
Abstract
The ability to detect peptides and proteins in single cells is vital for understanding cell heterogeneity in the nervous system. Capillary electrophoresis (CE) nanoelectrospray ionization (nanoESI) provides high-resolution mass spectrometry (HRMS) with trace-level sensitivity, but compressed separation during CE challenges protein identification by tandem HRMS with limited MS/MS duty cycle. Here, we supplemented ultrasensitive CE-nanoESI-HRMS with reversed-phase (RP) fractionation to enhance identifications from protein digest amounts that approximate to a few mammalian neurons. An ~1 to 20 μg neuronal protein digest was fractionated on a RP column (ZipTip), and 1 ng to 500 pg of peptides were analyzed by a custom-built CE-HRMS system. Compared with the control (no fractionation), RP fractionation improved CE separation (theoretical plates ~274,000 versus 412,000 maximum, resp.), which enhanced detection sensitivity (2.5-fold higher signal-to-noise ratio), minimized co-isolation spectral interferences during MS/MS, and increased the temporal rate of peptide identification by up to ~57%. From 1 ng of protein digest (<5 neurons), CE with RP fractionation identified 737 protein groups (1,753 peptides), or ~480 protein groups (~1,650 peptides) on average per analysis. The approach was scalable to 500 pg of protein digest (~a single neuron), identifying 225 protein groups (623 peptides) in technical triplicates, or 141 protein groups on average per analysis. Among identified proteins, 101 proteins were products of genes that are known to be transcriptionally active in single neurons during early development of the brain, including those involved in synaptic transmission and plasticity and cytoskeletal organization.

ᅟ
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The ability to measure gene expression in a small population of cells is critical for understanding molecular players of neuronal differentiation in the developing brain. Empowered by liquid chromatography and late-generation high-resolution mass spectrometry (HRMS), shotgun (bottom-up) proteomics now enables differentiation of neuronal cell types by quantifying the expression of ~13,000 protein groups from primary cell cultures [1]. To detect the encoded proteome in deeper coverage, ~10–100 μg of proteins are typically harvested from few to tens of millions of cells before analyzing ~100 ng to ~5 μg of protein digest per experiment by nano-flow (ultra) high-performance liquid chromatography (nanoLC) HRMS. Extension of proteomics to small populations of neurons would complement single-cell transcriptomics data [2,3,4], thus raising a potential to better understand molecular changes as neurons acquire functional phenotypes. Without technologies capable of amplifying the whole proteome, however, significant improvements are needed in the sensitivity of HRMS to detect proteins from sub-nanograms of total protein amounts expressed in neurons [5].
Separation is essential to deepen proteomic coverage by HRMS; separation simplifies molecular complexity along the temporal dimension, thus increasing the total number of tandem mass spectra that can be acquired to identify peptides regardless of limitations in MS/MS duty cycle (e.g., ~10–20 Hz). For example, the recent deep proteome maps of human tissues [6] and cell types in the brain [1] were made possible by cumulatively identifying ~300,000 peptides that were separated in multiple measurements with each employing 6-h-long gradient by nanoLC. Another way to improve identifications is by integrating technologies that employ orthogonal mechanisms of separation [7,8,9]. For example, the broadly successful multidimensional protein identification technology (MudPIT) is based on the on-line hyphenation of strong cation exchange and reversed-phase (RP) chromatographies into a single nanoLC platform [7]. Alternatively, the sample may be fractionated via ion exchange or RP chromatography followed by offline RP nanoLC under similar or different pH conditions prior to detection by HRMS [10,11,12,13]. While nanoLC serves as the mainstream separation approach for peptides, this technology typically requires ~1,000-fold higher protein digest amounts per measurement than the available protein content in single neurons. Recent developments in nanoLC tailored detection to substantially smaller protein amounts, such as the identification of ~3,700 proteins from 500 breast cancer cells (MCF-7), or 50 ng of total proteins using porous open-layer tubular columns (PLOT) [14] and ~6,000 proteins from ~500 ng digest of HeLa culture using an automated low-flow fractionator instrument [15]. Continuous advances in sample handling present a potential to approach protein amounts in single neurons using nanoLC.
Capillary electrophoresis (CE) is an alternative technology for limited samples. CE can achieve outstanding separation efficiency to resolve molecules in complex samples, offers various strategies of online preconcentration to enhance signal-to-noise ratio, and ensures compatibility with limited sample volumes (e.g., <1–10 nL) (reviewed in references [16,17,18,19]). We and others demonstrated CE electrospray ionization (ESI) HRMS to offer significant sensitivity gains for limited amounts of protein digests via bottom-up [20,21,22] or top-down [23, 24] workflows compared with nanoLC. Representative examples comprise the targeted detection of hemoglobins in single erythrocytes [25, 26] and attomoles of carbonic anhydrase from a few erythrocytes [27, 28] as well as the discovery detection of ~200 protein groups from 5 ng of Pyrococcus furiosus digest [29], ~100 protein groups from ~16 pg of Escherichia coli digest [30], and ~109 protein groups from 100 HeLa cells (~30 ng protein) [31]. By performing capillary zone electrophoresis, our custom-designed microanalytical CE-ESI-MS platform identified ~800 protein groups from 20 ng of protein digest from single identified cells in the 16-cell Xenopus laevis (frog) embryo, corresponding to ~0.1% of the total protein content of the cell [20, 22]. Most recently, we developed a nano-flow CE-ESI-MS interface capable of 260 zmol (156,000 copies) lower limit of detection. Using this technology, 217 protein groups were detected from 1 ng of protein digest from cultured mouse neurons, essentially approaching the total protein content of 1–5 neurons [5]. Remarkably, protein identifications in these CE-HRMS experiments by us and others relied on the fragmentation of peptide signals migrating across ~20–30 min, which is considerably shorter (compressed) compared with hours of separation typical of nanoLC. Owing to current limitations in MS/MS duty cycle, compressed separation is expected to hinder peptide detection, thus hampering the identification of proteins.
There is a significant interest in advancing peptide and protein identifications by microscale CE-ESI-MS [32]. In a similar fashion to nanoLC, multidimensional separation can reduce sample complexity prior to CE-ESI-MS. For example, ~3,430 proteins were identified after fractionating 1.5 mg of yeast protein digest into 182 aliquots using RP-nanoLC followed by CE-ESI-HRMS of each fraction [33]. Similar strategies recently identified 4,134 proteins by fractionating 600 μg of protein digest from ~50 Xenopus laevis eggs [34] and ~1,600 proteins by fractionating 50 μg of protein digest from the mouse brain [35]. Other strategies tailored sample handling to lower amounts of protein digests by streamlining fractionation on-line with CE analysis. For example, solid-phase microextraction (SPME) with CE-ESI-MS identified ~370 protein groups from 5 ng of digest from Pyrococcus furiosus [29], whereas strong cation exchange of 50 ng protein digest detected 799 protein groups from Escherichia coli [36] and ~1,000 protein groups from a whole Xenopus laevis zygote [37]. Alternatively, electrophoretic peak capacity can be enhanced by lengthening the separation time, albeit at the risk of diffusion-limited peak broadening deteriorating sensitivity. By minimizing/eliminating the electroosmotic flow using neutral-coated capillaries, a recent study identified ~250 proteins from 50 ng of protein digest from mouse brain, which was improved to ~1,600 protein identification by enabling the analysis of 50 μg of digest with 500 nL loading of sample directly on the separation capillary, remarkably on par with nanoLC MS [35]. However, to advance identifications in trace amounts of protein digests by fast CE, the rate of peptide separation and the duty cycles of MS/MS should ideally be matched.
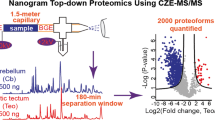
In this study, we evaluated the identification and quantification of peptides by capillary zone electrophoresis HRMS to measure proteins from digest amounts that approximate to ~1–5 neurons cultured from the mouse forebrain. Our microanalytical approach combined off-line RP fractionation with a custom-built CE-nanoESI-HRMS platform capable of ultrasensitive detection (260 zmol). Integration of these orthogonal separation mechanisms simplified compressed electrophoretic separation, improving separation performance and detection sensitivity. After processing 1–20 μg of protein digest from a neuron culture, the strategy identified 737 proteins in technical triplicates (~480 proteins/analysis on average) from ~1 ng of protein digest, approximating the total protein content from a few neurons. Furthermore, we demonstrated that this approach was scalable to identifying 225 protein groups in technical triplicates (141 proteins/analysis on average) from ~500 pg of digest, approaching single neuron protein content. Microscale RP fractionation with CE-HRMS raises sufficient sensitivity toward peptidomics and proteomics in single neurons to help elucidate molecular mechanisms responsible for the formation and maintenance of neuron-to-neuron heterogeneity in the brain.
Methods
Materials and Reagents
All materials were purchased at reagent grade or higher unless noted otherwise. Dithiothreitol (DTT), iodoacetamide (IAD), hydroxy methylaminomethane (Tris-base), Tris-hydrochloric acid (Tris-HCl), Tris-phosphate buffer solution, potassium chloride, and sodium hydroxide were obtained from Sigma-Aldrich (St. Louis, MO, USA). Papain dissociation system was from Worthington Biochemical Corporation (Lakewood, NJ, USA). Sodium dodecyl sulfate (SDS) was from Amresco (Solon, OH, USA). Ethylenediamine tetraacetic acid (EDTA), LC-MS grade acetonitrile (ACN), MS-grade trypsin protease, methanol (MeOH), formic acid (FA), acetic acid, and water (Optima) were from Fisher Scientific (Fair Lawn, NJ, USA). Pierce C18 Zip Tips (P/N 87784) were from Thermo Fisher Scientific (Waltham, MA, USA). Reagent-grade ammonium bicarbonate was from Avantor (Center Valley, PA, USA). Fused silica capillaries (20/90 μm inner/outer diameter (i.d./o.d.) for CE separation and 75/350 μm i.d./o.d. for sheath supply) were from Polymicro Technologies (Phoenix, AZ, USA) and used without modification. Stainless steel tapered-tip metal emitters (100/320 μm i.d./o.d.) were from New Objective (Woburn, MA, USA). Poly-L-ornithine, Hank’s balanced salt solution (HBSS), phosphate-buffered saline solution (PBS), minimum essential medium (MEM), neurobasal medium, glucose, penicillin-streptomycin, and fetal bovine serum were from Gibco (Grand Island, NY, USA). B27, N-2, Glutamax, and pyruvate supplements were from Thermo Fisher Scientific (Waltham, MA, USA). All standards were prepared in 500 μL or 2 mL LoBind protein microtubes from Eppendorf (Hauppauge, NY, USA).
Buffers and Solutions
Following protocols described elsewhere [38], the “neuronal plating medium” was prepared to contain MEM with 0.6% (w/v) D-glucose, 10% (v/v) horse serum, 1% glutamine, and 1% penicillin-streptomycin. The “neuronal maintenance media” was neurobasal supplemented with 2% B27, 1% N-2, 1% Glutamax, 1% penicillin-streptomycin, and 1% pyruvate. All media were filtered through a 0.2 μm porous mesh prior to usage. The cell “lysis buffer” contained 5 mM EDTA, 20 mM Tris-HCl, 35 mM NaCl, and 1% (v/v) SDS. The CE-nanoESI “sheath solution” contained 50% (v/v) MeOH with 0.1% (v/v) FA.
Neuron Culture
All procedures regarding the maintenance and handling of mice (Mus musculus) were approved by the George Washington University Institutional Animal Care and Use Committee (IACUC No. A274). Timed pregnant C57BL/6 dams were obtained from Charles River Laboratories (Wilmington, MA, USA). Primary cultures of mouse hippocampal neurons were prepared as described previously [38]. Briefly, hippocampal neurons were isolated from mouse embryos at Embryonic Day 16 and dissociated using papain solution for 30 min at 37 °C according to manufacturer recommendations (Worthington), followed by trituration and mixing with a plastic pipette in neuronal plating medium. Hippocampal cells were filtered with a 70 μm nylon mesh cell strainer (Falcon) and plated at 1 × 105 cell/cm2 on a 24-well plate previously coated with poly-L-ornithine (1 mg/mL) in plating medium. After 1 h, the medium was replaced with neurobasal maintenance medium and cultured for 14 d in vitro. Afterward, the cultured neurons were rinsed with ice-cold PBS to remove residual growth media, scraped, and the content of 2 wells was suspended in 150 μL PBS (Thermo Fisher Scientific). Cells were pelleted by centrifugation at 800 × g for 5 min at 4 °C, and stored at −80 °C until further processing for analysis by CE-HRMS.
To approximate the extractable total protein amount from an average single neuron in the culture, we combined cell counting and total protein assay. A 10 μL portion of the PBS-suspended cultured neurons were mixed with 10 μL of 0.4% trypan blue (Life Technologies, Washington, DC, USA), and stained cells were analyzed in a microfluidic cell counter (model Countess, Invitrogen, Carlsbad, CA, USA). The results revealed a 6.4 × 105 cells/mL cell density, and ~90% of the cells were quantifiably viable. This suspension contained 0.35 mg/mL protein based on the bicinchoninic acid assay (BCA, Thermo Scientific), which approximated to ~500 pg of total protein extractable from a neuron on average.
Bottom-Up Proteomic Workflow
The hippocampal neurons were processed following a standard bottom-up proteomic workflow [39]. In this study, the cultured neurons were lysed in 200 μL of lysis buffer with subsequent ultrasonication for 15 min in an ice-cold water bath to facilitate protein extraction. The resulting sample was reduced (4 μL of 1 M DTT, 30 min at 60 °C) and alkylated (8 μL of 1 M IAD, 15 min in the dark) before quenching the reaction (4 μL of 1 M DTT). To remove cell debris, the sample was centrifuged at 14,000 × g for 10 min at 4 °C, and the supernatant was transferred into a new 2 mL LoBind microcentrifuge tube. Next, proteins were purified by overnight precipitation in 1 mL chilled acetone (−20 °C), followed by centrifugation at 14,000 × g for 10 min at 4 °C. The resulting pellet was washed with chilled acetone (–20 °C), dried at room temperature, and reconstituted in 100 μL of ammonium bicarbonate (50 mM). Protein concentration was estimated based on BCA. A total of ~20 or 100 μg of protein amount were digested by trypsin at a 1:50 protein:enzyme ratio (0.4 μL or 2.0 μL of 1 μg/μL trypsin, respectively) for ~6 h at 37 °C. The resulting peptides were vacuum-dried and stored at –20 °C until analysis.
Reversed-Phase Peptide Separation
The dried peptides were reconstituted in 0.1% (v/v) FA to a 1 μg/μL peptide concentration, confirmed by the Colorimetric Peptide Assay (Thermo Scientific). An aliquot containing 20 μg (for method development) or 1 μg (for scalability test) peptides were fractionated on C18 ZipTip cartridges (100 μL or 10 µL format, Pierce, Thermo Scientific) following manufacturer instructions. Peptides were sequentially eluted using 10%, 20%, and 30% acetonitrile in water containing 0.1% FA. Each resulting fraction was dried and reconstituted in 20 μL (method development) or 2 μL (scalability test) of 75% (v/v) acetonitrile containing 0.05% (v/v) acetic acid (“sample”) before analysis by CE-HRMS.
CE-nanoESI-HRMS
Peptides were detected in a custom-built microanalytical CE-nanoESI-HRMS platform that we recently characterized (see reference [5]). In this study, capillary zone electrophoresis was performed on 1 nL of digest sample in a 90-cm capillary at ~250 V/cm field strength. Peptides were ionized in a CE-nanoESI interface operated in the cone-jet spraying mode for efficient ion generation using 50% MeOH (0.1% FA) at 300 nL/min flow rate as sheath liquid and 2.7 kV as electrospray potential. Peptide ions were mass-analyzed using a hybrid quadrupole Orbitrap mass spectrometer equipped with a higher-energy collision dissociation (HCD) cell (Q Exactive Plus, Thermo Scientific). Separating peptide features were surveyed between m/z 350 and 1,800 at 35,000 FWHM resolution (MS1) using the following settings: maximum IT, 50 ms; chromatography peak width (FWHM), 13 s; exclusion mass tolerance, 10.0 ppm; peptide matches, on; ion signals excluded below +2 charge state; ion signal intensity threshold, 1.5 × 103 counts; apex trigger, turned off. Ions that matched these criteria were selected for fragmentation in the HCD cell with the following settings: maximum IT, 60 ms; m/z isolation window for MS2, 1.0 Th; normalized collision energy, 28; MS2 resolution, 17,500 FWHM; TopN, 15; loop count, 15. Fragmented ions were dynamically excluded with 5 ppm accuracy for 9 s before being reconsidered for fragmentation.
Data Analysis
Raw mass spectrometric data were analyzed in MaxQuant ver. 1.5.7.4 (Max Planck Institute of Biochemistry) executing the Andromeda 1.5.6.0 search engine [40, 41] against the SwissProt mouse proteome database (downloaded from UniProt on November 11th, 2015) containing 16,792 entries. The search parameters were: digestion, tryptic; missed cleavages, maximum 2; minimum number of unique peptides, 1; fixed modification, carbamidomethylation of cysteines; variable modification, oxidation of methionines; main search peptide tolerance and MS/MS match tolerance, ±4.5 ppm and ±10 ppm, respectively; isotope match tolerance, 2 ppm; decoy mode, revert; label-free quantitation, enabled; fractionation, “no” for control (unfractionated samples) and “yes, three fractions” for fractionated samples. Proteins were identified with a false discovery rate (FDR) <1% against a reversed-sequence decoy database. For each identified peptide, the extent of co-isolation spectral interference was quantified in Proteome Discoverer 2.1 (Thermo Scientific, Waltham, MA. USA) using SEQUEST HT as the search engine against the mouse proteome with identical data processing settings as in MaxQuant. Following reporting guidelines in UniProt, we report proteins as groups based on the parsimony principle unless the MS-MS/MS data provide sufficient evidence to identify isoforms with known biological significance in SwissProt. Common contaminants were manually removed from the reported list of proteins. Label-free quantification (LFQ) intensities were calculated using the MaxLFQ algorithm [42] in MaxQuant with LFQ minimum ratio count set to 1 and fast LFQ disabled. For statistical analysis, p-value of less than 0.05 (Student’s t-test) was used to indicate statistical significance. Errors are reported as standard deviation (SD).
Safety Considerations
Fused silica capillaries and tapered-tip metal emitters, which pose needle-stick hazard, should be handled with care. Standard safety protocols were followed during the handling of chemicals. All electrically conductive parts of the CE-nanoESI system were grounded or isolated to prevent electrical shock hazard.
Results and Discussion
Sensitivity Needs for Limited Neuron Populations
The goal of this study was to advance protein identification from protein digest amounts that approximate to a few mammalian neurons to a single neuron. We and others recently demonstrated that CE-HRMS offers sensitivity benefits for limited amounts of peptides and proteins (see reviews [16,17,18,19]). For example, custom-built microanalytical CE has allowed us to identify ~800 proteins from ~20 ng of protein digest from single Xenopus laevis embryonic cells [20, 22] and 217 proteins from ~1 ng of protein digest from a cortical neuron culture of mouse [5]. These identifications relied on the detection of 829 [20, 22] and 525 [5] peptides, respectively, migrating over a 20–25 min window. In these experiments, compressed separation challenged the fragmentation of peptides with a limited MS/MS duty cycle, which in turn hindered protein identifications. To improve protein detection for limited populations of neurons, including single cells, using CE-HRMS, methodological or technological developments need to balance sample complexity, separation peak capacity, and/or the duty cycle of tandem mass spectrometry.
We proposed that multidimensional separation sufficiently simplifies molecular sample complexity for tandem mass spectrometry to enhance peptide detection from trace amounts of protein digests afforded from limited neuronal populations. Our working strategy adopted the principle that integration of orthogonal separation mechanisms improves the net peak capacity of the system. For example, RP fractionation of 600 μg of digest from fertilized X. laevis eggs by RP-LC with analysis of 1.5 μg digest by CE-ESI-MS recently identified ~4,100 protein groups [34]. However, to extend these measurements to significantly smaller numbers of neurons in this work, substantially smaller starting amounts of proteins and resulting protein digests must be processed. Indeed, by combining cell counting and total protein assay in a neuron culture (see Methods), we approximated an average neuron to yield ~500 pg of total protein extract. RP sorbent C18 columns present an attractive alternative to nanoLC for processing miniscule amounts of protein digest because this platform is scalable (e.g., protein binding up to 8 μg in 10 μL volume), operates sufficiently fast for manual sampling (10−15 min), and can be integrated off- or on-line with CE.
Improved Trace-Sensitive Peptide Detection
We evaluated the combination of RP fractionation and CE-nanoESI-HRMS for detecting proteins in trace amounts of protein digests. Schematics of the study are presented in Figure 1. After culturing primary hippocampal neurons from mouse, the cells were lysed in 1% SDS lysis buffer, and ~20–100 μg of proteins were processed via a bottom-up proteomic workflow (see Methods). A protein digest containing 20 μg of peptides was step-wise fractionated on a RP column (ZipTip) using 10%, 20%, and 30% (v/v) ACN containing 0.1% FA (see Figure 1). As no peptides were detectable upon additional elution with 40% ACN, 30% ACN was considered sufficient to recover peptides from the RP column. Eluted peptides were dried and reconstituted in 20 μL of 75% (v/v) ACN containing 0.05% (v/v) acetic acid, selected to perform on-column enrichment via field-amplified sample stacking for capillary zone electrophoresis in this work. An identical amount of protein digest served as “control” (without RP fractionation). To analyze digest amounts that approximate extractable proteins from <5 neurons (see Methods), we analyzed 1 ng of total peptides from the control and among the fractions. Last, we designed experiments to test scalability by RP fractionating 1 μg digest (approximating ~2,000 neurons) and measuring ~500 pg protein digest, essentially approximating protein amounts from a single neuron.

Experimental strategy for improving protein identifications from protein digest amounts approximating to 1–5 neurons. A ~20 μg of protein digest from cultured neurons was reversed-phase fractionated (ZipTip), and ~1 ng of total peptide mixture was analyzed by CE-nanoESI-HRMS. Unfractionated digest served as control. This approach was scalable for fractionating 1 μg protein digest (~2,000 neurons) before analyzing 500 pg of peptide mixture, essentially approaching the protein content of a single mammalian neuron
Without RP fractionation, peptide identification was constrained by compressed separation in the control (Figure 2). The rate of MS/MS events rapidly increased as migration unfolded with most tandem mass spectra resulting between 20–30 min and 33–37 min (see “MS/MS events”). A portion of these MS/MS events resulted in successful peptide spectral matches (PSM) against the mouse proteome (see “PSMs”). Most PSMs were acquired over a ~10-min window, revealing significantly shorter, or compressed, separation by capillary zone electrophoresis than is typical of nanoLC. Although the MS/MS duty cycle was not exhausted at any point of the separation, the rate of peptide identification was limited at ~100 PSMs/min at the apex of the electropherogram (22−28 min, see arrows); only ~45% of the acquired tandem mass spectra were successfully matched to PSMs in this region (Figure 2. top panel). Furthermore, the primary high-resolution MS-MS/MS data revealed notable spectral interference during fragmentation as a result of ion(s) that were co-isolated with the peptide precursor ion with a standard ±0.5 Th window. Co-isolation interference was quantifiable (>0%, see Methods) for ~65% of PSMs at the most compressed portion of the electropherogram (22–28 min) compared with ~50% of PSMs acquired with interference outside this compressed zone (14–22 min and 28–40 min). Combined, these data suggested possible improvements in protein identification by minimizing spectral interference during peptide separation.

Peptide detection by CE-nanoESI-HRMS. Distribution of separation time and hydrophobicity indices calculated for peptides that were detected in the control (unfractionated) digest revealed a zone of compressed separation, which in turn lowered the success rate of MS/MS events leading to peptide identifications. Each data point marks a different identified peptide
RP fractionation offered one such direction toward simplified peptide complexity. We calculated the hydrophobicity index of each identified peptide using the Sequence Specific Retention Calculator (ver. 3.x–2010) [43]. As shown in Figure 2, calculated indices suggested broad hydrophobic characteristics for the identified peptides. Additionally, the hydrophobicity indices were poorly correlated with migration time (R2 = 0.34, see Figure 2). This result confirmed orthogonality between hydrophobic retention on an RP column and electrophoretic migration using our custom-built CE-HRMS system. Therefore, we expected the integration of RP fractionation and electrophoretic separation to enhance the separation peak capacity of our custom-built CE-nanoESI-HRMS system.
We applied multidimensional separation for trace amounts of neuronal protein digests. We analyzed a total of 1 ng of peptides with fractionation and without (control) (Figure 3a); 1/3 ng of total peptides were measured from each of the three fractions (10%, 20%, or 30% ACN), confirmed by a total peptide assay (see Methods). The distribution mean of peptide hydrophobicity indices increased with eluent strength across the fractions (see left panel), thus validating the mechanism of retention as hydrophobic interaction with the stationary phase of the ZipTip column. In contrast, the distribution mean of migration times was indistinguishable between the control and the fractions (see right panel), revealing no detectable effects on electrophoretic mobility by RP fractionation. It follows that RP fractionation prior to CE minimized molecular complexity during capillary zone electrophoresis HRMS.

Multidimensional separation for enhancing peptide identifications. (a) Peptides were successfully separated on a C18 phase based on hydrophobicity differences (left panel), but not migration time (right panel). Key: *p < 0.05; ns, not significant. Each data point corresponds to a different identified peptide. (b) Cumulative peptide identifications rapidly increased during compressed CE separation; the combined fractions (“Net”) outperformed the control (left inset). Fractionation simplified peptide complexity, allowing for a greater portion of molecular features (MF) to be identified as peptide spectral match (PSM) (see PSM/MF, right inset)
Next, we characterized peptide identifications upon RP fractionation. Figure 3b monitors peptide identifications as migration unfolded. Cumulative identifications rapidly increased over the most compressed zone of separation. The identification rate, quantified as number of non-redundant peptides identified per unit time, was improved in the “Net Fraction” compared with the control (see left inset). The greatest improvement occurred during the most compressed portion of the electropherogram (see center), where the peptide identification rate was enhanced by 57%. The fragmentation data from these additional MS/MS transitions allowed a significantly greater portion of detected molecular features (MF) to be identified as PSMs in the Net Fraction compared with the control (see PSM/MF in right inset). Notably, ~90% of these PSMs were acquired with full C-trap fill time (60 ms) in the fractions or the control, revealing ion flux-limited conditions for the quadrupole Orbitrap mass spectrometer. Most recently, independent studies using nanoLC-HRMS found longer fill times (250 ms) advantageous for shotgun proteomics of limited protein digests (e.g., 1–10 ng) [44], suggesting that similar instrumental settings may also further peptide identifications during trace-sensitive CE-nanoESI-HRMS. While the number of identified peptides from the control (624 peptides) exceeded those from each of the fraction in our study (338, 227, 507 peptides in fractions 1, 2, and 3, resp.), the combined fractions with cumulative technical measurements identified substantially more peptides despite consuming an equal amount of total protein digest for analysis: 1,753 peptides were identified in union between the fractions (“Net Fraction”) versus 1,238 peptides in the control. Therefore, RP fractionation helped identify a greater number of peptides by CE-HRMS.
We also assessed the performance of peptide quantification upon fractionation (Figure 4). To account for biological variations between cell cultures, we repeated the analysis of 1 ng total protein digest using different neuron cultures (n = 3) upon fractionating 20 μg of protein digest from each replicate as described earlier (see also Figure 1). The results of technical replicates and cumulative identifications are presented in Figure 4a. Compared with the control, fractionation resulted in a 25% increase in peptide identification (left panel). We ascribe the observed sensitivity improvements to a combination of factors. Electrophoretic separation was notably improved in the fractions compared with the control. Using the 90-cm capillary, the mean theoretical plate number increased from ~274,000 in the control to ~373,000 in Fraction 1, ~412,000 in Fraction 2, and ~314,000 in Fraction 3 with these differences being significant (p = 2.8 × 10–3, 1.2 × 10–6, and 2.2 × 10–2, respectively). Additionally, the HRMS data revealed fractionation to effectively remove abundant salts and polar compounds from the culture media. Consequently, by lower salt concentration decreasing the conductivity of the sample, it is possible that field-amplified sample stacking was also improved in the fractionated samples. Furthermore, lower salt concentration in the fractionated samples is expected to have promoted ion generation by minimizing ionization interferences in the CE-nanoESI source. Indeed, signal-to-noise (S/N) ratios were improved by ~2-to-2.5-fold for several randomly selected peptides that migrated in the most compressed zone of the electropherogram, such as EIQTAVR with S/N = 127 ± 8 in Fraction #1 versus 50 ± 5 in the control, AVAVVVDPIQSVK with S/N = 143 ± 7 in Fraction #2 versus 60 ± 2 in the control, and SYELPDGQVITIGNER with S/N = 328 ± 8 in Fraction #3 versus 134 ± 3 in the control in the same experiment. Last, fractionation also minimized chemical complexity, thus likely allowing better utilization of the constrained MS/MS duty cycle to fragment trace-level peptide signals, which are otherwise deprioritized or triaged during data-dependent acquisition due to lower S/N.

Metrics of peptide and protein detection from 1 ng of protein digest. (a) Three-step reversed-phase fractionation with CE-nanoESI-HRMS identified more peptides (see “Net”), significantly improving protein identification than the control (left panel). Comparison of peptides identified in each fraction on the basis of three independent experiments (right panel). (b) Protein identifications against the mouse proteome (left panel). The mean label-free quantification (LFQ) intensity was indistinguishable for proteins between the fractionated and control samples (right panel). Proteins that were exclusively quantified by fractionation (“Excl.”) had significantly lower LFQ intensities, suggesting a sensitivity gain by fractionation. Error bars, standard deviation. Statistical significance, *p < 0.05
Improved Trace-sensitive Protein Detection
Higher-sensitivity peptide detection upon fractionation, in turn, improved the identification of proteins. As shown in Figure 4b, 31% more proteins were identifiable on average per technical replicate in the Net Fraction than the control with this difference being significant (p = 6.9 × 10–4). Cumulatively, triplicate analyses amounted to 737 protein groups identified in the Net Fraction versus 577 protein groups in the control. The distribution of LFQ intensities calculated for commonly detected proteins (see Methods) had indistinguishable means (see bottom panel), suggesting no quantifiable differences in the protein digest amounts that were recovered by fractionation or analyzed by CE-nanoESI-HRMS. Remarkably, proteins that were exclusively quantified by fractionation were found to populate the lower domain of the quantitative dynamic range (see right panel) with this difference being significant (p = 7.4 × 10–21). A list of identified and quantified proteins and their calculated LFQ intensity are tabulated in Supplementary Table 1. Combined, these results established higher qualitative and quantitative sensitivity to detect and identify peptides/proteins by supplementing CE-HRMS with RP fractionation, particularly for molecules of lower abundance.
Last, we tested the scalability of the approach to process protein digest amounts from a limited population of neurons and a single neuron. We RP-fractionated ~1 μg of peptides from the neuronal protein digest, confirmed by a total peptide assay, which approximates to proteins from ~2,000 neurons. Each fraction was dried and reconstituted in 2 μL of sample solvent in a microtube (see Methods). A total of 500 pg of peptide was analyzed by CE-HRMS between the fractions, corresponding to the content of a mammalian neuron. From equal amounts of total digest analyzed, triplicate analysis enabled 225 protein groups to be identified with fractionation compared to 141 protein groups from the control (Figure 5). A list of identified and quantified proteins is shown in Supplementary Table 2. Considering a 20-fold sample reduction during fractionation and a 2-fold sample reduction during detection (40-fold net reduction) in the workflow compared with the previous portion of this study, these identification numbers are encouraging toward trace-sensitive analysis of single neurons.

Case study of analyzing 500 pg of protein digest amount, approximating a single neuron. Products of many genes used as classical neuronal markers were identified by RP fractionation CE-nanoESI-HRMS. Representative examples are shown (left panel). Fractionation helped quantify proteins of lower-abundance (see red data points marking proteins, right panel). *Proteins from 1 ng digest are included
The identified proteins were enriched in products of many important neuronal genes. Our proteomics data identified 100 protein groups translated from 100 different genes that quantitative single-cell transcriptomics independently found to be differentially expressed during early neurogenesis [45]. A list of these proteins is tabulated in Supplementary Table 3. We identified a combination of proteins involved in neuronal differentiation and function, reflecting the developmental stage of the cultured neurons, which at 14 days in vitro have partially completed differentiation but are still extending processes and forming synapses [46, 47]. We found a high relative abundance of neuronal cytoskeletal protein such as Tubulin beta 3 chain (Tubb3), a well-established marker of differentiated neurons [48], and Tubulin alpha-3 chain (Tuba3a), which is involved in neuronal migration (Figure 5, right panel). Mutations of the TUBA3A gene in humans lead to lissencephaly, brain malformations caused by abnormal organization of the hippocampus and cortex [49]. Proteins linked to neuronal migration were identified, mostly belonging to the middle-to-lower domain of protein abundance in our dataset, including proteins critical for neurite differentiation and cytoskeletal dynamics: neural cell adhesion molecule 1 (Ncam1) [50, 51], microtubule associated protein 1B (Map1b) [52], and the serine-threonine kinase doublecortin-like and CAM kinase-like 1 (Dclk1) [53, 54]. Interestingly, Dclk1 and the microtubule-associated protein, doublecortin (Dcx), which is another classic neuronal marker, were only identified with the fractionation approach. Mutations or deletion in DCX also cause lissencephaly in humans [55], and Dclk1 is highly expressed in regions with active neurogenesis and critical for neuronal survival [54]. Other interesting family members associated with developmental processes are the isoforms collapsin-response-mediator proteins (CRMPs), CRMP-1 (Crmp) and CRMP-5 (Dpysl5), which are highly expressed in the developing brain and play a role in neurogenesis, axonal guidance, and neuronal physiology [56, 57].
A second set of the identified proteins are key for mature neuronal function and have been linked to cognition and neurodegenerative disorders, such as the neuronal sodium/potassium-transporting ATPase α3 subunits (Atp1a3). Mutations in ATP1A3 have been found in individuals with rapid-onset dystonia-parkinsonism [58]. The calcium/calmodulin-dependent protein kinase type II subunit alpha (Camk2a) [59] and microtubule-associated protein tau (Mapt) have been linked to Alzheimer’s disease. Finally, we identified proteins at the core of synaptic function regulation [60,61,62], such as synaptosomal-associated protein 25 (Snap25), syntaxin-1B (Stx1b), synaptotagmin-1 (Syt1), synaptophysin (Syp), and synapsin-2 (Syn2). In addition to Syn2 and Syp, lower-abundance protein like vesicle-associated membrane protein 2, also known as synaptobrevin-2 (Vamp2), was only identified by fractionation, likely amounting to the simplification of the sample complexity to identify proteins in the lower domain of the dynamic range of protein concentrations (see Figure 5, right panel).
Overall, these results showed that measurement of 500 pg to 1 ng of total peptides after fractionating 20 to 1 μg of protein digest is sensitive enough to identify neuronal markers associated with the development or disease of hippocampal neurons. Our findings also reveal sensitivity gains by fractionation to enable the detection and quantification of a greater number of proteins, including many of lower abundance (Figure 5), from trace amounts of protein digests. The agreement between data from ultrasensitive proteomics from this study and single-cell transcriptomics can help query molecular mechanisms during early neuronal differentiation.
Conclusions
Single-neuron measurements raise a powerful potential to better understand molecular processes as neurons develop in the brain. However, protein amounts contained by single neurons are 1,000- to 10,000-fold smaller (~500 pg/cell) than are typically detectable by standard nanoLC-HRMS. Here, we demonstrated that RP fractionation of 20 to 1 μg of protein digest in a ZipTip column improved the detection and quantification of proteins and peptides from 1 ng to 500 pg protein digests using CE-nanoESI-HRMS. This approach attains sufficient sensitivity toward characterizing protein digest amounts that single mammalian neurons are approximated to yield. We ascribe these improvements in sensitivity and quantification to a combination of factors, including reduced sample complexity, improved separation performance during CE, enhanced ionization efficiency during nanoESI, and higher peak capacity benefiting peptide fragmentation with a limited MS/MS duty cycle. As an example, we demonstrated the identification of 480 proteins/experiment from 1 ng protein digest (737 proteins by technical triplicates) and 141 proteins per experiment from 500 pg protein digest (225 proteins by technical triplicates), essentially approximating proteins extractable from ~1 to 5 neurons. However, to extend these measurements directly to small populations of neurons or single neurons, continuous advances are needed at nearly all stages of the proteomic workflow, beginning with the handling of cells, followed by the efficient extraction and processing of their limited protein content, before trace-sensitive characterization of proteins and peptides. The presented work lays down encouraging detection sensitivity by microanalytical RP fractionation with CE-nanoESI-HRMS to open a gate to study mechanisms of gene translation in limited populations of neurons to single neurons in the brain.
References
Sharma, K., Schmitt, S., Bergner, C.G., Tyanova, S., Kannaiyan, N., Manrique-Hoyos, N.: Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831 (2015)
Johnson, M.B., Wang, P.P., Atabay, K.D., Murphy, E.A., Doan, R.N., Hecht, J.L.: Single-cell analysis reveals transcriptional heterogeneity of neural progenitors in human cortex. Nat. Neurosci. 18, 637–647 (2015)
Lengler, J., Jug, F., Steger, A.: Reliable neuronal systems: the importance of heterogeneity. PLoS One. 8, (e80694), (2013)
Shekhar, K., Lapan, S.W., Whitney, I.E., Tran, N.M., Macosko, E.Z., Kowalczyk, M.: Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell. 166, 1308–1323 (2016)
Choi, S.B., Zamarbide, M., Manzini, M.C., Nemes, P.: Tapered-tip capillary electrophoresis nano-electrospray ionization mass spectrometry for ultrasensitive proteomics: the mouse cortex. J. Am. Soc. Mass Spectrom. 28, 597–607 (2017)
Kim, M.S., Pinto, S.M., Getnet, D., Nirujogi, R.S., Manda, S.S., Chaerkady, R.: A draft map of the human proteome. Nature. 509, 575–581 (2014)
Washburn, M.P., Wolters, D., Yates, J.R.: Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247 (2001)
Dowell, J.A., Frost, D.C., Zhang, J., Li, L.J.: Comparison of two-dimensional fractionation techniques for shotgun proteomics. Anal. Chem. 80, 6715–6723 (2008)
Zhang, X., Fang, A.Q., Riley, C.P., Wang, M., Regnier, F.E., Buck, C.: Multi-dimensional liquid chromatography in proteomics. Anal. Chim. Acta. 664, 101–113 (2010)
Wisniewski, J.R., Zougman, A., Mann, M.: Combination of FASP and StageTip-based fractionation allows in-depth analysis of the hippocampal membrane proteome. J. Proteome Res. 8, 5674–5678 (2009)
Wang, H., Sun, S.N., Zhang, Y., Chen, S., Liu, P., Liu, B.: An off-line high pH reversed-phase fractionation and nano-liquid chromatography-mass spectrometry method for global proteomic profiling of cell lines. J. Chromatogr. B. 974, 90–95 (2015)
Gilar, M., Olivova, P., Daly, A.E., Gebler, J.C.: Two-dimensional separation of peptides using RP-RP-HPLC system with different pH in first and second separation dimensions. J. Sep. Sci. 28, 1694–1703 (2005)
Gokce, E., Andrews, G.L., Dean, R.A., Muddiman, D.C.: Increasing proteome coverage with offline RP HPLC coupled to online RP nanoLC-MS. J. Chromatogr. B. 879, 610–614 (2011)
Li, S.Y., Plouffe, B.D., Belov, A.M., Ray, S., Wang, X.Z., Murthy, S.K.: An integrated platform for isolation, processing, and mass spectrometry-based proteomic profiling of rare cells in whole blood. Mol. Cell. Proteom. 14, 1672–1683 (2015)
Kulak, N.A., Geyer, P.E., Mann, M.: Loss-less nano-fractionator for high sensitivity, high coverage proteomics. Mol. Cell. Proteom. 16, 694–705 (2017)
Romanova, E.V., Aerts, J.T., Croushore, C.A., Sweedler, J.V.: Small-volume analysis of cell-cell signaling molecules in the brain. Neuropsychopharmacology. 39, 50–64 (2014)
Harstad, R.K., Johnson, A.C., Weisenberger, M.M., Bowser, M.T.: Capillary electrophoresis. Anal. Chem. 88, 299–319 (2016)
Dawod, M., Arvin, N.E., Kennedy, R.T.: Recent advances in protein analysis by capillary and microchip electrophoresis. Analyst. 142, 1847–1866 (2017)
Slampova, A., Mala, Z., Gebauer, P., Bocek, P.: Recent progress of sample stacking in capillary electrophoresis (2014–2016). Electrophoresis. 38, 20–32 (2017)
Lombard-Banek, C., Moody, S.A., Nemes, P.: Single-cell mass spectrometry for discovery proteomics: quantifying translational cell heterogeneity in the 16-cell frog (Xenopus) embryo. Angew. Chem. Int. Ed. 55, 2454–2458 (2016)
Sun, L.L., Hebert, A.S., Yan, X.J., Zhao, Y.M., Westphall, M.S., Rush, M.J.P.: Over 10,000 peptide identifications from the HeLa proteome by using single-shot capillary zone electrophoresis combined with tandem mass spectrometry. Angew. Chem. Int. Ed. 53, 13931–13933 (2014)
Lombard-Banek, C., Reddy, S., Moody, S.A., Nemes, P.: Label-free quantification of proteins in single embryonic cells with neural fate in the cleavage-stage frog (Xenopus laevis) embryo using capillary electrophoresis electrospray ionization high-resolution mass spectrometry (CE-ESI-HRMS). Mol. Cell. Proteom. 15, 2756–2768 (2016)
Han, X., Wang, Y., Aslanian, A., Bern, M., Lavallee-Adam, M., Yates, J.R.: Sheathless capillary electrophoresis-tandem mass spectrometry for top-down characterization of pyrococcus furiosus proteins on a proteome scale. Anal. Chem. 86, 11006–11012 (2014)
Zhao, Y.M., Sun, L.L., Zhu, G.J., Dovichi, N.J.: Coupling capillary zone electrophoresis to a Q Exactive HF mass spectrometer for top-down proteomics: 580 proteoform identifications from yeast. J. Proteome Res. 15, 3679–3685 (2016)
Hofstadler, S.A., Severs, J.C., Smith, R.D., Swanek, F.D., Ewing, A.G.: Analysis of single cells with capillary electrophoresis electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 10, 919–922 (1996)
Mellors, J.S., Jorabchi, K., Smith, L.M., Ramsey, J.M.: Integrated microfluidic device for automated single cell analysis using electrophoretic separation and electrospray ionization mass spectrometry. Anal. Chem. 82, 967–973 (2010)
Valaskovic, G.A., Kelleher, N.L., McLafferty, F.W.: Attomole protein characterization by capillary electrophoresis mass spectrometry. Science. 273, 1199–1202 (1996)
Moini, M., Demars, S.M., Huang, H.L.: Analysis of carbonic anhydrase in human red blood cells using capillary electrophoresis/electrospray ionization-mass spectrometry. Anal. Chem. 74, 3772–3776 (2002)
Wang, Y., Fonslow, B.R., Wong, C.C.L., Nakorchevsky, A., Yates, J.R.: Improving the comprehensiveness and sensitivity of sheathless capillary electrophoresis-tandem mass spectrometry for proteomic analysis. Anal. Chem. 84, 8505–8513 (2012)
Sun, L.L., Zhu, G.J., Zhao, Y.M., Yan, X.J., Mou, S., Dovichi, N.J.: Ultrasensitive and fast bottom-up analysis of femtogram amounts of complex proteome digests. Angew. Chem. Int. Ed. 52, 13661–13664 (2013)
Chen, Q., Yan, G.Q., Gao, M.X., Zhang, X.M.: Direct digestion of proteins in living cells into peptides for proteomic analysis. Anal. Bioanal. Chem. 407, 1027–1032 (2015)
Comi, T.J., Do, T.D., Rubakhin, S.S., Sweedler, J.V.: Categorizing cells on the basis of their chemical profiles: progress in single-cell mass spectrometry. J. Am. Chem. Soc. 139, 3920–3929 (2017)
Faserl, K., Kremser, L., Muller, M., Teis, D., Lindner, H.H.: Quantitative proteomics using ultra low flow capillary electrophoresis-mass spectrometry. Anal. Chem. 87, 4633–4640 (2015)
Yan, X.J., Sun, L.L., Zhu, G.J., Cox, O.F., Dovichi, N.J.: Over 4100 protein identifications from a Xenopus laevis fertilized egg digest using reversed-phase chromatographic prefractionation followed by capillary zone electrophoresis-electrospray ionization-tandem mass spectrometry analysis. Proteomics. 16, 2945–2952 (2016)
Chen, D., Shen, X., Sun, L.: Capillary zone electrophoresis-mass spectrometry with microliter-scale loading capacity, 140 min separation window and high peak capacity for bottom-up proteomics. Analyst. 142, 2118–2127 (2017)
Zhang, Z.B., Yan, X.J., Sun, L.L., Zhu, G.J., Dovichi, N.J.: Detachable strong cation exchange monolith, integrated with capillary zone electrophoresis and coupled with pH gradient elution, produces improved sensitivity and numbers of peptide identifications during bottom-up analysis of complex proteomes. Anal. Chem. 87, 4572–4577 (2015)
Zhang, Z.B., Sun, L.L., Zhu, G.J., Cox, O.F., Huber, P.W., Dovichi, N.J.: Nearly 1000 protein identifications from 50 ng of Xenopus laevis zygote homogenate using online sample preparation on a strong cation exchange monolith based microreactor coupled with capillary zone electrophoresis. Anal. Chem. 88, 877–882 (2016)
Kaech, S., Banker, G.: Culturing hippocampal neurons. Nat. Protoc. 1, 2406–2415 (2006)
Zhang, Y.Y., Fonslow, B.R., Shan, B., Baek, M.C., Yates, J.R.: Protein analysis by shotgun/bottom-up proteomics. Chem. Rev. 113, 2343–2394 (2013)
Cox, J., Mann, M.: MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008)
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R.A., Olsen, J.V., Mann, M.: Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 (2011)
Cox, J., Hein, M.Y., Luber, C.A., Paron, I., Nagaraj, N., Mann, M.: Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteom. 13, 2513–2526 (2014)
Spicer, V., Grigoryan, M., Gotfrid, A., Standing, K.G., Krokhin, O.V.: Predicting retention time shifts associated with variation of the gradient slope in peptide RP-HPLC. Anal. Chem. 82, 9678–9685 (2010)
Sun, B., Kovatch, J.R., Badiong, A., Merbouh, N.: Optimization and modeling of quadrupole orbitrap parameters for sensitive analysis toward single-cell proteomics. J. Proteome Res. 16, 3711–3721 (2017)
Pollen, A.A., Nowakowski, T.J., Shuga, J., Wang, X.H., Leyrat, A.A., Lui, J.H.: Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat. Biotechnol. 32, 1053–1058 (2014)
Grassi, D., Plonka, F.B., Oksdath, M., Guil, A.N., Sosa, L.J., Quiroga, S.: Selected SNARE proteins are essential for the polarized membrane insertion of igf-1 receptor and the regulation of initial axonal outgrowth in neurons. Cell Discov. 1, 15023 (2015)
Jurado, S., Goswami, D., Zhang, Y.S., Molina, A.J.M., Sudhof, T.C., Malenka, R.C.: LTP requires a unique postsynaptic SNARE fusion machinery. Neuron 77, 542–558 (2013)
Menezes, J.R.L., Luskin, M.B.: Expression of neuron-specific tubulin defines a novel population in the proliferative layers of the developing telencephalon. J. Neurosci. 14, 5399–5416 (1994)
Keays, D.A., Tian, G., Poirier, K., Huang, G.J., Siebold, C., Cleak, J.: Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 128, 45–57 (2007)
Doherty, P., Cohen, J., Walsh, F.S.: Neurite outgrowth in response to transfected N-CAM changes during development and is modulated by polysialic acid. Neuron 5, 209–219 (1990)
Bukalo, O., Fentrop, N., Lee, A.Y.W., Salmen, B., Law, J.W.S., Wotjak, C.T.: Conditional ablation of the neural cell adhesion molecule reduces precision of spatial learning, long-term potentiation, and depression in the CA1 subfield of mouse hippocampus. J. Neurosci. 24, 1565–1577 (2004)
Gonzalez-Billault, C., Jimenez-Mateos, E.M., Caceres, A., Diaz-Nido, J., Wandosell, F., Avila, J.: Microtubule-associated protein 1B function during normal development, regeneration, and pathological conditions in the nervous system. J. Neurobiol. 58, 48–59 (2004)
Francis, F., Friocourt, G., Chafey, P., Vinet, M.C., Koulakoff, A., Cheesman, E.: Doublecortin is a developmentally regulated microtubule-associated protein expressed in migrating and differentiating neurons. Neuron 23, 247–256 (1999)
Nawabi, H., Belin, S., Cartoni, R., Williams, P.R., Wang, C., Latremoliere, A.: Doublecortin-like kinases promote neuronal survival and induce growth cone reformation via distinct mechanisms. Neuron 88, 704–719 (2015)
Des Portes, V., Francis, F., Pinard, J.M., Desguerre, I., Moutard, M.L., Snoeck, I.: Doublecortin is the major gene causing X-linked subcortical laminar heterotopia (SCLH). Hum. Mol. Genet. 7, 1063–1070 (1998)
Etherton, M., Foldy, C., Sharma, M., Tabuchi, K., Liu, X.R., Shamloo, M.: Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl. Acad. Sci. USA. 108, 13764–13769 (2011)
Bretin, S., Reibel, S., Charrier, E., Maus-Moatti, M., Auvergnon, N., Thevenoux, A.: Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J. Comp. Neurol. 486, 1–17 (2005)
Heinzen, E.L., Arzimanoglou, A., Brashear, A., Clapcote, S.J., Gurrieri, F., Goldstein, D.B.: Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 13, 503–514 (2014)
Ghosh, A., Giese, K.P.: Calcium/calmodulin-dependent kinase II and Alzheimer's disease. Mol. Brain. 8, 78 (2015)
Deak, F., Schoch, S., Liu, X.R., Sudhof, T.C., Kavalali, E.T.: Synaptobrevin is essential for fast synaptic-vesicle endocytosis. Nat. Cell Biol. 6, 1102–1108 (2004)
Chapman, E.R., An, S., Barton, N., Jahn, R.: Snap-25, a t-snare which binds to both syntaxin and synaptobrevin via domains that may form coiled coils. J. Biol. Chem. 269, 27427–27432 (1994)
Sudhof, T.C.: Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 (2013)
Acknowledgments
This work was supported by the Arnold and Mabel Beckman Foundation Young Investigator Grant (to P.N.), the American Society for Mass Spectrometry Research Award (to P.N.), the DuPont Young Professor Award (to P.N.), and Cosmos Club Foundation Fellowships (to S.B.C. and C.L-B.).
Author information
Authors and Affiliations
Contributions
P.N., S.B.C., and C.L-B. designed the research; P.M-L. and M.C.M. provided the neuron cultures; S.B.C. and C.L-B. processed the samples; S.B.C. measured the samples; P.N. and S.B.C. analyzed the data; P.N. and S.B.C. wrote the manuscript. All authors commented on the manuscript.
Corresponding author
Electronic supplementary material
ESM 1
(XLSX 326 kb)
Rights and permissions
About this article

Cite this article
Choi, S.B., Lombard-Banek, C., Muñoz-LLancao, P. et al. Enhanced Peptide Detection Toward Single-Neuron Proteomics by Reversed-Phase Fractionation Capillary Electrophoresis Mass Spectrometry. J. Am. Soc. Mass Spectrom. 29, 913–922 (2018). https://doi.org/10.1007/s13361-017-1838-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-017-1838-1