-based Tool for Counting Nuclei (ITNC) from Image J software (NIH) to determine the percentage of apoptotic neurons. A significant increment of apoptotic neurons is present at 12 dpi (*p < 0.05). A significant decrease of apoptosis is observed in neurons that received the microglial medium pretreated with the specific cathepsin B inhibitor (CA-074). Specificity of cathepsin B effects to significantly decrease apoptosis was further demonstrated in neurons that received microglia HIV-infected supernatants pretreated with cathepsin B antibody at 1:500 (*p < 0.05). A two-way ANOVA was used for statistical analysis under 95 % confidence interval. This is a representative experiment of HIV-infected microglia from three different donors
The role of CHME-5 microglia-derived cathepsin B in neuronal apoptosis was tested using confluent SK-N-SH neuronal cells incubated in serum-free CHME-5-conditioned medium from uninfected and HIV-infected cultures and collected at 3, 6, and 12 dpi. Neuronal apoptosis was measured using TUNEL, and images analyzed by confocal microscopy. The results show an increase of neuronal apoptosis at 6 and 12 dpi (Supplementary Figure 5A). As observed with primary neurons, apoptosis can be blocked with CHME-5 microglial conditioned medium containing the cathepsin B inhibitor (CA-074) or pretreated medium with anti-cathepsin B antibody. A significant increase in the percentage of apoptotic neurons was observed at 6 dpi (p < 0.01) and 12 dpi (p < 0.001) compared to neurons treated with uninfected CHME-5 microglia condition media. However, inhibition of cathepsin B by CA-074 (Supplementary Figure 5B) decreased significantly the percentage of apoptotic neurons at 6 dpi (p < 0.05) and 12 dpi (p < 0.001) compared to neurons treated with HIV-infected CHME-5 condition media. Likewise, CHME-5 microglia supernatant pretreated with cathepsin B antibody reverted significantly the percentage of apoptotic neurons at 6 dpi (p < 0.01) and 12 dpi (p < 0.001) (Supplementary Figure 5C).
Cathepsin B, cystatin B, and cystatin C are increased in microglia and perivascular macrophages from patients with HIVE
To assess microglial and macrophage expression of cathepsin B and cystatin C in HIV infection, frontal cortex tissues from 5 HIVE cases and 3 control cases (no HIV no CNS alterations) were co-immunolabeled with the microglial/macrophage-specific marker IBA-1 and either cathepsin B or cystatin C. In control cases, cells labeled with IBA-1 (green) typical ramified microglia morphology were observed (Fig. 5a, b) with very low levels of cathepsin B detected (red). In contrast, HIVE cases showed increased cathepsin B (red) in IBA-1 (green) immunoreactive cells with morphologies characteristic of activated microglia (Fig. 5c, d) and macrophages (Fig. 5e, f) associated with small blood vessels (Fig. 5e, f). Moreover, in HIVE, co-labeling of IBA-1 and cathepsin B in the same cell or closely adjacent cells was detected in microglia or macrophages. Some cathepsin B immunoreactive cells were also positive for caspase-3, a marker for apoptosis, indicating that these cells were undergoing programmed cell death (Fig. 5g, h). Of note, some cathepsin B-positive cells were not co-labeled with caspase-3 and vice-versa (Fig. 5g, h), indicating that some bystander cells not expressing detectable cathepsin B were undergoing apoptosis. These results suggest that in HIVE, expression of cathepsin B in IBA-1-immunoreactive cells is increased, compared to uninfected control and that its expression is associated with microglia and macrophages. To assess expression of cystatin C, a cathepsin B inhibitor in IBA immunoreactive cells, 3 control and 5 HIVE cases were assessed. As expected, IBA-1 was detected in macrophages/microglia in all control and HIVE cases (Fig. 6a–d, green). Cystatin C expression was observed in IBA-1-positive cells with typical microglial morphology (Fig. 6a, d, c, arrowheads) and in IBA-1-positive cells resembling macrophages and associated with small vessels (Fig. 6e, arrow). Cystatin B immunoreactivity was also analyzed in human post-mortem tissues from control or HIV-positive cases, with increased expression levels in microglia from HIV positives (***p<0.001) and subsyndromics over HIV+ with normal cognition (*p<0.05) (Fig. 7). Taken together, these results show that in HIVE, cathepsin B, cystatin C, and cystatin B levels are increased compared to uninfected control cases. Cathepsin B, cystatin C, and cystatin B are expressed in microglial cells and perivascular macrophages. Evidence of apoptosis is detected not only in cathepsin B expressing IBA-positive cells but in bystander cells as well.

a–i Cells with typical microglial morphology are shown in red. d Cells associated with small vessels with typical macrophage morphology are shown in red. g, h Tissues are immunolabeled with caspase-3 (green) and cathepsin B (red). Cells were immunoreactive for cathepsin B or caspase but not for both. Colocalization of cathepsin B and caspase-3 is shown in yellow. i Lumens of vessels are shown

Double immunofluorescence labeling of the frontal cortex tissue from representative control and HIVE patients with cystatin C. a, b control. c, d HIVE. Tissues are immunolabeled with IBA (green) and cystatin C (red). Arrowheads indicate cells with typical microglial morphology and arrows indicatecells associated with small vessels with typical macrophage morphology. d, e Co-localization is indicated in yellow. Asterisks indicate the lumens of vessels. Nuclei are stained in blue with DAPI

Double immunofluorescence labeling of the frontal cortex tissue from representative control and HIV-positive patients with cystatin B. a HIV-negative, b HIV+ normal cognition, c HIV sub-syndromic, d HIVE and HAD. Tissues are immunolabeled with IBA (red) for microglia and macrophages, and cystatin B (green). Nuclei are stained in blue with DAPI. Co-localization is indicated in yellow. e Quantification of the expression of cystatin B from each condition was determined using IMARIS program. A significant increase in cystatin B expression was observed in HIV+ brain tissue compared to HIV− (***p < 0.001). A significant increase in cystatin B expression was observed in HIV+ subsyndromic over HIV+ with normal cognition (*p < 0.05)
Discussion
Resident microglia and infiltrating macrophages are the principal producers of HIV-1 in the brain and key players in induction of neuronal damage during HAND (Glass et al. 1995). Proteomic analysis of HIV-infected macrophages has revealed that HIV infection can induce profound alterations in its normal cell physiology, potentially contributing to neuronal dysfunction [as reviewed by Melendez and collaborators, 2011; (Melendez et al. 2011). These alterations include not only the production of neurotoxins but also the modulation of normal cellular processes. However, the difficulty and complexity of studying the human brain ex vivo is an obstacle to the understanding of HAND (Jayadev and Garden 2009). With that in mind, we compared the role of microglia cathepsin B and its inhibitors in HIV-induced neuronal injury. Our study demonstrated that HIV-infected microglia secreted neurotoxic levels of cathepsin B.
Although microglia are the sentinels of the brain, they are susceptible to HIV infection and consequently play a pivotal role in HAND. Investigation of HIV infection of microglia has continued since the discovery of HIV in the early 1980s (Barré-Sinoussi et al. 1983). Microglia are commonly believed to originate from myeloid-monocytic cells precursors and express CD4, CCR5, and other essential co-receptors for HIV infection (He et al. 1997; Shieh et al. 1998). It is also well known that these cells are productively infected by HIV (González-Scarano and Baltuch 1999) and thus act as reservoirs of low but persistent virus in the brain (Alexaki et al. 2008).
In vitro infection of both fetal and adult human microglia has been demonstrated by several groups (Lee et al. 1993; Ioannidis et al. 1995); however, research with human primary microglia is laborious and a subject of controversy as fetal cells may not behave as adult microglia cells. We compared the expression of cathepsin B and its inhibitors in human primary microglia obtained from our Temple University CNAC to a human microglia cell line (CHME-5), obtained through transfection with SV-40 T-antigen (Janabi et al. 1995), and explored the effect of HIV-1 infection. The initial characterization of CHME-5 by Janabi and collaborators (1995) reported a lack of expression of the monocytic marker, CD4. In our study, however, we found that CHME-5 cells expressed low levels of the CD4 receptor. It has been demonstrated that CHME-5 microglia expresses the CCR5 chemokine receptor, which is known to be one of the principal co-receptors of HIV-1 in the brain (He et al. 1997; Shieh et al. 1998). CCR5 is also the predominant co-receptor used by several neurotropic HIV-1 isolates, such as HIV-1DS-br, HIV-1RC-br, and HIV-1YU-2 (Albright et al. 1999). We found that CHME-5 expressed both receptors and were productively infected by HIVADA, a macrophage-tropic isolate that uses CCR5 as a co-receptor. This viral isolate was originally isolated from peripheral blood mononuclear cells of an AIDS patient with Kaposi’s sarcoma (Gendelman et al. 1988) and is known to infect primary microglia (Ghorpade et al. 1998b). Several groups have previously used CHME-5 microglia as a model for CNS inflammation and infectious diseases including HIV (Atanassov et al. 1995; Lachance et al. 1998; Arbour et al. 1999; Chugh et al. 2008; Lucas et al. 2010). These groups have transfected CHME-5 with HIV proteins, in contrast to direct infection of CHME-5 microglia cells (Chugh et al. 2008; Lucas et al. 2010).
The main goal of this study was to determine if human primary microglia-derived cathepsin B could play a role in HAND by comparing its intracellular and extracellular expression and to determine if CHME-5 microglia cell line can model for primary human microglia in these effects. The expression and activity of cathepsin B and its inhibitors were also measured in post-mortem brain tissue microglia in relation to HIV-1 infection.
Cysteine proteases, such as cathepsin B, are ubiquitous host proteins involved mainly in nonselective intracellular protein degradation in lysosomes (Barrett and Kirschke 1981). Outside the lysosomes, cathepsins are tightly regulated by the cystatins (Kopitar-Jerala 2006). Until recently, cathepsins were thought to be completely inactive at neutral pH, but several groups have provided evidence that associates cytoplasmic and secreted cathepsin B with inflammation (Stoka et al. 2001; Schurigt et al. 2005; Nagaraj et al. 2006; Tsai et al. 2009) and apoptosis (Guicciardi et al. 2000; Kingham and Pocock 2001; Gan et al. 2004; Li and Pober 2005; Nagaraj et al. 2006; Wang et al. 2008; Tran et al. 2009). It is known that HIV infection triggers TNF-α and IL-1 inflammatory pathways (Albright 2002; O’Donnell et al. 2006; Ghafouri et al. 2006), and this action is associated with increased oxidative stress (Guicciardi et al. 2000; Li and Pober 2005) and antioxidant dysfunction (Velazquez et al. 2009) during HAND. At low concentrations, cytoplasmic cathepsin B can be modulated by cystatins. Increased TNF and IL-1 promote the release of cathepsin B from the lysosomes (Guicciardi et al. 2000). When cathepsin B released from lysosomes exceeds the amount of available cystatins, the protease inhibitor system is disrupted and free active cathepsin B can indiscriminately degrade essential proteins and/or be secreted from the cell (Li and Pober 2005).
As both perivascular macrophages and microglia express CD4 and chemokine receptors and are susceptible to HIV-1, we asked if microglia cells would modulate cathepsin B and its inhibitors as we have observed for MDM (Rodriguez-Franco et al. 2012). In the present study, we found that HIV-infected primary microglia and CHME-5 expressed significantly higher levels of intracellular cathepsin B compared to their uninfected controls. Results indicated that HIV-1ADA replicated productively in primary microglia cells. Primary microglia expressed an increased extracellular cathepsin B at 6 and 12 dpi compared with uninfected controls suggesting that HIV-1 infection promotes the secretion of cathepsin B. We also showed that cystatins B and C had very different patterns of expression. Cystatin B was found in primary microglia that remained at similar levels in HIV-infected and uninfected cells. In contrast, very low but also similar levels of cystatin B were found in both HIV-infected and uninfected CHME-5 cells. In primary microglia, cystatin C expression is higher in than CHME-5 and increased significantly at 3 dpi in HIV-1 infection compared with uninfected controls. We did not found any difference of intracellular expression of cathepsin B and its inhibitors in primary microglia. These results point out similarities in the intracellular expression of cystatin B in primary microglia and CHME-5 cells. We found that CHME-5 produce low levels of HIV p24 and undetectable cystatin B in supernatants which correlates with the low level of HIV-1 replication found in these cells as well as other tissue macrophages including placental macrophages (Luciano-Montalvo et al. 2008; Luciano-Montalvo and Melendez 2009). In contrast high levels of secreted cystatin B at 12 dpi correlates with HIV-1 infection in MDM as reported in previous studies (Meléndez et al. 2011; Rivera-Rivera et al. 2012).
Regarding the secretion of cathepsin B and its inhibitors, we found that cathepsin B secretion was higher primary microglia (25 ng/ml) compared to CHME-5 (0.5 ng/ml), but its secretion was significantly increased in HIV-infected primary microglia and CHME-5 cells as compared with uninfected controls. These results indicate that cathepsin B expression is increased in response to HIV infection in both types of microglia albeit at different levels.
Interestingly, the levels of secreted-bioactive cathepsin B were much higher in HIV-infected and control CHME-5 cells compared to primary microglia. Following TUNEL assay experiments, we found that HIV-infected primary microglia and CHME-5 conditioned media both induce neuronal apoptosis and that can be prevented with the addition of cathepsin B inhibitor CA-074. These findings indicate that cathepsin B is also an important neurotoxic factor in microglia as reported for MDM (Rodriguez-Franco et al. 2012), and this detrimental effect can be prevented using the cathepsin B inhibitor, CA-074, or anti-cathepsin B antibody.
Higher levels of cystatins B and C were produced by primary microglia that diminished cathepsin B activity significantly but did not prevent its secretion and neurotoxicity. In CHME-5 cells, cystatin B appears not to have a crucial role in inhibiting cathepsin B due to its low expression; instead, our results suggest that cystatin C is the main enzyme regulating intracellular cathepsin B. However, high levels of cystatin C prevented the secretion of cathepsin B but did not diminish its activity. These findings are confirmed by studies that reported the sequestration of cystatin C in the cytoplasm, increased levels of intracellular enzyme, and a concomitant decrease in extracellular levels during late HIV infection (Yoshii et al. 2011). This could be a mechanism of cellular protection against increments in cytoplasmic cathepsin B released from lysosomes after infection. Cystatin C has been associated with neuroprotection against increased oxidative stress (Pennacchio et al. 1996; Nagai et al. 2000; Wakasugi et al. 2004; Laspiur et al. 2007).
Studies of post-mortem brain tissue confirmed the identification of microglia and perivascular macrophages as cellular sources for increased cathepsin B, cystatin C, and cystatin B in five HIVE cases. The secretion of cathepsin B was associated with increased caspase-3 in microglia, macrophages, and surrounding cells. These results support the role of cathepsin B in apoptosis during HIVE and suggest the development of strategies to control the secretion of this enzyme.
In conclusion, our study shows that HIV infection of primary microglia and CHME-5 cells induce secretion of bioactive intracellular cathepsin B and neurotoxicity. This study also demonstrated that human primary cells and CHME-5 differ in basal levels of cathepsin B and its activity. The opposite occurs in primary microglia compared to CHME-5 cells, increased secretion and low activity. However, both CHME-5 and primary microglia mediate neuronal apoptosis in response to HIV infection (Huang et al. 2011). In addition, our data indicates that microglia-derived cathepsin B is an important mediator of neuronal apoptosis and suggest that cystatin C limits cathepsin B activity but is not sufficient to prevent neuronal apoptosis. Our data indicate that the regulation of cathepsin B and its inhibitors is unique for each cell type but together with studies of post-mortem brain tissue supports the role of microglia and perivascular macrophages derived cathepsin B in neuronal injury, as demonstrated recently in MDM (Rodriguez-Franco et al. 2012). The regulation of these enzymes in response to drugs of abuse has recently been studied and results demonstrate that cocaine increases further cathepsin B secretion and activity from HIV-infected MDM increasing the levels of neuronal apoptosis (Zenón et al. 2014). Since both primary microglia and CHME-5 increase cathepsin B-induced neuronal apoptosis after HIV infection, CHME-5 cells could be used for further studies of regulation by other CNS cells in response to HIV infection and drug of abuse.
References
Albright A (2002) Winning at losing: a guide to healthy weight loss. Best and worst exercises for weight loss. What works and what does not for shedding pounds through physical activity. Diabetes Forecast 55:69–70, 72, 74.
Albright AV, Shieh JT, Itoh T et al (1999) Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol 73:205–13
Alexaki A, Liu Y, Wigdahl B (2008) Cellular reservoirs of HIV-1 and their role in viral persistence. Curr HIV Res 6:388–400
Arbour N, Ekandé S, Côté G et al (1999) Persistent infection of human oligodendrocytic and neuroglial cell lines by human coronavirus 229E. J Virol 73:3326–37
Atanassov CL, Muller CD, Dumont S et al (1995) Effect of ammonia on endocytosis and cytokine production by immortalized human microglia and astroglia cells. Neurochem Int 27:417–24
Baratchi S, Kanwar RK, Kanwar JR (2011) Survivin mutant protects differentiated dopaminergic SK-N-SH cells against oxidative stress. PLoS One 6:e15865. doi:10.1371/journal.pone.0015865
Barré-Sinoussi F, Chermann JC, Rey F et al (1983) Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220:868–71
Barrett AJ, Kirschke H (1981) Cathepsin B, cathepsin H, and cathepsin L. Methods Enzymol 80 Pt C:535–61
Biedler JL, Helson L, Spengler BA (1973) Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture1. 2643–2652.
Biedler JL, Roffler-tarlov S, Schachner M, Freedman LS (1978) Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. 3751–3757.
Cantres-Rosario Y, Plaud-Valentín M, Gerena Y et al (2013) Cathepsin B and cystatin B in HIV-seropositive women are associated with infection and HIV-1-associated neurocognitive disorders. AIDS 27:347–56. doi:10.1097/QAD.0b013e32835b3e47
Chugh P, Bradel-Tretheway B, Monteiro-Filho CMR et al (2008) Akt inhibitors as an HIV-1 infected macrophage-specific anti-viral therapy. Retrovirology 5:11. doi:10.1186/1742-4690-5-11
Gan L, Ye S, Chu A et al (2004) Identification of cathepsin B as a mediator of neuronal death induced by Abeta-activated microglial cells using a functional genomics approach. J Biol Chem 279:5565–72. doi:10.1074/jbc.M306183200
Gendelman HE, Baca LM, Husayni H et al (1990) Macrophage-HIV interaction: viral isolation and target cell tropism. AIDS 4:221–8
Gendelman HE, Orenstein JM, Martin MA et al (1988) Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med 167:1428–41
Ghafouri M, Amini S, Khalili K, Sawaya BE (2006) HIV-1 associated dementia: symptoms and causes. Retrovirology 3:28. doi:10.1186/1742-4690-3-28
Ghorpade A, Xia MQ, Hyman BT et al (1998a) Role of the beta-chemokine receptors CCR3 and CCR5 in human immunodeficiency virus type 1 infection of monocytes and microglia. J Virol 72:3351–61
Ghorpade A, Nukuna A, Che M et al (1998b) Human immunodeficiency virus neurotropism: an analysis of viral replication and cytopathicity for divergent strains in monocytes and microglia. J Virol 72:3340–50
Glass JD, Fedor H, Wesselingh SL, McArthur JC (1995) Immunocytochemical quantitation of human immunodeficiency virus in the brain: correlations with dementia. Ann Neurol 38:755–62. doi:10.1002/ana.410380510
González-Scarano F, Baltuch G (1999) Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci 22:219–40. doi:10.1146/annurev.neuro.22.1.219
Guicciardi ME, Deussing J, Miyoshi H et al (2000) Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 106:1127–37. doi:10.1172/JCI9914
He J, Chen Y, Farzan M et al (1997) CCR3 and CCR5 are co-receptors for HIV-1 infection of microglia. Nature 385:645–9. doi:10.1038/385645a0
Honey K, Rudensky AY (2003) Lysosomal cysteine proteases regulate antigen presentation. Nat Rev Immunol 3:472–82. doi:10.1038/nri1110
Huang Y, Zhao L, Jia B et al (2011) Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1-associated neurocognitive disorders. J Neurosci 31:15195–204. doi:10.1523/JNEUROSCI.2051-11.2011
Ioannidis JP, Reichlin S, Skolnik PR (1995) Long-term productive human immunodeficiency virus-1 infection in human infant microglia. Am J Pathol 147:1200–6
Janabi N, Peudenier S, Héron B et al (1995) Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci Lett 195:105–8
Jayadev S, Garden GA (2009) Host and viral factors influencing the pathogenesis of HIV-associated neurocognitive disorders. J Neuroimmune Pharmacol 4:175–89. doi:10.1007/s11481-009-9154-6
Kingham PJ, Pocock JM (2001) Microglial secreted cathepsin B induces neuronal apoptosis. J Neurochem 76:1475–84
Kopitar-Jerala N (2006) The role of cystatins in cells of the immune system. FEBS Lett 580:6295–301. doi:10.1016/j.febslet.2006.10.055
Lachance C, Arbour N, Cashman NR, Talbot PJ (1998) Involvement of aminopeptidase N (CD13) in infection of human neural cells by human coronavirus 229E. J Virol 72:6511–9
Laspiur JP, Anderson ER, Ciborowski P et al (2007) CSF proteomic fingerprints for HIV-associated cognitive impairment. J Neuroimmunol 192:157–170. doi:10.1016/j.jneuroim.2007.08.004
Lecointe D, Dugas N, Leclerc P et al (2002) Human cytomegalovirus infection reduces surface CCR5 expression in human microglial cells, astrocytes and monocyte-derived macrophages. Microbes Infect 4:1401–8
Lee SC, Hatch WC, Liu W et al (1993) Productive infection of human fetal microglia in vitro by HIV-1. Ann N Y Acad Sci 693:314–6
Li JH, Pober JS (2005) The cathepsin B death pathway contributes to TNF plus IFN-gamma-mediated human endothelial injury. J Immunol 175:1858–1866
Lucas A, Kim Y, Rivera-Pabon O et al (2010) Targeting the PI3K/Akt cell survival pathway to induce cell death of HIV-1 infected macrophages with alkylphospholipid compounds. PLoS One. doi:10.1371/journal.pone.0013121
Luciano-Montalvo C, Ciborowski P, Duan F et al (2008) Proteomic analyses associate cystatin B with restricted HIV-1 replication in placental macrophages. Placenta 29:1016–1023. doi:10.1016/j.placenta.2008.09.005
Luciano-Montalvo C, Melendez LM (2009) Cystatin B associates with signal transducer and activator of transcription 1 in monocyte-derived and placental macrophages. Placenta 30:464–467. doi:10.1016/j.placenta.2009.03.003
Melendez LM, Colon K, Rivera L et al (2011) Proteomic analysis of HIV-infected macrophages. J Neuroimmune Pharmacol 6:89–106. doi:10.1007/s11481-010-9253-4
Nagai A, Murakawa Y, Terashima M et al (2000) Cystatin C and cathepsin B in CSF from patients with inflammatory neurologic diseases. Neurology 55:1828–32
Nagaraj NS, Vigneswaran N, Zacharias W (2006) Cathepsin B mediates TRAIL-induced apoptosis in oral cancer cells. J Cancer Res Clin Oncol 132:171–83. doi:10.1007/s00432-005-0053-9
Nguyen MD, Julien J-P, Rivest S (2002) Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat Rev Neurosci 3:216–27. doi:10.1038/nrn752
Nishida Y, Adati N, Ozawa R et al (2008) Identification and classification of genes regulated by phosphatidylinositol 3-kinase- and TRKB-mediated signalling pathways during neuronal differentiation in two subtypes of the human neuroblastoma cell line SH-SY5Y. BMC Res Notes 1:95. doi:10.1186/1756-0500-1-95
Nixon RA, Cataldo AM, Mathews PM (2000) The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: a review. Neurochem Res 25:1161–72
O’Donnell LA, Agrawal A, Jordan-Sciutto KL et al (2006) Human immunodeficiency virus (HIV)-induced neurotoxicity: roles for the NMDA receptor subtypes. J Neurosci 26:981–990. doi:10.1523/JNEUROSCI.4617-05.2006
Pennacchio LA, Lehesjoki AE, Stone NE et al (1996) Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science 271:1731–4
Peudenier S, Hery C, Montagnier L, Tardieu M (1991) Human microglial cells: characterization in cerebral tissue and in primary culture, and study of their susceptibility to HIV-1 infection. Ann Neurol 29:152–61. doi:10.1002/ana.410290207
Rivera-Rivera L, Perez-Laspiur J, Colón K, Meléndez LMM (2012) Inhibition of interferon response by cystatin B: implication in HIV replication of macrophage reservoirs. J Neurovirol 18:20–9. doi:10.1007/s13365-011-0061-2
Rock RB, Gekker G, Hu S et al (2004) Role of microglia in central nervous system infections. Clin Microbiol Rev 17:942–64. doi:10.1128/CMR.17.4.942-964.2004, table of contents
Rodriguez-Franco EJ, Cantres-Rosario YM, Plaud-Valentin M et al (2012) Dysregulation of macrophage-secreted cathepsin B contributes to HIV-1-linked neuronal apoptosis. PLoS One 7:e36571. doi:10.1371/journal.pone.0036571
Schurigt U, Stopfel N, Hückel M et al (2005) Local expression of matrix metalloproteinases, cathepsins, and their inhibitors during the development of murine antigen-induced arthritis. Arthritis Res Ther 7:R174–88. doi:10.1186/ar1466
Shieh JT, Albright AV, Sharron M et al (1998) Chemokine receptor utilization by human immunodeficiency virus type 1 isolates that replicate in microglia. J Virol 72:4243–9
Stoka V, Turk B, Schendel SL et al (2001) Lysosomal protease pathways to apoptosis. Cleavage of bid, not pro-caspases, is the most likely route. J Biol Chem 276:3149–57. doi:10.1074/jbc.M008944200
Tambuyzer BR, Ponsaerts P, Nouwen EJ (2009) Microglia: gatekeepers of central nervous system immunology. J Leukoc Biol 85:352–70. doi:10.1189/jlb.0608385
Tran TM, Temkin V, Shi B et al (2009) TNFalpha-induced macrophage death via caspase-dependent and independent pathways. Apoptosis 14:320–32. doi:10.1007/s10495-009-0311-4
Tsai H-T, Wang P-H, Tee Y-T et al (2009) Imbalanced serum concentration between cathepsin B and cystatin C in patients with pelvic inflammatory disease. Fertil Steril 91:549–55. doi:10.1016/j.fertnstert.2007.12.076
Velazquez I, Plaud M, Wojna V et al (2009) Antioxidant enzyme dysfunction in monocytes and CSF of Hispanic women with HIV-associated cognitive impairment. J Neuroimmunol 206:106–111. doi:10.1016/j.jneuroim.2008.10.013
Voyvodic JT (1996) Cell death in cortical development: how much? Why? So what? Neuron 16:693–6
Wakasugi K, Nakano T, Morishima I (2004) Association of human neuroglobin with cystatin C, a cysteine proteinase inhibitor. Biochemistry 43:5119–25. doi:10.1021/bi0495782
Wang T, Gong N, Liu J et al (2008) Proteomic modeling for HIV-1 infected microglia-astrocyte crosstalk. PLoS One 3:e2507. doi:10.1371/journal.pone.0002507
Yoshii H, Kamiyama H, Goto K et al (2011) CD4-independent human immunodeficiency virus infection involves participation of endocytosis and cathepsin B. PLoS One 6:e19352. doi:10.1371/journal.pone.0019352
Zenón F, Segarra AC, Gonzalez M, Meléndez LM (2014) Cocaine potentiates cathepsin B secretion and neuronal apoptosis from HIV-infected macrophages. J Neuroimmune Pharmacol 9:703–15. doi:10.1007/s11481-014-9563-z
Acknowledgments
We thank Dr. Randall L. Davis from the Oklahoma State University Center for Health Sciences for kindly providing the CHME-5 microglia cell line. We thank Dr. Howard Gendelman from the University of Nebraska Medical Center for providing HIV-ADA isolates. We would like to thank Rafael Romeu for his help with cell cultures and Dr. Yamil Gerena for the advice in flow cytometry. We would like to acknowledge Eduard Guerrero, Carol Torres from the RCMI Emergent Disease Core Laboratories, and Dr. Juliana Perez Laspiur and Yolanda Rodriguez from the Proteomics Core Laboratory for all their excellent technical support. Part of this work was previously presented as a poster at the ISNV. May 28-June 2, 2012, NY. Part of this work was also presented at the Annual Biomedical Research Conference for Minority Students (ABRCMS) meeting, November 13–16, 2013, Nashville, TN, USA
Funding
This work was supported in part by grants from the National Institutes of Health R01MH083516 (to LMM), U54NS4301 (to LMM), R25GM061838 (to FZ, ER, YC), GM08224, and NIMHHD8G12-MD007600 (Emergent Diseases Core and Translational Proteomics Center), and SC1GM11369-01. We acknowledge the facilities from grants ISI0 RR-13705-01 and DBI-0923132 to establish and upgrade the Confocal Microscopy Facility at the University of Puerto Rico (CIF-UPR). UPR Vice President (M.P.). UPR Medical Sciences Campus Chancellor, and the Associate Deanship of Biomedical Sciences provided additional funding to complete this study. This publication was made possible from NIH funding through the NIMH and NINDS Institutes by the following grants: Manhattan HIV Brain Bank U01MH083501, R24MH59724; Texas NeuroAIDS Research Center U01MH083507, R24 NS45491; National Neurological AIDS Bank 5U01MH083500, NS 38841; California NeuroAIDS Tissue Network U01MH083506, R24MH59745; and Statistics and Data Coordinating Center U01MH083545, N01MH32002. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NNTC or NIH.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 77584 kb)
Rights and permissions
About this article

Cite this article
Zenón, F., Cantres-Rosario, Y., Adiga, R. et al. HIV-infected microglia mediate cathepsin B-induced neurotoxicity. J. Neurovirol. 21, 544–558 (2015). https://doi.org/10.1007/s13365-015-0358-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-015-0358-7