
Abstract
Absolute quantification of protein targets using liquid chromatography-mass spectrometry (LC-MS) is a key component of candidate biomarker validation. One popular method combines multiple reaction monitoring (MRM) using a triple quadrupole instrument with stable isotope-labeled standards (SIS) for absolute quantification (AQUA). LC-MRM AQUA assays are sensitive and specific, but they are also expensive because of the cost of synthesizing stable isotope peptide standards. While the chemical modification approach using mass differential tags for relative and absolute quantification (mTRAQ) represents a more economical approach when quantifying large numbers of peptides, these reagents are costly and still suffer from lower throughput because only two concentration values per peptide can be obtained in a single LC-MS run. Here, we have developed and applied a set of five novel mass difference reagents, isotopic N,N-dimethyl leucine (iDiLeu). These labels contain an amine reactive group, triazine ester, are cost effective because of their synthetic simplicity, and have increased throughput compared with previous LC-MS quantification methods by allowing construction of a four-point standard curve in one run. iDiLeu-labeled peptides show remarkably similar retention time shifts, slightly lower energy thresholds for higher-energy collisional dissociation (HCD) fragmentation, and high quantification accuracy for trypsin-digested protein samples (median errors <15%). By spiking in an iDiLeu-labeled neuropeptide, allatostatin, into mouse urine matrix, two quantification methods are validated. The first uses one labeled peptide as an internal standard to normalize labeled peptide peak areas across runs (<19% error), whereas the second enables standard curve creation and analyte quantification in one run (<8% error).

ᅟ
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Protein candidate biomarker discovery generates long lists of proteins that must be further verified by targeted absolute quantification methods before being examined in clinical applications. The considerable monetary and personnel investment devoted to creating these candidate lists has thus far returned only a few biomarkers currently used clinically [1]. One major reason for the lack of clinical biomarkers is the difficulty in verifying the majority of candidates generated by discovery studies [1–3]. Immunoassay techniques, particularly the enzyme-linked immunosorbent assay (ELISA), are commonly used approaches for biomarker verification. This highly specific and sensitive assay uses a pair of antibodies against a protein to quantify candidates from below the ng/mL range [1]. Unfortunately, suitable antibodies for immunoassays exist for less than 10% of the proteins that comprise the human proteome [4]. High costs, ranging from $50,000 to $100,000, and development times of up to a year to develop an ELISA sufficient to validate a single candidate solidify the impracticalities of this method for large-scale candidate verification [2, 4].
Quantification with liquid chromatography-mass spectrometry (LC-MS) is an attractive alternative to immunoassays. The absolute quantification (AQUA) technique using stable isotope-labeled peptide standards was first demonstrated in 1996 [5] and later expanded by the Gygi group in 2003 [6]. The AQUA approach incorporates a known concentration of stable isotope-labeled peptide to a protein digest that yields the same unlabeled sequence. Abundances of the peptides are then quantified by LC-multiple reaction monitoring (MRM), typically with a triple quadrupole instrument, and peptide concentration is determined according to a previously constructed standard curve. Peptides are used as surrogates representing absolute protein abundance levels and can be quantified at sub-femtomole levels [6, 7]. Protein quantification using the LC-MRM AQUA approach is still frequently used because of its simplicity, accuracy, sensitivity, and ability to multiplex for measurement of multiple peptides in one run [8–11], and it has even been applied to non-tryptic peptides [12]. Unfortunately, the high cost of custom AQUA peptide synthesis (~$500 for 5 nmol) can limit the number of candidates analyzed by financially conscientious research groups.
Chemical derivatization of target peptide standards through mass difference labeling produces peptide pairs with identical retention times but different precursor ion masses, which can be used for quantification or to generate hybrid labeling approaches like the combined precursor isotopic labeling and isobaric tagging (cPILOT) technique [13]. The analysis of MRM transitions at different concentrations of a light-labeled peptide while maintaining a constant amount of a heavy-labeled peptide establishes a normalized calibration curve of peptide abundance. Tryptic digests from samples of interest are then labeled with the light reagent, and the internal standard peptide is spiked in at a known quantity. After MRM, the normalized abundance of the candidate peptide is calculated from the calibration curve and correlated back to the protein. Absolute quantification via mass difference labels has been demonstrated in multiple studies using AB SCIEX’s mass differential tags for relative and absolute quantification (mTRAQ) [14–18]. mTRAQ reagents label the N-terminus of a peptide as well as primary or secondary amines through an N-hydroxysuccinimide (NHS) ester moiety. Incorporation of a triplex mTRAQ label adds ~141, 145, or 149 Da to the precursor peptide mass depending on whether the light, medium, or heavy reagent is used [19]. AB SCIEX designed mTRAQ labels to be non-isobaric so that sequence-specific fragment ions can be used as MRM transitions in absolute quantification. This feature avoids the quantitative suppression that sometimes occurs in reporter ion quantification attributable to co-isolated precursor ions. Some groups have also used mTRAQ for candidate discovery in relative quantification studies [20–22], but one convincing report showed that although mTRAQ quantification is more accurate than AB SCIEX’s isobaric label, iTRAQ, it also has a greater standard deviation [23]. Furthermore, multiplexing samples with the triplex mTRAQ kit creates samples with three times as many precursor ions to select for MS2 events. A greater redundancy in MS2 scanning events results in more complete targeted analysis, but discovery experiments in complex protein mixtures suffer from under-sampling of the proteome [23]. Although mTRAQ multiplexing is not ideal for global studies, it is an effective and significantly more cost-efficient approach for biomarker verification over the AQUA method when large numbers of peptides are examined. However, mTRAQ labels are still quite expensive, costing ~ $134 per peptide candidate. Some studies quantify peptides using labeled peptide internal standards as a single point calibration [14], but higher accuracy results can be obtained by constructing a calibration curve accounting for response ratios between the labeled pair over a wide concentration range [17]. The triplex chemistry of the mTRAQ reagent allows for at most two calibration points to be determined per LC-MS run since these values must be normalized by the third labeled peptide, restricting throughput.
Cost and throughput disadvantages of mTRAQ could be solved by the existence of an easily produced mass difference label with a greater multiplexing capability. A 5-plex isotope dimethyl labeling method has recently been reported, but this method has not been applied to absolute quantification, and singly labeled peptides differ by only 2 Da [24]. Increasing the “plex” of mass difference labels to five would allow for collection of up to four peptide concentration values per LC-MS run, significantly improving the amount of quantitative data captured per run and reducing the number of runs for calibration curve construction. Labeled tryptic peptides could also be confidently quantified in one LC-MS run by spiking in a concentration range of synthetic peptide standards labeled by the remaining four reagent channels prior to sample clean-up.
The theoretical benefits of a 5-plex mass difference label have inspired our research team to design and synthesize the isotopic N,N-dimethyl leucine (iDiLeu) reagent. The general structure of the iDiLeu label is the same as our custom isobaric label, DiLeu [25], with a few key exceptions. Stable 13C, 15 N, 2H, and 18O isotopes are placed onto the iDiLeu reagent backbone by formaldehyde dimethylation and 18O exchange to maximize the mass differences between labels, but the carbonyl group does not function as a balance group that adjusts for the reporter region’s increasing mass. The resulting iDiLeu set is composed of five dimethylated leucines that target the N-terminus and lysine side chains of peptides through an amine-reactive triazine ester. Benefits of the original DiLeu label that are maintained in the iDiLeu variant include its synthetic simplicity, high yield (85%), modest mass, high intensity reporter ion production, and efficient peptide labeling [25]. In this report, we explore the use of 5-plex iDiLeu for absolute quantification of peptides and proteins using four labels to build a peptide calibration curve and the fifth to either label peptides in a complex sample or as a labeled internal standard. A Thermo Q-Exactive Orbitrap was used to demonstrate iDiLeu quantification using precursor extracted ion chromatogram peak areas, reporter ion intensities, and higher-energy collisional dissociation (HCD) fragment ions. Fragmentation ramp tests compared peptide identifications from a yeast tryptic digest as a function of normalized collision energy (NCE) for iDiLeu-, iTRAQ-, and TMT-labeled peptides using a Thermo Orbitrap Elite.
2 Experimental
2.1 Chemicals
Isotopic reagents for label synthesis including leucines (L-leucine, L-leucine-1-13C, 15 N, and L-leucine-1-13C), heavy formaldehydes (CD2O and 13CD2O), sodium cyanoborodeuteride (NaBD3CN), 97% 18O water (H2 18O), and deuterium water (D2O) were obtained from ISOTEC Inc. (Miamisburg, OH, USA). Sodium cyanoborohydride (NaBH3CN), formaldehyde (CH2O), hydrogen chloride gas (HCl), iodoacetamide (IAA), tris-hydrochloride, reagent-grade formic acid (FA), triethylammonium bicarbonate (TEAB), N,N-dimethylformamide (DMF), 4-(4, 6-dimethoxy-1, 3, 5-triazin-2-yl)-4-methylmorpholinium tetrafluoroborate (DMTMM), N-methylmorpholine (NMM), and bovine serum albumin (BSA) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Neuropeptide standard allatostatin I (AST-I, GDGRLYAFGL-NH2) was purchased from American Peptide Company (Sunnyvale, CA, USA). Sequencing grade trypsin was acquired from Promega (Madison, WI, USA). Urea, ammonium bicarbonate, ACS grade methanol (MeOH), dichloromethane (DCM, CH2Cl2), and acetonitrile (ACN, C2H3N) were purchased along with Optima LC/MS grade ACN, water, and formic acid from Fisher Scientific (Pittsburgh, PA, USA). The 8-plex iTRAQ reagents were provided by AB SCIEX (Foster City, CA. USA) while TMT0 was provided by Thermo Pierce (Rockford, IL, USA).
2.2 Synthesis of Isotopic N,N-Dimethylated Leucine (iDiLeu)
Dimethylation (Scheme 1) was accomplished by suspending either L-leucine or isotopic L-leucine (L-leucine-1-13C, 15 N, and L-leucine-1-13C) in H2O or D2O with a 2.5 molar excess of sodium cyanoborohydride (NaBH3CN) or sodium cyanoborodeuteride (NaBD3CN). The reaction vial was sealed and placed in an ice-water bath. Light (CH2O, 37% w/w) or heavy formaldehyde (CD2O and 13CD2O, 20% w/w) was then added at a 2.5 molar excess, and the reaction mixture was stirred for 30 min. Dimethylation progress was monitored by ninhydrin staining on a thin-layer chromatography (TLC) plate. Dimethyl leucines were purified by flash column chromatography (DCM/MeOH) and dried with a Büchi RE 111 Rotovapor (Flawil, Switzerland).

Synthetic steps required to produce each iDiLeu reagent
Caution: Formaldehyde and sodium cyanoborohydride are toxic by inhalation, in contact with skin, or if swallowed, and may cause cancer and heritable genetic damage. These chemicals and reactions must be handled in a fume hood.
2.3 18O Exchange
The d12 label requires an 18O exchange (Scheme 1) before formaldehyde dimethylation. L-leucine-1-13C, 15 N was dissolved in 1 N HCl H2 18O solution (pH 1) and stirred at 65°C for 4 h. After HCl evaporation, leftover acid was removed with StratoSpheres PL-HCO3 MP resin (Agilent Technologies, Santa Clara, CA, USA).
2.4 iDiLeu Activation to Triazine Ester Form
One milligram of each iDiLeu label was dissolved in 50 μL of anhydrous DMF and combined in a 0.9× molar ratio with DMTMM and NMM. Activation occurred at room temperature by vortexing the reaction mixture for 1 h. Peptide labeling directly after iDiLeu activation produces optimal results.
2.5 Protein Reduction, Alkylation, and Digestion
Saccharomyces cerevisiae whole-protein extract provided by Promega (Madison, WI, USA), mouse urine proteins provided by the Marker lab, and BSA were digested separately using the same method. Cysteine residues were reduced by adding 5 mM DTT and incubating for 1 h at 37°C. Free thiol groups were alkylated in the presence of 15 mM IAA for 15 min in the dark at room temperature. The alkylation reaction was quenched by 5 mM DTT. Each protein sample was diluted with 50 mM Tris HCl (pH 8) to reduce the urea concentration to ≤1 M before adding enough trypsin required for a 50:1 protein:enzyme ratio. Proteins were digested for 16 h in a 37°C water bath. Digests were quenched by acidifying to pH <3 using TFA, desalted with either SepPak C18 SPE cartridges (Waters, Milford, MA, USA) or C18 OMIX pipette tips (Agilent Technologies), and dried down by vacuum centrifugation with a Savant SC 110 SpeedVac concentrator (Thermo Scientific, Waltham, MA, USA). Peptides were dissolved in 0.5 M TEAB before labeling.
2.6 Peptide Labeling
Bovine serum albumin tryptic peptide and AST-I aliquots were separately labeled in triplicate with a 10× w/w excess of iDiLeu, whereas yeast tryptic peptides were labeled with a 4× w/w excess of iDiLeu. Anhydrous DMF was added to the reaction mixture until the organic:aqueous ratio was ~75%. Yeast tryptic peptides were also labeled with TMT and iTRAQ according to the manufacturer’s protocols. After vortexing the reactions for 2 h, labeling was quenched by adding hydroxylamine to a concentration of 0.25% the total sample volume. Labeled BSA aliquots were combined in ratios of 1:1:1:1:1, 10:0:5:1:10, and 1:2:5:8:10, whereas AST-I aliquots were combined in ratios of 1:3:7:10 and 1:10:50:100. Combined AST-I samples were then serially diluted to concentration ranges of 0.1–1000 fmol (amount injected onto column) and combined with mouse urine tryptic peptide background already containing the fifth labeled AST-I standard. All labeled samples were then dried using a SpeedVac.
2.7 Strong Cation Exchange
Labeled BSA peptides were resuspended in strong cation exchange (SCX) resuspension buffer (Protea Biosciences, Morgantown, WV, USA). Residual iDiLeu reaction byproducts were removed from each digest using SCX spintips according to the manufacturer’s instructions. Yeast peptides labeled with iDiLeu, iTRAQ, and TMT0 were dissolved in 10 mM KH2PO4, 20% ACN (v/v), pH 3. Peptides were fractionated with SCX liquid chromatography using a polySULFOETHYL A 200 mm × 2.1 mm, 5 μm, 300 Å column (PolyLC, Columbia, MD, USA). Buffer A consisted of 10 mM KH2PO4, 20% ACN (v/v), pH 3, and buffer B was 10 mM KH2PO4, 500 mM KCl, 20% ACN (v/v), pH 3. After peptide loading, buffer B was increased from 3% to 48% in 90 min at a flow rate of 0.2 mL/min. Fractions were collected every 1.5 min and combined into eight vials determined by the UV-Vis trace at a wavelength of 215 nm. All samples were then dried using a SpeedVac before another desalting step.
2.8 LC-MS and LC-MS2 Acquisition
Labeled BSA, AST-I, and yeast peptides were dissolved in 0.1% formic acid(aq) (FA) (v/v) and separated using a Waters nanoAcquity UPLC system before introduction into either a Thermo Q-Exactive Orbitrap or Orbitrap Elite. Mobile phase A was 0.1% FA, and mobile phase B was ACN in 0.1% FA. The BSA and AST-I peptides were injected and loaded onto a Symmetry C18 nanoAcquity trap column (180 μm × 20 mm, 5 μm) in 99% A at a flow rate of 5 μL/min for 5 min. Peptides were separated on a 1.7 μm BEH C18 column (75 μm × 100 mm, 130 Å) at a flow rate of 300 nL/min with a 30 min gradient ramping from 3% to 35% B. Precursor quantification experiments were performed in profile mode using full MS scans ranging from m/z 380 to 2000 at a resolution of 140 K. The automatic gain control (AGC) target was set to 1 × 106, while the maximum injection time (IT) was 100 ms. Identification of labeled BSA peptides occurred by selecting the top 10 precursor ions from m/z 380 to 2000 and then placing those ions on a 25 s exclusion list. Precursor scan resolution was 70 K with an AGC of 1 × 106, a maximum IT of 100 ms, and an isolation window of 2.0 Th. Tandem mass spectrometry parameters on the Q-Exactive included a resolution of 17.5 K, an AGC target of 1 × 105, a maximum IT of 150 ms, an NCE of 28, and a fixed first mass at m/z 110. Multiplexed (MSX) targeted MS2 (t-MS2) experiments were acquired at a resolution of 17.5 K, an AGC target of 1 × 105, a maximum IT of 200 ms, an MSX count of five, isolation windows of 0.5–2.0 Th, and an NCE of 28.
Yeast digest peptides were injected in triplicate and loaded onto a column fabricated with an integrated emitter as previously described [26]. The 15 cm, 75 μm i.d. column was packed with 1.7 μm 150 Å, BEH C18 material extracted from a Waters UPLC column (part no. 186004661). Peptides were loaded onto this column in 100% A (0.1% FA(aq)) for 14.29 min at a flow rate of 0.350 μL/min, which was also the flow rate for the entire LC-MS run. Solvent B (ACN, 0.1% FA) was increased from 0% to 5% over 0.1 min, and then to 30% over the next 80 min. Normalized collision energy ramp tests were conducted by operating the Orbitrap Elite at an MS resolution of 30 K, MS2 resolution of 15 K, and an isolation width of 2 Th. The most abundant ion from m/z 380 to 1800 in each cycle was isolated 10 times to be fragmented with normalized HCD energies of 5, 10, 15, 20, 25, 30, 35, 40, 45, and 50.
2.9 Data Analysis and Isotopic Interference Corrections
Identification of BSA tryptic peptides was performed with Proteome Discoverer (1.4.0288, Thermo Scientific). Raw files were searched against the Bos taurus proteome database from Uniprot using the Sequest HT algorithm. Peptides produced by a tryptic digest with a maximum of two missed cleavages were matched using precursor and fragment mass tolerances of 50 ppm and 0.02 Da, respectively. iDiLeu labeling of N-termini and lysine residues (+141.1154, +144.1313, +147.1409, +150.1631, and +153.1644 Da) and carbamidomethylation of cysteine residues (+57.0215 Da) were chosen as static modifications. Methionine oxidation (+15.9949 Da) was chosen as a dynamic modification.
The Coon OMSSA (Open Mass Spectrometry Search Algorithm) Proteomic Analysis Software Suite [27] (COMPASS) was used for identification and HCD energy comparisons of iDiLeu, TMT0, and iTRAQ labeled yeast peptides. The Uniprot reference database for Saccharomyces cerevisiae was searched using OMSSA with the enzyme trypsin and two missed cleavages allowed. Precursor and fragment mass tolerances of 50 ppm and 0.01 Da were used with static modifications accounting for cysteine carbamidomethylation and N-terminus/lysine incorporation of iDiLeu (+141.1154 Da), TMT0 (+224.1525 Da), or iTRAQ (+304.1990 Da). Methionine oxidation was once again chosen as a dynamic modification. Tandem mass scans were grouped by activation energy and the unique peptide false discovery rate was set to ≤1%.
Raw quantitative data was processed manually in Thermo Xcalibur 2.2 using either peak areas generated by the Genesis peak detection algorithm or ion intensities. A series of correction factors were applied in Microsoft Excel using a script developed in PTC Mathcad 14 (Needham, MA, USA) to rectify raw quantitative values from both precursor and fragment data using a previously reported method [28]. Calibration curves and box plots were constructed in Microsoft Excel and Graphpad Prism (v. 6.0) software.
3 Results and Discussion
3.1 iDiLeu Reagent Synthesis
Mass difference reagents differ from their isobaric counterparts by introducing unique mass additions to the labeled peptide’s mass. The iDiLeu tags each consist of an isotopic variant of N,N-dimethyl leucine and exploit the amine reactivity of the triazine ester to selectively label the N-terminus and lysine side chains of a peptide. The choice of a triazine ester amine-reactive group over NHS ester has been previously discussed but is primarily due to its high reactivity with dimethylated leucine, resulting in a rapid activation time of 1 h [25]. iDiLeu labels were synthesized and activated in two to three steps. Dimethylation of light and heavy leucine gave yields of 85%–90% whereas the 18O-exchange rate was nearly quantitative due to the high excess of H2 18O to leucine. iDiLeu can be activated to the amine-reactive form in 1 h with DMTMM/NMM in anhydrous DMF. Although light and heavy dimethylated leucines have been reported to maintain stability for years when kept in dry conditions, the amine-reactive reagent should be used immediately upon activation for highest labeling efficiency (>99.99%) and accuracy [25]. All isotopic leucine variants and other reagents involved in iDiLeu synthesis are commercially available, and the cost of a 5-plex labeling with 100 μg of a peptide standard or digest is under $5.00, which represents a significant cost advantage over previously discussed absolute quantification experiments.
Mass additions of 141.1, 144.1, 147.1, 150.2, and 153.2 Da are efficiently incorporated [25] into peptides by the d0, d3, d6, d9, and d12 labels (Scheme 2). Each label adds a relatively modest mass and modifies the peptide sequence by only one or two dimethyl leucine residues. iDiLeu labels are comparatively light to minimize drastic elevations in peptide hydrophobicity that negatively impact the chromatographic separation and number of peptide identifications in an LC-MS2 experiment. For instance, it has been shown that the largest number of peptides was identified using the lighter 4-plex iTRAQ in comparison to 6-plex TMT (229.1629 Da) and 8-plex iTRAQ (304.1990 Da) [29].

Peptide labeling and mixing with iDiLeu to produce a standard curve based on peak areas
3.2 Chromatographic Behavior of iDiLeu-Labeled Peptides
A common misconception involving chemical labels is that deuterium incorporation, regardless of location, will shift retention times of peptides labeled with deuterium-based reagents far enough from those labeled with hydrogen-based reagents to render quantification unreliable. Methods to control deuterium effects have been published since 2002 and include grouping deuterium atoms around polar functional groups, minimizing the number of deuterium substituents, and reducing the number of derivatization sites [30]. In another study, it was revealed that the bias in ratio calculation caused by retention time differences between H2- and D2-formaldehyde labeled peptides is only about 3% [31]. iDiLeu and DiLeu labels group deuterium around the more polar amine group of leucine. According to solvophobic theory, the probability of deuterium interacting with a column’s stationary phase is reduced by placing it next to a hydrophilic group that has little to no affinity to the stationary phase [30]. This theory is corroborated by our own iDiLeu labeling results. Scheme 1 shows that the structures of the 5-plex iDiLeu reagents incorporate from zero to six deuterium atoms at the dimethylated amine, and Figure 1 is a chromatogram of iDiLeu-labeled BSA peptides combined in ratios of 1:2:5:8:10. Visual inspection of the nine randomly chosen peptides from the chromatogram reveals that no appreciable retention time differences exist between peptides labeled by d0 (0 2H) and d12 (6 2H). Since iDiLeu quantification values consist of either the area under entire peaks or intensity sums, it is unlikely that miniscule retention time differences could impact the iDiLeu method’s accuracy. When the higher cost of 13C and 15 N isotopes is taken into account, strategic 2H positioning becomes the preferred method of heavy isotope incorporation.

iDiLeu-labeled BSA in ratios of 1:2:5:8:10. Nine peptides were randomly selected to show that retention times are not significantly shifted from one another because of 2H incorporation. Strategic placement of 2H around polar functional groups reduced the probability of them interacting with C18 stationary phases. Retention time differences between d0- and d12-labeled peptides are displayed above each selected peptide extracted ion chromatogram
3.3 HCD Collision Energy Comparisons Between iDiLeu, iTRAQ, and TMT
As previously stated, mass difference and isobaric labels must be designed so that their incorporation onto peptides does not hinder chromatographic separation or peptide spectral matches (PSMs). We compared the fragmentation and number of PSMs generated by HCD fragmentation of yeast tryptic peptides labeled by iDiLeu, 8-plex iTRAQ, and 6-plex TMT by using an Orbitrap Elite to isolate the most abundant precursor ions 10 times, acquiring MS2 spectra at HCD normalized collision energies of 5, 10, 15, 20, 25, 30, 35, 40, 45, and 50. The maximum number of target PSMs, although significantly lower for iTRAQ labeled peptides (2549), was comparable for both iDiLeu and TMT labeled peptides (3492 and 3661), as displayed in Figure 2a–c. The optimal number of target PSMs after TMT and iTRAQ labeling required a higher normalized HCD collision energy than what is typical for peptide fragmentation. In contrast, iDiLeu labeling performed better using slightly reduced collision energies and an adequate range of collision energies for acceptable numbers of target PSMs. The difference in collision energies recommended for use with iDiLeu and iTRAQ or TMT is represented by the MS2 spectra of the tryptic peptide, ADVLTAFLNK (Supplemental Figure 1). At a normalized energy of 25, the iDiLeu-labeled peptide fragmented into a dimethylated leucine immonium ion and a multitude of b- and y-ions useful for identification. Neither TMT- nor iTRAQ-labeled peptides produced adequate fragmentation at this lower HCD energy. iDiLeu suffered from secondary fragmentation at the higher collision energy of 35, resulting in mostly dimethyl immonium ion and fewer peptide backbone fragments. In contrast, TMT and iTRAQ showed complete precursor fragmentation, intense reporter ion production, and more b- and y-ions at this higher HCD energy. The different range for optimal normalized collision energies for iDiLeu (25–30) in comparison to the two commercial tags (30–35) may stem from the fact that iDiLeu-labeled peptides are structurally similar to normal peptides, only differing by an added dimethylated leucine and/or labeled lysine.

Number of target PSMs identified from eight fractions of iDiLeu- (a), TMT0- (b), and 8-plex iTRAQ- (c) labeled yeast tryptic peptides according to NCE. Although TMT and iTRAQ incorporation elevates the HCD energy required to generate sufficient fragmentation for identification, most IDs using iDiLeu are found at an NCE of 25
3.4 iDiLeu Quantification and Correction Factors
iDiLeu-tagged peptides differ from one another by 3, 6, 9, and 12 Da. A peptide of charge +2 that is labeled by 5-plex iDiLeu solely at its N-terminus will, therefore, produce five monoisotopic precursor peaks shifted from one another by 1.5 m/z. Peptides with multiple labeling sites enjoy little isotopic envelope interference in their monoisotopic signals, but those with only one iDiLeu modification experience varying amounts of interference. Examples of interference in peptides with charges of +2 and +3 and one or two attached iDiLeu labels are shown by Supplemental Figure 2. The use of algorithms to correct for isotopic interference in mass difference labels has been reported previously [19], and our lab has already implemented the i-Tracker method for reporter ion corrections into our DiLeu quantitative workflow [26, 30]. These corrections were extended to iDiLeu-labeled peptides to account for interferences and variations in the isotopic distribution of a peptide resulting from incorporation of distinct isotopic labels (Supplemental Figure 2). Using the i-Tracker method, intensity values from the isotopic distribution of a precursor peptide were added to the monoisotopic peak’s raw signal and interfering peaks were subtracted from it [28]. Construction of an equation for each labeled peptide results in the following system of equations:
The S variable is the uncorrected signal of a monoisotopic peptide peak while x, y, and z are fractional contributions of a peptide’s total intensity, I, and represent the percentages of a peptide’s distribution existing as a monoisotopic peak, interference with the heavier mass-labeled peptide, or interference with the lighter mass-labeled peptide, respectively. The equations show that the raw signal of each peptide is a sum of its monoisotopic peak intensity and interferences from neighboring peptides. The system of equations was symbolically rearranged to solve for I using MathCad and transferred to Excel to process data more efficiently. Transformed equations are shown in Supplemental Figure 3. Raw signal values are obtained in any iDiLeu experiment, but the fractional contributions must be experimentally determined for each peptide of interest by labeling digest aliquots with each reagent and running the labeled BSA samples separately. Each labeled peptide requires a tailored correction factor at the MS level because its isotopic distribution is unique due to charge, number of amino acids, amino acid combination, and number of times labeled.
iDiLeu’s quantitative performance was first characterized by labeling tryptic BSA peptides in triplicate at molar ratios of 1:1:1:1:1 and analyzing the precursor ratios, normalized to d0, with LC-MS. Across 35 peptides (nine runs, 315 values), the median uncorrected ratio values were 1.00:1.00:1.10:0.85:0.90 compared with the median corrected values of 1.00:1.03:1.06:0.85:1.06. Altough both datasets show acceptable relative quantification accuracy under 15% error, the d12:d0 channel ratio is substantially higher after correction. Another quantitative test considered the channels that typically experience the most interference (d6, d9, and d12) at ratios of 5:1:10 and normalized these values to d0-labeled peptide intensities. The importance of correction factors was highlighted in this data set, showing uncorrected medians of 5.48:0.91:7.37 versus corrected medians of 5.32:0.87:8.56. In this case, the d12-tagged channel’s median error was 26.3% in the uncorrected data, but the curated ratios were once again all within 15% error. Channel d9 appears to be more accurate in the uncorrected data, but interference peaks from d6 and d12 contribute to the d9 monoisotopic peak intensity, artificially raising its normalized value before correction. Peptides labeled with d12 have much lower precursor intensities before correction because their distributions consist of a lower fraction of monoisotopic ion and an isotopic envelope that extend to m/z values below the peptide’s exact mass (Supplemental Figure 2). Figure 3a and b display Tukey boxplots constructed from the log2-transformed precursor ratios discussed above. Each ratio is normalized to the d0 channel, representing combinations at 1:1:1:1:1 (Figure 3a) and 10:0:5:1:10 (Figure 3b). Ratio values from 35 peptides labeled and injected in triplicate constitute each plot. Medians show high accuracy (<15% error) and high precision, corroborating previous data showing that ratio compression is nonexistent when quantification is based on precursor values [23]. High-resolution and accurate mass (HR-AM) mass spectrometers like the Q-Exactive Orbitrap instrument used for this work ensure an extra level of selectivity for accurate relative or absolute quantification when an MS scan resolution of 140 K is selected.

Tukey boxplots of log2-transformed fold changes for iDiLeu-labeled BSA tryptic peptides at ratios of 1:1:1:1:1 (a), (c) and 1:0:5:1:10 (b), (d). Precursor quantitation from triplicate injections was used for relative quantification in (a) and (b). A 5-plex t-MS2 method targeted 24 BSA peptides over 1 Th isolation windows from the most accurate and precise labeling replicate (c), (d). Ratios were compressed in (c) and (d) because of significant co-isolation of interference ions along with targeted precursors. Reporter ion channel ratios acquired from the t-MS2 method were plotted versus precursor channel ratios in (e). The t-MS2 method suffered from higher variability and hindered quantification by compressing ratios
Targeted-MS2 (t-MS2) applications, such as parallel reaction monitoring (PRM) [32] using quadrupole-equipped HR-AM mass spectrometers like the Q-Exactive or Orbitrap Fusion, have gained popularity in the last 2 y [33–37]. Multiplexed t-MS2 methods accumulate product ions from selected precursor masses before transferring these ions to the Orbitrap mass analyzer. iDiLeu reagents can be fragmented by CID or HCD to form dimethylated immonium reporter ions distinguished by m/z values of 114.1, 116.1, 119.1, 122.2, and 123.2. Equations accounting for reporter ion interferences were implemented for each dimethylated immonium ion using purity-correction calculations shown in Supplemental Figure 4. We extended a multiplexed (5-plex) t-MS2 method to target 24 random iDiLeu-labeled BSA tryptic peptides placed on an inclusion list and compared quantitative results using reporter ions versus precursor ions. Figure 3c and d show Tukey boxplots derived from theoretical log2-transformed reporter ion ratios of 1:1:1:1:1 and 10:0:5:1:10. At ratios of 1:1:1:1:1 (Figure 3c), reporter ion quantification appeared to be accurate but less precise than the precursor example. Difficulties arose when labeled peptides at ratios of 10:0:5:1:10 (Figure 3d) were analyzed. Multiple 5-plex clusters of iDiLeu-labeled peptides complicate spectra considerably, suggesting that co-isolation of different peptides in t-MS2 analyses is common. In fact, about half of the chosen peptides were isolated with interfering peptide clusters, consequentially yielding significant ratio compression among peptide channels. Techniques to minimize or eliminate isobaric co-isolation exist but were not explored because of instrumental and practical considerations [38–41]. When reporter ion channel ratios were plotted according to precursor channel ratios (Figure 3e), it was found that reporter ion quantification is less accurate and less precise compared with precursor quantification values. An example of t-MS2 co-isolation is provided by Figure 4a and b. In Figure 4a, precursor ions of equal ratios were fragmented using five isolation widths of 1.6 Th. Significant co-isolation of a d9- and d12-labeled +2 peptide skewed reporter ion intensities and adversely affected quantitative accuracy (Figure 4b). Various instances of co-isolation increase the variability in iDiLeu quantification and compress peptide ratios. Peptide co-isolation can also hamper identification and quantification from b- and y-ion intensities by either complicating spectra with extra fragments or completely masking peaks of interest. The b- and y-ion clusters in Figure 4c were undetected at an isolation width of 1.6 Th but were revealed after lowering the window to fragment fewer +2 interference peaks. In this case, lowering the isolation window to 0.5 Th produced fragment ion clusters in Figure 4c that can be used for peptide quantification in the absence of HR-AM mass spectrometers.

Example of an iDiLeu-labeled +2 peptide cluster interfering with the isolation of an iDiLeu-labeled +3 peptide cluster. In (a), t-MS2 method using an isolation window of 1.6 Th selected five iDiLeu-labeled BSA peptides along with two co-isolated interference peptides. The MS2 spectrum in (b) shows co-isolated +2 peptides had a substantially negative effect on iDiLeu reporter ion quantification, increasing the d9 and d12 channels greatly in comparison to d0, d3, and d6. Fragments of interfering +2 peptides rendered quantification using b- and y-ions impossible until the isolation window was lowered. The MS2 spectrum in (c) was acquired with an isolation width of 0.5 Th, allowing for quantification using b-ion fragments or sometimes y-ion fragments if a C-terminal lysine is present
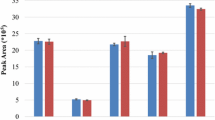
Previously, we discussed how a digest of interest should be labeled with each iDiLeu reagent and run separately to determine unique correction factors for each peptide. These labeled digests can then be combined into ratios of increasing concentration to determine the most linear and accurate peptide surrogates for protein quantification. Figure 5a plots actual iDiLeu-labeled BSA channel ratios (1:2:5:8:10) normalized to d0 against the theoretical ratios injected onto the column. The linear relationship is characterized by a slope close to unity and a small y-intercept. Three peptides were chosen in Figure 5b to represent BSA abundance, CASIQK, AWSVAR, and GACLLPK, based on their linearity (R2 = ~0.990) and accuracy at each channel (<15% error). In an absolute quantification study, peptides can be chosen experimentally to represent all candidate proteins based on their accuracy, variability, and linearity across a range determined by the researcher.

Representation of peptide selection from a BSA digest with channel ratios of 1:2:5:8:10. Plotting the actual iDiLeu channel ratios against the theoretical iDiLeu ratios revealed a linear response comprising all identified BSA peptides (a). Three peptides were chosen as surrogate peptides to represent BSA concentrations in (b). Each of these peptides was characterized by signal response versus concentration being close to unity and high linearity
3.5 Allatostatin I Quantification in Mouse Urine Peptide Matrix
We envision two methods of absolute peptide/protein quantification using iDiLeu (Supplemental Figure 5). The first technique requires labeling a peptide standard with all five iDiLeu reagents. Four labeled standards are then serially diluted to construct a standard curve across an order of magnitude. These peptides can then be further diluted into aliquots to span a wider concentration range. The fifth labeled peptide is added at a constant concentration to each aliquot, acting as an internal standard for normalization across multiple runs. Each concentration range aliquot is then added to an unlabeled digest before any sample cleanup steps occur. Once a response curve has been constructed across multiple LC-MS runs, the labeled internal standard peptide can be added to a protein digest modified by one iDiLeu reagent. After this sample has been run, peptide concentration can be determined from the normalized peak area according to the pregenerated calibration curve. We demonstrated this concept’s proof-of-principle by preparing a solution of AST-I labeled with d0, d3, d6, and d9 mixed to deliver amounts of 100, 300, 700, and 1000 fmol on-column. Labeling efficiency of AST-I was determined to be > 99.7% across iDiLeu reactions, meaning that inefficient or unequal labeling of peptides was not a significant source of error in this experiment. This stock solution was serially diluted twice for an overall concentration range of 1–1000 fmol and combined with 100 fmol of d12-labeled peptide before being spiked into mouse urine tryptic peptide background. Allatostatin-I modified with d0 was spiked into mouse urine peptide matrix at on-column amounts of 8.00, 25.0, and 500 fmol and combined with 100 fmol of d12-labeled AST-I. After three LC-MS runs, a 12-point calibration curve was built from 1–1000 fmol based on precursor peak areas and normalized to d12 (Figure 6a). The resulting curve showed high linearity (R2 = 0.998) and small variation from each injection. From this curve, peptide amounts from triplicate injections of d0-labeled AST-I were back-calculated and displayed in Table 1. This method was found to be quite accurate, showing errors less than 19% (with average error at ~7%), and demonstrating comparable precision between injected amounts with RSDs under 11%.

Calibration curves constructed from three injections of three AST-I peptide labeling mixtures (n = 9 total) spiked into mouse urine peptide background. The curve in (a) was constructed from 1–1000 fmol (amount injected on column), utilizing the d12 channel as an internal standard to normalize precursor signal responses in ratios of (1:3:7:10) across three LC-MS runs. Peak areas in (b) at ratios of (1:10:50:100) were used to make response curves from 0.1–10, 1–100, and 10–1000 fmol (amount injected on column). The d0 channel was spiked into mouse urine peptides along with each calibration curve solution in order to quantify a predetermined concentration of AST-I in a single run
The second quantification method consists of making single calibration curve solutions of iDiLeu-labeled peptide standards spanning two orders of magnitude and spiking that mixture into a prelabeled digest. We demonstrated this technique by labeling an AST-I standard with the d3, d6, d9, and d12 labels and combining the labeled peptides so that their quantities on column were 10, 100, 500, and 1000 fmol. This solution was diluted twice to aliquots of 1–100 fmol and 0.1–10 fmol. Labeled AST-I (d0) was separately combined with mouse urine peptide aliquots in the following amounts: 0.250, 2.50, 7.50, 25.0, 75.0, 250, and 750 fmol. Calibration curve solutions were then matched with mouse urine digests containing AST-I concentrations within their range so that curve generation and quantification were accomplished in one run. Figure 6b shows the resulting AST-I response curves normalized to the d12 channel. Each range shows high linearity and responses close to unity, but variability increases somewhat in the 0.1–1 fmol range. Femtomole amounts below 0.1 fmol ceased to follow a linear trend, meaning the experimental linear range for AST-I quantification is from ~0.1 fmol to >1000 fmol. The iDiLeu method’s linear range, limit of quantification, and limit of detection will vary depending on each peptide’s ionization efficiency because of factors like hydrophobicity and basicity. Table 1 shows calculated amounts of AST-I from the “one-run” quantification method. The accuracy at 0.250 fmol, 83.7%, was 8.8% lower than the least accurate AST-I measurement from 1–1000 fmol, 92.5%. These results suggest that AST-I measurements with the highest accuracy occur between 1 and 1000 fmol. Both iDiLeu methods are viable quantification methods, but the “one-run” method is more accurate with higher throughput. The higher accuracy of this method in comparison to the first is most likely due to the fact that the response curve and peptide surrogate are analyzed in one LC-MS run, eliminating errors caused by building a standard curve from multiple acquisitions.
4 Conclusions
Mass spectrometry’s role in the absolute quantification of candidate proteins has increased due to its sensitivity, throughput, and avoidance of antibody development. Approaches with LC-MRM AQUA assays and mTRAQ quantification have substantially increased the confidence in LC-MS quantification, but their associated costs may limit the number of experiments or candidate proteins analyzed. Our iDiLeu reagent is a cost-effective alternative solution to targeted quantification. The label efficiently couples to peptides and increases the throughput of targeted quantification assays by allowing a four-point calibration curve to be constructed in one LC-MS run. Characterization of iDiLeu revealed the following: (1) incorporated 2H atoms do not skew retention times of labeled peptides; (2) efficient fragmentation for characterization and quantification is achieved at lower NCE values; (3) while peptide fragment ions can be selected for quantification, precursor quantification of iDiLeu-labeled peptides with HR-AM mass spectrometers is selective, accurate and precise over a concentration range of 1–1000 fmol, outperforming similar experiments using iDiLeu reporter ions from a t-MS2 method. iDiLeu’s low cost, ease of production, and accurate performance should foster its use in proteomics and peptidomics workflows. Our lab is currently implementing protein quantification strategies where relative quantification with DiLeu is used in candidate protein biomarker discovery, and targets are validated using the iDiLeu technique.
References
Huttenhain, R., Malmstrom, J., Picotti, P., Aebersold, R.: Perspectives of targeted mass spectrometry for protein biomarker verification. Curr. Opin. Chem. Biol. 13, 518–525 (2009)
Makawita, S., Diamandis, E.P.: The bottleneck in the cancer biomarker pipeline and protein quantification through mass spectrometry-based approaches: current strategies for candidate verification. Clin. Chem. 56, 212–222 (2010)
Rifai, N., Gillette, M.A., Carr, S.A.: Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat. Biotechnol. 24, 971–983 (2006)
Addona, T.A., Shi, X., Keshishian, H., Mani, D.R., Burgess, M., Gillette, M.A., Clauser, K.R., Shen, D., Lewis, G.D., Farrell, L.A., Fifer, M.A., Sabatine, M.S., Gerszten, R.E., Carr, S.A.: A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat. Biotechnol. 29, 635–643 (2011)
Barr, J.R., Maggio, V.L., Patterson, D.G., Cooper, G.R., Henderson, L.O., Turner, W.E., Smith, S.J., Hannon, W.H., Needham, L.L., Sampson, E.J.: Isotope dilution-mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin. Chem. 42, 1676–1682 (1996)
Gerber, S.A., Rush, J., Stemman, O., Kirschner, M.W., Gygi, S.P.: Absolute quantification of proteins and phosphoproteins from cell lysates by tandem MS. Proc. Natl. Acad. Sci. U. S. A. 100, 6940–6945 (2003)
Kirkpatrick, D.S., Gerber, S.A., Gygi, S.P.: The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods 35, 265–273 (2005)
Zhao, Y., Brasier, A.R.: Applications of selected reaction monitoring (SRM)-mass spectrometry (MS) for quantitative measurement of signaling pathways. Methods 61, 313–322 (2013)
Warnken, U., Schleich, K., Schnölzer, M., Lavrik, I.: Quantification of high-molecular weight protein platforms by AQUA mass spectrometry as exemplified for the CD95 death-inducing signaling complex (DISC). Cells 2, 476–495 (2013)
Wildsmith, K.R., Schauer, S.P., Smith, A.M., Arnott, D., Zhu, Y., Haznedar, J., Kaur, S., Mathews, W.R., Honigberg, L.A.: Identification of longitudinally dynamic biomarkers in alzheimer’s disease cerebrospinal fluid by targeted proteomics. Mol. Neurodegener. 9, 22 (2014)
Kuzyk, M.A., Smith, D., Yang, J., Cross, T.J., Jackson, A.M., Hardie, D.B., Anderson, N.L., Borchers, C.H.: Multiple reaction monitoring-based, multiplexed, absolute quantitation of 45 proteins in human plasma. Mol. Cell. Proteomics 8, 1860–1877 (2009)
Sturm, R., Sheynkman, G., Booth, C., Smith, L.M., Pedersen, J.A., Li, L.: Absolute quantification of prion protein (90-231) using stable isotope-labeled chymotryptic peptide standards in a LC-MRM AQUA workflow. J. Am. Soc. Mass Spectrom. 23, 1522–1533 (2012)
Robinson, R.A.S., Evans, A.R.: Enhanced sample multiplexing for nitrotyrosine-modified proteins using combined precursor isotopic labeling and isobaric tagging. Anal. Chem. 84, 4677–4686 (2012)
DeSouza, L.V., Taylor, A.M., Li, W., Minkoff, M.S., Romaschin, A.D., Colgan, T.J., Siu, K.W.M.: Multiple reaction monitoring of mTRAQ-labeled peptides enables absolute quantification of endogenous levels of a potential cancer marker in cancerous and normal endometrial tissues. J. Proteome Res. 7, 3525–3534 (2008)
DeSouza, L.V., Romaschin, A.D., Colgan, T.J., Siu, K.W.M.: Absolute quantification of potential cancer markers in clinical tissue homogenates using multiple reaction monitoring on a hybrid triple quadrupole/linear ion trap tandem mass spectrometer. Anal. Chem. 81, 3462–3470 (2009)
Zhou, L., Wei, R., Zhao, P., Koh, S.K., Beuerman, R.W., Ding, C.: Proteomic analysis revealed the altered tear protein profile in a rabbit model of Sjögren’s syndrome-associated dry eye. Proteomics 13, 2469–2481 (2013)
Zhang, S., Wen, B., Zhou, B., Yang, L., Cha, C., Xu, S., Qiu, X., Wang, Q., Sun, H., Lou, X., Zi, J., Zhang, Y., Lin, L., Liu, S.: Quantitative analysis of the human AKR family members in cancer cell lines using the mTRAQ/MRM approach. J. Proteome Res. 12, 2022–2033 (2013)
Yin, H., Zhang, L., Xie, L., Huang, L.: Hyperplex-MRM: a hybrid multiple reaction monitoring method using mTRAQ/iTRAQ labeling for multiplex absolute quantification of human colorectal cancer. J. Proteome Res. 12, 3912–3919 (2013)
Yoon, J.Y., Yeom, J., Lee, H., Kim, K., Na, S., Park, K., Paek, E., Lee, C.: High-throughput peptide quantification using mTRAQ reagent triplex. BMC Bioinformatics 12(Suppl 1), S46 (2011)
DeSouza, L.V., Krakovska, O., Darfler, M.M., Krizman, D.B., Romaschin, A.D., Colgan, T.J., Siu, K.W.M.: mTRAQ-based quantification of potential endometrial carcinoma biomarkers from archived formalin-fixed paraffin-embedded tissues. Proteomics 10, 3108–3116 (2010)
Hwang, C.Y., Kim, K., Choi, J.Y., Bahn, Y.J., Lee, S.-M., Kim, Y.K., Lee, C., Kwon, K.-S.: Quantitative proteome analysis of age-related changes in mouse gastrocnemius muscle using mTRAQ. Proteomics 14, 121–132 (2014)
Carneiro, L.G., Nouh, H., Salih, E.: Quantitative gingival crevicular fluid proteome in health and periodontal disease using stable isotope chemistries and mass spectrometry. J. Clin. Periodontol. 41, 733–747 (2014)
Mertins, P., Udeshi, N.D., Clauser, K.R., Mani, D.R., Patel, J., Ong, S., Jaffe, J.D., Carr, S.A.: iTRAQ labeling is superior to mTRAQ for quantitative global proteomics and phosphoproteomics. Mol. Cell. Proteomics 11, M111.014423, 1–12 (2012)
Wu, Y., Wang, F., Liu, Z., Qin, H., Song, C., Huang, J., Bian, Y., Wei, X., Dong, J., Zou, H.: Five-plex isotope dimethyl labeling for quantitative proteomics. Chem. Commun. (Camb.) 50, 1708–1710 (2014)
Xiang, F., Ye, H., Chen, R., Fu, Q., Li, L.: N,N-dimethyl leucines as novel isobaric tandem mass tags for quantitative proteomics and peptidomics. Anal. Chem. 82, 2817–2825 (2010)
Lietz, C.B., Yu, Q., Li, L.: Large-scale collision cross-section profiling on a traveling wave ion mobility mass spectrometer. J. Am. Soc. Mass Spectrom. (2014). doi:10.1007/s13361–014–0920–1
Wenger, C.D., Phanstiel, D.H., Lee, M.V., Bailey, D.J., Coon, J.J.: COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics 11, 1064–1074 (2011)
Shadforth, I.P., Dunkley, T.P.J., Lilley, K.S., Bessant, C.: I-Tracker: for quantitative proteomics using iTRAQ. BMC Genomics 6, 145 (2005)
Pichler, P., Köcher, T., Holzmann, J., Mazanek, M., Taus, T., Ammerer, G., Mechtler, K.: Peptide labeling with isobaric tags yields higher identification rates using iTRAQ 4-Plex compared to TMT 6-plex and iTRAQ 8-plex on LTQ Orbitrap. Anal. Chem. 82, 6549–6558 (2010)
Zhang, R., Sioma, C.S., Thompson, R.A., Xiong, L., Regnier, F.E.: Controlling deuterium isotope effects in comparative proteomics. Anal. Chem. 74, 3662–3669 (2002)
Boutilier, J.M., Warden, H., Doucette, A.A., Wentzell, P.D.: Chromatographic behavior of peptides following dimethylation with H2/D2-formaldehyde: implications for comparative proteomics. J. Chromatogr. B. 908, 59–66 (2012)
Peterson, A.C., Russell, J.D., Bailey, D.J., Westphall, M.S., Coon, J.J.: Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 (2012)
Gallien, S., Duriez, E., Crone, C., Kellmann, M., Moehring, T., Domon, B.: Targeted proteomic quantification on quadrupole-Orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709–1723 (2012)
Tsuchiya, H., Tanaka, K., Saeki, Y.: The parallel reaction monitoring method contributes to a highly sensitive polyubiquitin chain quantification. Biochem. Biophys. Res. Commun. 436, 223–229 (2013)
Maerkens, A., Kley, R.A., Olive, M., Theis, V., van der Ven, P.F.M., Reimann, J., Milting, H., Schreiner, A., Uszkoreit, J., Eisenacher, M., Barkovits, K., Guettsches, A.K., Tonillo, J., Kuhlmann, K., Meyer, H.E., Schroeder, R., Tegenthoff, M., Fuerst, D.O., Mueller, T., Goldfarb, L.G., Vorgerd, M., Marcus, K.: Differential proteomic analysis of abnormal intramyoplasmic aggregates in desminopathy. J. Proteome 90, 14–27 (2013)
Zarth, A.T., Cheng, G., Zhang, Z., Wang, M., Villalta, P.W., Balbo, S., Hecht, S.S.: Analysis of the benzene oxide-DNA adduct 7-phenylguanine by liquid chromatography-nanoelectrospray ionization-high resolution tandem mass spectrometry-parallel reaction monitoring: application to DNA from exposed mice and humans. Chem. Biol. Interact. 215, 40–45 (2014)
Tang, H., Fang, H., Yin, E., Brasier, A.: Multiplexed parallel reaction monitoring (PRM) targets histone modifications on the Q-Exactive mass spectrometer. Anal. Chem. 86, 5526–5534 (2014)
Sturm, R.M., Lietz, C.B., Li, L.: Improved isobaric tandem mass tag quantification by ion mobility mass spectrometry. Rapid Commun. Mass Spectrom. 28, 1051–1060 (2014)
Ting, L., Rad, R., Gygi, S.P., Haas, W.: MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8, 937–940 (2011)
Wenger, C.D., Lee, M.V., Hebert, A.S., McAlister, G.C., Phanstiel, D.H., Westphall, M.S., Coon, J.J.: Gas-phase purification enables accurate, multiplexed proteome quantification with isobaric tagging. Nat. Methods 8, 933–935 (2011)
McAlister, G.C., Nusinow, D.P., Jedrychowski, M.P., Wühr, M., Huttlin, E.L., Erickson, B.K., Rad, R., Haas, W., Gygi, S.P.: MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 86, 7150–7158 (2014)
Acknowledgments
The authors acknowledge support for this work by the National Institutes of Health grant (1R01DK071801). The Q-Exactive Orbitrap and Orbitrap Elite instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531). C.B.L. acknowledges an NIH-supported Chemistry Biology Interface Training Program Predoctoral Fellowship grant (T32-GM008505) and an NSF Graduate Research Fellowship (DGE-1256259). L.L. acknowledges a H. I. Romnes Faculty Research Fellowship. The authors also acknowledge the generous gifts of yeast protein extracts from Promega (Madison, WI), mouse urine samples from the Paul Marker group at UW-Madison, and TMT0 reagent from Thermo Fisher (Rockford, IL).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 354 kb)
Rights and permissions
About this article

Cite this article
Greer, T., Lietz, C.B., Xiang, F. et al. Novel isotopic N,N-Dimethyl Leucine (iDiLeu) Reagents Enable Absolute Quantification of Peptides and Proteins Using a Standard Curve Approach. J. Am. Soc. Mass Spectrom. 26, 107–119 (2015). https://doi.org/10.1007/s13361-014-1012-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13361-014-1012-y