
Abstract
To improve the therapeutic activity of inhaled glucocorticoids and reduce potential side effects, we designed a formulation combining the advantages of nanoparticles, which have an enhanced uptake by alveolar cells, allow targeted delivery and sustained drug release, as well as limited drug systemic passage, with those of microparticles, which display good alveolar deposition. Herein, a polymer-drug conjugate, poly(malic acid)-budesonide (PMAB), was first synthesized with either 11, 20, 33, or 43 mol% budesonide (drug:polymer from 1:8 to 3:4), the drug creating hydrophobic domains. The obtained conjugates self-assemble into nanoconjugates in water, yielding excellent drug loading of up to 73 wt%, with 80–100 nm diameters. In vitro assays showed that budesonide could be steadily released from the nanoconjugates, and the anti-inflammatory activity was preserved, as evidenced by reduced cytokine production in LPS-activated RAW 264.7 macrophages. Nanoconjugates were then embedded into microparticles through spray-drying with L-leucine, forming nano-embedded microparticles (NEMs). NEMs were produced with an aerodynamic diameter close to 1 µm and a density below 0.1 g.cm−3, indicative of a high alveolar deposition. NEMs spray-dried with the less hydrophobic nanoconjugates, PMAB 1:4, were readily dissolved in simulated lung fluid and were chosen for in vivo experiments to study pharmacokinetics in healthy rats. As it was released in vivo from NEMs, sustained distribution of budesonide was obtained for 48 h in lung tissue, cells, and lining fluid. With high loading rates, modulable release kinetics, and low cytotoxicity, these nanoconjugates delivered by NEMs are promising for the more efficient treatment of pulmonary inflammatory diseases.
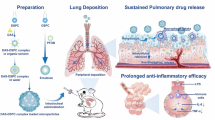
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Inhaled glucocorticoids (GCs) are widely used as anti-inflammatory and immunosuppressive drugs in chronic lung inflammatory diseases, particularly in chronic obstructive pulmonary disease (COPD) [1, 2]. COPD is orchestrated by alveolar macrophages activated by continuous atmospheric pollution, eventually associated with tobacco [3]. The disease is characterized by chronic inflammation in the pulmonary tissue where inflammatory cells produce pro-inflammatory cytokines, inducing chronic bronchitis and emphysema in more advanced cases of acute exacerbation [4,5,6]. In this latest case, budesonide is given as a dry powder inhaled dosage form associated with long-acting muscarinic antagonists and long-acting β2-agonists [7].
Indeed, local administration of GCs is preferred as it reduces adverse effects related to systemic exposure, such as adrenal suppression, osteoporosis, myopathy, hyperglycemia, dyslipidemia, weight gain, and growth suppression [8]. Although inhalation successfully decreases the drug dose and systemic exposure [9], the efficacy remains limited because of the poor deposition of the drug in deep lungs and its rapid elimination by pulmonary clearance and passage to the systemic circulation. For that reason, nanoparticles were considered for improving pulmonary drug bioavailability [10]. Due to their high tropism for inflammatory cells [11], their use could allow better delivery at the site of action and increase the drug cellular pool [12,13,14]. However, despite their efficacy, nanoparticles are mostly exhaled after inhalation due to their low density [15].
Several formulations of nanoparticles encapsulating GCs have been developed during the last decade [16,17,18,19,20,21]. Unfortunately, most of these formulations suffer from poor drug loading and burst release upon contact with biological fluids attributed to the physical entrapment of the drug that is loosely bound to the nanoparticle matrix [22,23,24]. Drug loading was shown to be improved by formulating nanoparticles from prodrugs where the drug is covalently bound to a carrier. This "chemical loading" approach significantly enhances drug stability and loading and reduces burst release [25]. Recently, our group has shown that nanoparticles made of dexamethasone palmitate could be obtained in the presence of DSPE-PEG with a drug loading up to 50 wt% [26]. Still, drug release after hydrolysis of the ester bond remained too fast [26].
Polymer conjugates are particularly interesting in slowing down drug release, as they can achieve high drug loading, encapsulate hydrophobic compounds, and progressively deliver the drug, ensuring a sustained release [25, 27, 28]. We have designed a nanoconjugate by coupling budesonide to a hydrophilic polymer, poly(malic acid) (PMA), through its numerous carboxylic acid functions. PMA is a biocompatible polymer with hydrolyzable ester bonds within its polymer backbone, allowing its degradation into biocompatible metabolites [29]. In a preliminary study, the same chemical loading strategy was successfully applied to pyrazinoic acid [30].
To deliver nanoconjugates to the deep lung and alveoli, they should be included in particles with an aerodynamic diameter lower than 5 µm and greater than 1 µm to avoid being exhaled [31]. For this purpose, nano-embedded microparticles (NEMs) produced by spray-drying have been designed to improve aerosolization and deposition properties in the respiratory tract [31,32,33,34,35,36]. NEMs represent an optimal solution combining the enhanced deposition of powder in the alveolar region and the cellular uptake and sustained drug release properties of nanoparticles.
This publication reports on developing NEMs containing poly(malic acid)-budesonide nanoconjugates. The physicochemical characteristics of nanoconjugates were evaluated along with in vitro testing to assess their efficacy and safety towards macrophages. The study then focused on the formulation of NEMs and the characterization of physicochemical and aerodynamic properties. Finally, the pharmacokinetics and biodistribution of budesonide delivered by NEMs were investigated.
Materials and methods
Materials
Budesonide, dexamethasone, and dexamethasone palmitate were purchased from Chemos GmbB & Co. KG (Germany). L-(-)-malic acid, N,N'-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), L-leucine, rhodamine B, bovine serum albumin (BSA), Dulbecco's Modified Eagle's Medium (DMEM), penicillin–streptomycin, methylthiazolyldiphenyl-tetrazolium bromide (MTT) and lipopolysaccharides (LPS) from Escherichia coli were obtained from Sigma-Aldrich (France). Fetal bovine serum (FBS) was supplied by Thermo Fischer Scientific (USA), and the Cytometric Bead Array (CBA) Mouse Inflammation kit was acquired from BD Biosciences (USA). Rhodamine B piperazine was synthesized using a method already described by Nguyen and Francis [37].
Synthesis and characterization of poly (α,β-malic acid) and conjugates
Synthesis of poly(α,β-malic acid)
The polymerization of malic acid was carried out by a polycondensation reaction. 5 g of L-malic acid was added in a Schlenk tube and stirred at 110 °C for 72 h under a 6 mmHg vacuum. The crude polymer was then dissolved in tetrahydrofuran (THF) and precipitated in a mixture of diethyl ether:petroleum ether (3:1). The purified polymer was dissolved in water before freeze-drying for 48 h using an Alpha-1–2 LD apparatus (Christ, France), leading to a white powder. L-malic acid conversion was 80% (1H NMR in DMSO-d6).
Synthesis of poly(α,β -malic acid)-budesonide conjugates
PMA-budesonide conjugates were synthesized by esterifying PMA with budesonide using different molar ratios of budesonide:PMA: 1:8, 1:4, 1:2, and 3:4. 0.5 g of PMA (4.31 mmol of malic acid units) and different amounts of budesonide were purged under argon and then dissolved with anhydrous THF before being mixed. DMAP (0.43 mmol) and DCC (2 molar equivalents of budesonide) were purged together and dissolved by anhydrous THF before being added dropwise to the PMA/budesonide mixture at 0 °C for 15 min [38]. The mixture was allowed to stir at room temperature overnight and was subsequently filtered to remove dicyclohexylurea (DCU). The PMA-budesonide conjugate (PMAB) was then purified by precipitation in a diethyl ether:petroleum ether mixture (1:1), followed by dialysis in water (regenerated cellulose membrane with 1000 g/mol MWCO from Spectrum) and then freeze-dried to provide purified and dried PMAB.
Polymer characterization
1H NMR and 13C NMR spectroscopies were carried out on a Bruker Avance-400 MHz spectrometer. Differential scanning calorimetry (DSC) was performed using a DSC Q1000 from TA Instruments. The molar mass and dispersity of polymers were measured by size-exclusion chromatography (SEC). Measurements were performed at 30 °C with two columns from Malvern Panalytical (Viscotek LT4000L Mixed, Low 300 × 8 mm), a triple detection system (Viscotek 270 Dual Detector and Waters 2414 Refractive Index Detector) coupled with a Waters 515 HPLC pump and Waters 717 plus Autosampler. THF was used as the eluent with a flow rate of 1 mL/min, and data was analyzed using OmniSEC 4.0 software.
Preparation of PMAB nanoconjugates
PMAB nanoconjugates were obtained by a nanoprecipitation method [39]. 10 mg of PMAB dissolved in 0.5 mL of acetone was slowly injected into 10 mL of milliQ water under magnetic stirring. After 5 min of stirring, the solvent was removed by rotary evaporation (1 h, 100 mbar). The volume was adjusted to 10 mL if necessary to maintain concentration at 1 mg/mL.
Preparation of rhodamine-labeled PMAB nanoconjugates
First, PMAB-rhodamine conjugates were synthesized by the same method as PMAB conjugates. Briefly, 200 mg of PMAB 1:4 (with 0.8 mmol of free malic acid units) and 0.2 molar equivalents of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) were purged by argon and dissolved by dimethylformamide (DMF). Then, 0.1 molar equivalents of rhodamine B piperazine and DMAP dissolved in DMF were added to the PMAB and EDC solution. The reaction was left at room temperature overnight. Then, the product was dialyzed in water and freeze-dried. Nanoconjugates were also obtained by nanoprecipitation but were composed of 5% PMAB-rhodamine, the rest being unlabeled PMAB. The size of these nanoconjugates was 83 nm (PDI = 0.18), and their zeta potential was -56 mV.
Physicochemical characterization of nanoconjugates
The mean diameter, polydispersity index (PDI), and zeta potential of nanoconjugates were measured with a Zetasizer NanoZS (Nanoseries, Malvern Instruments, France) at an angle of 173°. Three measurements were carried out at 25 °C for each sample, diluted in water for size measurements or NaCl 1 mM for zeta potential at a 1/10 dilution, corresponding to a 0.1 mg/mL concentration.
Transmission electron microscopy (TEM) was performed at I2BC (Gif-sur-Yvette, France) using a JEOL JEM-1400 operating at 80 kV. 10 µL of purified suspensions of nanoconjugates were deposited for 1 min on a glow-discharged copper grid. Samples were then stained using 2% phosphotungstic acid solution for 30 s. The excess solution was blotted off using filter paper. Images were acquired using an Orius camera (Gatan Inc., USA).
The amount of budesonide in nanoconjugates was determined by 1H NMR spectroscopy from PMAB conjugates in CDCl3.
In vitro release of budesonide from nanoconjugates
The in vitro release rate of budesonide was studied in a cell culture medium (DMEM) containing 10% fetal bovine serum (FBS). For this purpose, PMAB 1:4 and 3:4 nanoconjugates at a concentration of 15 µg/mL of budesonide were introduced into different vials to obtain 3 vials per predetermined sampling time, each containing 2 mL of solution. The vials were then stirred at 100 rpm in an incubator at 37 °C. At determined time intervals, 3 vials per sample were taken out and frozen at -20 °C until analysis was performed. For HPLC analysis, budesonide was extracted using the following method. 100 µL of each sample was collected and mixed with 100 µL of internal standard (20 µg/mL of dexamethasone in ethanol). 3 mL of CHCl3/MeOH (9/1) was then added to the mixture, which was vortexed for 3 min before being centrifuged at 1700 G at 4 °C for 15 min. The organic phase was collected and transferred to a vial to be analyzed by HPLC. The system consisted of a Water 1525 Binary HPLC Pump, a Waters 2707 Autosampler injector, a UV–visible Waters 2998 Photodiode Array Detector equipped with a reverse-phase Waters Symmetry Shield RP18 column (5 µm, 4.6 × 250 mm). The analysis was performed with a mobile phase of acetonitrile/water (55/45) at a flow of 1 mL/min and a temperature of 35°C. Detection was carried out at 244 nm, the maximum determined wavelength for budesonide.
Cell culture
The Murine RAW 264.7 macrophage cell line was obtained from the American Type Culture Collection (ATCC, USA). The cells were cultured in DMEM supplemented by 10% heat-inactivated FBS, 50 U/mL of streptomycin, and 50 U/mL of penicillin. They were maintained at 37 °C in a humidified atmosphere of 5% CO2 and were sub-cultured every 3–4 days.
Uptake of nanoconjugates by RAW 264.7 cells
The uptake kinetics of nanoconjugates by RAW 264.7 cells were studied by flow cytometry and confocal microscopy.
Flow cytometry
4 × 104 cells/mL were seeded in 12-well plates and incubated for 24 h at 37 °C in a 5% CO2-humidified atmosphere. Then, the medium was removed, replaced by a new one, and incubated for 24 h. Cells were treated by removing half of the old medium and replacing it with either a new medium for the control wells or by a medium containing PMAB 1:4, 1:2, or 3:4 nanoconjugates labeled with 5% of PMAB-rhodamine, diluted until reaching a concentration of budesonide of 10 µg/mL. The treatment incubated cells for 48, 24, 8, 5, 3, or 1 h. Control wells were incubated for 24 or 48 h only. Cells were washed with cold phosphate-buffered saline solution (PBS) after removing the medium and scraped from the bottom of the wells. After centrifugation at 300 g at 4 °C for 5 min, cells were washed in cold PBS, and flow cytometry was used to quantify the internalization of nanoconjugates over time. The mean fluorescence intensity (MFI) ratio of treated cells compared with the control MFI was calculated for each time point.
Confocal microscopy
RAW 264.7 cells were seeded at a density of 4 × 104 cells/mL in 6-well plates containing round glass coverslips and incubated at 37 °C in a 5% CO2 humidified atmosphere for 24 h before adding treatment. Nanoconjugates composed of PMAB 1:4 and 5% of PMAB-rhodamine were prepared as described above and diluted in fresh cell culture medium to obtain a 10 µg/mL concentration of budesonide.
The cells were treated and incubated for up to 24 h at 37 °C with the treatment before being observed with a Carl Zeiss LSM-510 confocal microscope (Carl Zeiss, Germany) using a 63X/1.4 Plan-Apochromat objective lens.
Cell viability assay
RAW 264.7 cells (8 × 103 cells/well) were cultured in 96-well plates for 24 h until 80–90% confluence. Then, they were treated with free budesonide (previously dissolved in ethanol and then diluted in PBS) or by nanoconjugates with an equivalent concentration of encapsulated budesonide. Free budesonide and nanoconjugates composed of PMAB 1:4, 1:2, and 3:4 were tested at concentrations of budesonide ranging from 0.1 to 100 µg/mL. PMA was tested at higher concentrations than nanoconjugates (100 to 750 µg/mL) due to its low toxicity toward macrophages [40].
The cell viability was determined after 24 or 48 h of incubation with the treatments using a colorimetric MTT assay (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) dissolved in PBS[41]. 20 µL of MTT solution was added to each well and incubated at 37 °C for 1 h. The medium was then discarded, and 200 µL of DMSO was added to each well. The plates were shaken for 30 min and protected from light to dissolve crystals. They were then submitted to a microplate reader (Multiskan MS, LabSystems, Finland) at a wavelength of 570 nm for absorbance measurements. The viability was determined by the ratio of treated wells absorbance to the absorbance of the control wells. All conditions were reproduced in triplicate and multiplied three times.
Multiplex bead-based cytokine assay
The anti-inflammatory activity of nanoconjugates or free budesonide was assessed using a multiplex bead-based cytokine assay kit. Cells (4 × 104 cells/well) were seeded in 24-well plates and incubated for 48 h. They were then exposed to 0.1 µg/mL of LPS for 3 h. After 3 h, the cells were treated with 10 µg/mL of free budesonide or nanoconjugates containing the same concentration of grafted budesonide by adding 100 µL of treatment with a 100 µg/mL concentration to the treated wells. 100 µL of fresh medium was added to the control wells. After 24 h, the supernatant was collected and frozen at -20 °C until analysis. Untreated cells were used as a negative control, and cells incubated with LPS only were used as a positive control. Each condition was done in triplicate.
Cytokines released from RAW 264.7 cells were quantified in the cell culture supernatants with a multiplex immunoassay method using a Cytometric Bead Array (CBA) mouse inflammation kit from BD Biosciences. Results are summarized from three independent experiments and expressed with mean ± SD. Statistical analysis was performed by two-way analysis of variance (ANOVA), and p < 0.05 was considered to have a significant difference.
NEMs formulation
NEMs were prepared by spray-drying with a B-290 Mini Spray Dryer (Büchi, Switzerland) equipped with a fluid nozzle with a 0.7 mm diameter. Six g/L of L-leucine dissolved in water were mixed with nanoconjugates at a concentration of 0.3, 0.6, or 0.9 g/L corresponding to 5, 10 or 15 wt% of L-leucine. All samples were prepared using the optimized spray-drying conditions. Pre-formulation experiments were carried out (data not shown) to optimize spray-drying conditions. Then, all samples were prepared using these optimized conditions: inlet temperature of 190 °C, outlet temperature of 59 ± 2 °C, feed flow rate of 13.1 ± 0.9 ml/min, air flow rate of 473 L.h–1 (40% Q flow) and aspirator setting of 100%. After spray-drying, NEMs were stored under vacuum at room temperature to avoid moisture uptake. The yield was calculated by dividing the powder mass gathered in the collector by the initial mass in the solutions before spray-drying.
To quantify the amount of powder in each stage of the multistage liquid impinger, NEMs containing rhodamine B were prepared. Briefly, 6 mg/L (0.1 wt%) of rhodamine was added to the L-leucine solution before mixing with the nanoconjugates solution.
Unloaded microparticles were prepared by spray-drying a L-leucine solution of 6 g/L in the same conditions as NEMs. They were also used to prepare the formulation for the control group in the in vivo experiments.
Microparticle size distribution
The particle size distribution of NEMs was measured by light diffraction using a Mastersizer 2000 equipped with a Scirocco 2000 dry disperser (Malvern Instruments, France). The particle refractive index used was the same as L-leucine's: 1.46. Data obtained were expressed in terms of particle diameter at 10%, 50%, and 90% of the total volume of material in the sample (D10, D50, and D90). The span showing the width of the size distribution was also measured.
Scanning electron microscopy
Scanning Electron Microscopy (SEM) of NEMs was performed using a Merlin FEG-SEM (Carl Zeiss, Germany) operating at 0.4 kV. Powder samples were deposited on a carbon conductive double-sided tape (Euromedex, France). They were coated with a palladium layer of 4 nm using a Cressington sputter-coater 208HR with a rotary planetary-tilt stage equipped with an MTM-20 thickness controller.
Tap density
The powder tap density of NEMs was measured with a Tap Density Tester PT-TD1 (Pharma Test, Germany). A 5 mL graduated cylinder was filled with an accurately weighed powder sample, and the powder volume was measured after 1000 taps (adapted from Chapter 2.9.34 Eur. Pharm.) [42].
Aerosolization characterization
Aerodynamic particle size distribution was evaluated using a MultiStage Liquid Impinger (MSLI) from Copley Scientific (Switzerland) and was conducted following the European Pharmacopoeia guidelines (Chapter 2.9.18, Eur. Pharm.) [42]. Each stage of the MSLI was filled with 20 mL of a solution composed of acetonitrile/water (55/45 v/v), and a 1.0 µm glass microfiber filter (70 mm GF/B grade, Whatman, UK) was placed in the bottom stage. Five hydroxypropyl methylcellulose capsules (size 3, Laboratoire LGA, France) were loaded with an average of 10 mg of powder each. The capsules were then placed in a dry powder inhalation device (Aerolizer®, Novartis, Switzerland), and the test was carried out at 60 L/min for 4 s with a pressure P3/P2 < 0.5 ratio. The solutions were collected for each of the four stages, and then the stages were rinsed with the ACN/water solution. The filter, DPI, adapter, and tube were also rinsed with ACN/water. The solutions rinsing the DPI, adapter, and tube were pooled together, but the solutions were collected in separate graduated flasks for each stage and the filter.
The powder deposition in each part was indirectly determined by measuring rhodamine B's fluorescence using an F-2000 Hitachi spectrofluorometer (Hitachi, Japan) with excitation/emission wavelengths of 553/576 nm. The emitted fraction (EF) of powder was calculated as the difference between the weight of filled capsules and the weight of emptied capsules after the test. The fine particle fraction (FPF) and the alveolar fraction (AF), corresponding to the amount of particles with an aerodynamic diameter smaller than 5 µm or 3.1 µm, respectively, were determined by interpolation from the cumulative amount of each stage from the bottom up. The Mass Median Aerodynamic Diameter (MMAD) was also interpolated from the cumulative distribution of mass as the diameter at which 50% of particles by mass are larger and 50% are smaller.
Dispersion of nanoconjugates from microparticles
The ability of nanoconjugates to redisperse from NEMs L-leucine matrix was studied by dissolving the powder in a simulated lung fluid (SLF) with a composition representative of the physiological conditions [43]. Briefly, 10 mg of powder was mixed with 10 mL of SLF and put in an incubator at 37 °C with a mixing plate for 30 min. Dispersed nanoconjugates' mean diameter and size distribution were measured with a Zetasizer NanoZS (Nanoseries, Malvern Instruments, France) at a 173° angle laser. Each sample had a concentration close to 0.1 mg/mL of nanoconjugates after dispersion, and each experiment was repeated three times. The morphology of nanoconjugates, after dispersion, was observed by TEM as described in paragraph 1.5.
Animals
6-weeks male Sprague–Dawley rats (n = 31) with an average weight of 269 ± 17 g from Janvier Laboratories (Le Genest-St-Isle, France) were used in this study. Animals were maintained on a 12/12 h light/dark cycle at constant temperature and humidity (22 ± 2 °C and 50 ± 15%, respectively) with free access to food and water. Animal experiments were carried out following the Guide for Care and Use of Laboratory Animals as recommended by the European Parliament and Council (Directive 2010/63/EU, September 22, 2010). The protocol for the present experiment (APAFIS#14993–2018050714402162 v1) was approved by the local ethics committee.
Intratracheal administration of microparticles and sample collection
The powder was administered to the rats by intratracheal insufflation with a Dry Powder Insufflator (DP-4 model, Penn-Century Inc., USA). The amount of powder expelled from the dry powder insufflator was evaluated before the in vivo experiments to adjust the mass needed to administer 60 µg of budesonide. It was determined that 70% of the powder was discharged after one or two pulses of 2 cm3 of air. It was established that the sample chamber needed to be filled with 2 mg of NEMs (composed of 10% of PMAB 1:4 nanoconjugates) to deliver 60 ± 13 µg of budesonide in both cases.
Animals were treated with NEMs and were randomly formed with six collection time-points, each time-point with n = 5 rats. Rats were anesthetized by isoflurane inhalation followed by an injection of zoletil (10 mg.kg−1) mixed with xylazine (10 mg.kg−1). They were placed in a supine position and maintained by the upper incisors on a rodent work stand (Hallowell EMC, USA) inclined at a 45° angle. The insufflator's tube was inserted into the trachea. The powder was administered through the insufflator by rapidly pushing a 2 cm3 bolus of air.
Blood from the caudal vein was collected in heparin tubes after 15 min, 2 h, 4 h, and 18 h by anesthetizing the rats with isoflurane inhalation. Rats were euthanized by the administration of a high dose of Dolethal (150 mg.kg−1 IP) after 30 min, 1, 3, 6, 24, and 48 h post-administration of powder, after which blood was collected through cardiac puncture and bronchoalveolar lavage (BAL) was performed with the following procedure. After blood collection, the trachea was exposed, and a catheter was inserted through an incision in the upper part of the trachea. Rats were then placed in a supine position, and 1 mL of phosphate-buffered saline (PBS) solution was injected, followed by aspiration of the maximum volume. This procedure was repeated 3 times to obtain BAL fluid samples. Afterward, the lungs were collected and frozen at -80 °C. Shortly after collection, blood samples were centrifuged at 2000 g for 10 min to retrieve the plasma fraction, and BAL fluid was centrifuged at 489 g for 10 min to separate the supernatant from the cells, forming a pellet. All samples were frozen and stored at -80 °C until further analysis.
Extraction of budesonide from tissue samples
Plasma samples were thawed at 37 °C to avoid the advent of cryoprecipitate, and the other samples were thawed at room temperature. Alveolar cell samples were prepared by adding 1 mL of PBS to each tube and sonicating at 20% (Digital Sonifier, Branson, Netherlands) for 1 min to break down the cells. Lungs were each weighed, then cut into small pieces and transferred to a 15 mL polypropylene tube with 1 mL of PBS per gram of lung tissue added. The samples were then homogenized with an overhead stirrer (RZR 2021, Heidolph Instruments, Germany) equipped with a macro-pestle (Rotilabo, Carl Roth) for 3 min at 800 rpm.
The extraction of budesonide from samples was performed as follows. In PTFE centrifuge tube, 100 µL of plasma, BAL fluid, or PBS containing alveolar cell fragments were mixed with 100 µL of the internal standard and then vortexed for 30 s. The internal standard was composed of a 50/50 mixture of dexamethasone (DXM)/dexamethasone palmitate (DXP) solutions in acetonitrile with a concentration of 37.5 ng/mL each.
About 100 mg of lung homogenate was weighed and mixed with 100 µL of the internal standard, then vortexed for 30 s. On each tube, 3 mL of a CHCl3/MeOH (9/1 v/v) mixture was added, followed by vortexing the tube for 3 min. The samples were centrifuged at 1690 g for 30 min (Sorvall ST16R centrifuge, TX-400 rotor, Thermo Scientific, France). The organic phase was collected, transferred to a glass vial, and evaporated under a stream of nitrogen at 35 °C. The residue was then solubilized with 250 µL of acetonitrile.
LC–MS/MS method for budesonide quantification
Determination of budesonide concentration in plasma, BAL fluid, alveolar cell lysate, or lungs was performed by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS). The system consisted of an Acquity UPLC-TQD (triple quadrupole detector) from Waters piloted by the MassLynx 4.1 software. Chromatography was performed on an Acquity BEH C8 column (2.1 × 50 mm, 1.7 µm) from Waters maintained at 40 °C. The flow rate was 0.6 mL.min-1, following an increasing gradient between a mobile phase of 0.1% (vol/vol) formic acid in water and 0.1% (vol/vol) formic acid in acetonitrile. The injection volume was 5 µL. The mass spectrometer was operated with positive electrospray ionization (ESI +). The optimal MS/MS setup parameters were a 3.6 kV capillary voltage, 3 V extractor, a 150 °C source temperature, an 800 L.h−1 and 450 °C desolvation gas (N2) flow and temperature, and a 50 L.h−1 cone gas (N2) flow. The triple quadrupole analyzer was used in multiple-reaction monitoring (MRM) mode to analyze the ions according to the parameters in Table 1. Argon was used as collision gas at 4.5 × 10–3 mBar.
Four nine-point calibration standard curves between 0.1 and 200 ng.mL−1 were prepared by spiking plasma, BAL fluid, alveolar cell lysate, and homogenized lung tissue from an untreated rat with budesonide and budesonide palmitate. 100 µL or 100 µg of each type of sample was mixed with 25 µL of budesonide (BUD) and 25 µL of budesonide palmitate (BP) in acetonitrile for each point of the standard curve. An internal standards mixture of 50 µL of DXM and DXP was added with a concentration of 75 ng.mL−1 each, and extraction was done following the same procedure as previously described. Samples and standards were filtered on 0.22 µm PVDF filters before analysis by LC/MS.
The limit of quantification (LOQ) was defined as the lowest concentration in the calibration curves at which the accuracy and precision were within ± 20%. The accuracy and precision were within ± 15% at all other concentrations.
Determination of budesonide concentration in ELF and alveolar cells
The concentration of budesonide (CELF) in epithelial lining fluid (ELF) was calculated after correcting the concentration of budesonide in BAL fluid (CBAL) with a dilution factor, according to the following Eq. 1 [44]:
Quantification of urea in BAL fluid (UreaBAL) and plasma (Ureaplasma) allowed the determination of this dilution factor since the urea concentration in ELF is equal to its concentration in plasma [45]. Thus, the volume of ELF (VELF) could also be estimated from the volume of BAL (VBAL) after correction for dilution, according to Eq. 2:
The concentration of budesonide in alveolar cells (CAC as ng/µg of protein) was calculated as the amount of budesonide per µg of protein in the cell lysate. After quantifying the concentration of budesonide (CBud as ng.mL−1) and the concentration of proteins (Cprot as µg.mL−1) in each sample, the concentration of budesonide in alveolar cells (CAC) was determined with the following Eq. 3 :
Quantification of urea in BAL fluid and plasma
Urea concentration in BAL fluid and plasma was determined using a urea nitrogen colorimetric detection kit (ThermoFischer Scientific, Courtabœuf, France). Seven-point calibration curves were prepared by serial dilution of the kit's urea nitrogen standard in deionized water at concentrations between 1.56 and 100 µg/mL. Samples were thawed and centrifuged with a MiniSpin centrifuge at 6708 g for 5 min. 20 µL of plasma samples were diluted in 180 µL of water (1/10 dilution), and 100 µL of BAL fluid samples were diluted in 100 µL of water (1/2 dilution), then 50 µL of each was added in 96-well plates. 75 µL of the two-color reagents were added to the samples and standards, then incubated for 30 min at room temperature. The plates were read with a microplate reader at a wavelength of 450 nm.
Mean concentrations of urea in BAL fluid and plasma were quantified at 4.1 ± 1.8 µg.mL−1 and 131.1 ± 29.1 µg.mL−1 (n = 31), respectively, corresponding to a dilution factor of 35.2 ± 10.2. ELF was estimated with an average volume of 93 ± 30 µL. The dilution factor corresponds to the plasma urea concentration divided by the BAL urea concentration.
Since budesonide in ELF would be diluted in the same manner as urea, the dilution factor for each rat allowed the determination of budesonide concentration in ELF (Eq. 1).
Quantification of protein in alveolar cells
Following the extraction of budesonide from sonicated alveolar cell samples, protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad, USA), based on the Bradford protein assay [46]. A six-point calibration curve from 4 to 60 µg/mL was prepared with bovine serum albumin solubilized in PBS. Samples were diluted between a factor of 1/2 up to 1/8 for higher concentrations of proteins, and 10 µL of each was pipetted in 96-well plates. Two hundred µL of 1/5th diluted Dye Reagent was thoroughly mixed with each sample and incubated at room temperature for 15 min. The plates were read with a microplate reader at a wavelength of 595 nm. Mean protein concentration was measured at 90 ± 86 µg.mL−1, with values spanning from 21 to 354 µg.mL−1.
Pharmacokinetic modeling
Pharmacokinetic parameters of budesonide in plasma and epithelial lining fluid were evaluated according to a non-compartmental model of a dose-normalized concentration-versus-time profile, conducted with the PK Solver add-on for Excel [47].
Results and discussion
Syntheses and characterization of budesonide conjugates
PMA-budesonide conjugates were synthesized using the two-step approach depicted in Fig. 1. First, malic acid was polycondensed to get PMA, a hydrophilic biodegradable polymer, followed by the conjugation of budesonide on the carboxyl side groups of PMA, which are readily available to form an ester bond with the free OH on C-21 of budesonide. Different amounts of budesonide were grafted onto the polymer by modulating the ratios of budesonide per PMA unit from 1:8 to 3:4. The successful preparation of conjugates was confirmed by NMR (Figs. S1 and S2) and characterized by DSC and SEC (Table 2).

Synthesis of Budesonide PMA (PMAB) conjugate
As observed in Table 2, the amount of conjugated budesonide could be easily modulated by the initial feed ratio of budesonide to malic acid units from 24 to 73 wt%, demonstrating an optimal grafting efficiency. Furthermore, the increasing incorporation of budesonide onto PMA induces a shift in the molar mass of the conjugates (from 4000 g/mol for PMAB 1:8 to 12,100 g/mol for PMAB 3:4) and glass transition temperature (from 44 °C for PMAB 1:8 to 102 °C for PMAB 3:4) (Table 2), which is consistent with the introduction of a heavy group onto the polymer side chain.
Nanoconjugate formulation
PMAB nanoconjugates were successfully prepared in demineralized water from all conjugates (PMAB 1:8 to PMAB 3:4) using a simple nanoprecipitation method without any surfactant. The hydrophobic budesonide linked to the polymer triggered the formation of the nanoconjugates to minimize the interfacial energy. Thus, the moieties composed of budesonide will form the hydrophobic core of the nanoconjugates, which is not readily accessible to degradation. In contrast, the outer layer would mainly be composed of PMA chains with the hydrophilic carboxylic acid functions oriented outwards from the nanoconjugate's surface.
The resulting nanoconjugates were characterized by dynamic light scattering (DLS) to measure the average hydrodynamic size and polydispersity, as summarized in Table 3. The nanoconjugate sizes were independent of the PMAB ratio, with an uniform size distribution around 90 nm regardless of the amount of budesonide. This value is higher than the 70 nm limit, allowing nanoconjugates to gain access to the macrophages and not be cleared by the capillary blood flow [48, 49].
The surface charges of all nanoconjugates were strongly negative due to the ionized carboxylic acid functions of PMA, which provides colloidal stability to the nanoconjugates by avoiding aggregation. The negative charge also gives an advantage to the nanoconjugates for pulmonary administration since, as reported, they have better retention in the lungs compared to positively charged particles [50].
The nanoconjugates were also analyzed by TEM, showing spherical morphologies and sizes in agreement with those determined by DLS (Fig. 2). PMAB 1:8 nanoconjugate could not be imaged by TEM because of its low stability when exposed to the heat generated by the electron beam from the microscope due to its much lower Tg than the other conjugates. This nanoconjugate, with its lower drug loading and stability, was not considered further in this study.

Transmission electron microscopy (TEM) images of PMAB 1:4 (A), PMAB 1:2 (B) and PMAB 3:4 (C) nanoconjugates
Compared to conventional drug delivery systems, an essential feature of nanoconjugates is their ability to yield high drug loading. Since PMAB forms the nanoconjugates exclusively without adding surfactants, the loading is equivalent to the experimental results obtained for the grafting rate of budesonide with PMA (Table 2). Therefore, the loading of budesonide in the nanoconjugates is 24%, 41%, 51%, and 73% by mass, respectively for PMAB 1:8, 1:4, 1:2 and 3:4. This is far much higher than what was previously described with PLGA nanoparticles encapsulating only 1 wt% of dexamethasone [22], and more recently also with PLGA nanoparticles encapsulating budesonide with a loading efficiently of at best 10% [51].
In vitro release of budesonide from the nanoconjugates
Sustained drug release from nanoconjugates is a prerequisite for local lung therapy to achieve high drug concentration at the disease site and avoid early release. Budesonide release was, therefore, followed up to 72 h in a cell culture medium to mimic biological fluids with the presence of esterase capable of hydrolyzing the ester bond between budesonide and polymer. PMAB 1:4 and 3:4 nanoconjugates were selected for their stability and distinct hydrophilic/hydrophobic ratios. As shown in Fig. 3, the budesonide release rate from the nanoconjugates is progressive, up to 24% for PMAB 1:4 and 13% for PMAB 3:4 at 72 h. On the other hand, the concentration of free budesonide in the same medium was monitored as a control, and a 25% decrease in the concentration over time was observed, reflecting the poor stability of the drug in the medium (Fig. S3). This could be due to various causes such as the degradation of the drug by the aqueous medium [52], binding to plasmatic proteins of FBS, or esterification of budesonide, adding a lipidic chain to the molecule, transforming it into a fatty acid ester [53]. Therefore, this raises the question of how much budesonide was released from the nanoconjugates since budesonide metabolites were hardly quantifiable. Matter et al. [52] have shown that dexamethasone released from PLGA implants degraded at the same rate as dexamethasone released from the implant. Consequently, a plateau was observed where only 30% of the drug was released, whereas quantification of the various degradation products by LC–MS/MS showed that 80% was released. It seems clear that the amount of released budesonide in our condition is underdosed. Nevertheless, the nanoconjugates manage to avoid a burst release where a large amount of the loaded drug is released in the first few hours of contact with an external medium. Burst release is a common problem for nanoformulations [54], especially those where the drug is physically entrapped in the carrier. Taking advantage of the covalent binding of budesonide to PMA, nanoconjugates could release a higher amount of budesonide preferentially inside macrophages, thus avoiding undesirable and premature extracellular delivery.

Release kinetics of budesonide from nanoconjugates of PMAB 1:4 (■) or 3:4 (●) in cell culture medium containing 10% of FBS at 37 °C (n = 3)
Noteworthily, the release of budesonide from PMAB 3:4 nanoconjugates was slower than from the PMAB 1:4 nanoconjugates (13% and 24%, respectively). The more hydrophobic core of PMAB 3:4, which is densely packed with budesonide, significantly slows down the release of the drug compared to the less loaded PMAB 1:4, providing higher accessibility of the ester bonds between budesonide and polymer backbone on the nanoconjugate core.
It is essential to mention that the medium used to evaluate the release differs from the one expected inside endo-lysosomes of alveolar macrophages [55]. In these endo-lysosomes, the esterase concentration and acidity would be much higher and should allow a higher and faster PMA degradation, providing a quicker release of budesonide.
Cellular internalization and in vitro cytotoxicity
Confocal microscopy and flow cytometry were applied to assess the kinetic of nanoconjugate internalization in RAW 264.7 macrophage cell line. For that purpose, macrophages were first activated by LPS to stimulate an inflammatory response, then incubated with rhodamine-labeled nanoconjugates, and fluorescence was measured by flow cytometry at different time points.
All tested nanoconjugates display a rapid cellular uptake, as evidenced by flow cytometry experiments (Fig. 4A). After only 1 h of incubation, the nanoconjugates were observed in cells (Fig. 4A), and the maximum fluorescence was reached around 16 h. The gradual uptake of the nanoconjugates for up to 24 h could allow the anti-inflammatory treatment to be spread throughout the day so that a single dose administration could be sufficient.

A: Kinetics of rhodamine-loaded nanoconjugates uptake by RAW 264.7 macrophages expressed as mean fluorescence intensity (MFI) ratio of treated cells compared to untreated cells. Two-way ANOVA comparing MFI ratio for each time point with the control for the respective nanoconjugate. *: p-value < 0.1, **: p-value < 0.01, ***: p-value < 0.001 and ****: p-value < 0.0001. B: Confocal microscopy imaging of RAW 264.7 cells incubated for 24 h with PMAB1:4 nanoconjugates labeled with rhodamine (scale bar 20 µm). C: Viability rate of RAW 264.7 cells treated with free budesonide or nanoconjugates (NC) after 24 h of incubation. D: quantification of cytokines released by LPS-activated RAW 264.7 macrophages treated by free budesonide or by PMAB 1:4, 1:2, or 3:4 nanoconjugates, compared to negative control (C-) (cells without LPS stimulation), or positive control (C +) (cells stimulated by LPS).**: p < 0.01 and ****: p < 0.0001
The intracellular fate of the nanoconjugates after uptake by activated macrophages was visualized by confocal microscopy (Fig. 4B). The rhodamine-labeled nanoconjugates progressively penetrated in incubated cells for 16 h. They were localized intracellularly. A z-stack visualization (Fig. S4) showed an absence of nanoconjugates inside the cellular membrane or the nucleus. They appear to be located mainly in the endosomes of the cells, which should fuse with lysosomes containing esterases that can hydrolyze the bonds between budesonide and PMA, allowing the drug to be released into the cytoplasm where it could bind to its receptor [56].
The cytotoxicity of budesonide, PMA, and nanoconjugates was also studied with RAW 264.7 macrophages as the target cells for budesonide (Figs. 4C and S5). First, the cellular viability of pure PMA is maintained at over 70% after incubating the polymer with the cells for 24 h, even at high concentrations (Fig. S5). The polymer is, therefore, not cytotoxic, which was expected thanks to its high biodegradability with quickly metabolized L-malic acid units through the Krebs cycle [57, 58]. The absence of cytotoxicity was comparable to what was observed on the same cell line with poly(DL-lactide-co-glycolide)-polyethylene glycol (PLGA-PEG) nanoparticles [59].
Incubation of budesonide and the different nanoconjugates showed little effect on the cellular viability of RAW264.7 cells, even at the highest concentrations tested (Fig. 4C). In all cases, cell viability was consistently above 70%, demonstrating the safety of the nanoconjugates on macrophages regardless of the amount of budesonide grafted. Therefore, PMA represents a polymer of choice for the design of budesonide nanoconjugates.
Anti-inflammatory activity of nanoconjugates
The capacity of nanoconjugates and free budesonide to modulate the expression of typical pro-inflammatory factors such as TNF-α, MCP-1, IL-6, and IL-10 was evaluated on LPS-activated macrophages RAW 264.7 [60] with free budesonide or the different nanoconjugates at a concentration of 10 µg/mL. Preliminary assays performed without stimulating the macrophage with LPS showed no significant amount of cytokines released compared to the negative control, confirming that free budesonide or nanoconjugates do not induce an inflammatory response in the absence of LPS (Fig. S6). For the LPS-stimulated macrophages, a significant reduction in the release of TNF-α, MCP-1, IL-6, and IL-10 was observed with cells treated by free budesonide or nanoconjugates compared to the positive control (Fig. 4D). Interestingly, all three nanoconjugates exhibited the same anti-inflammatory activities to free budesonide after 24 h of treatment, implying that the covalent conjugation of budesonide to PMA did not compromise the anti-inflammatory efficacy.
Although the release rates of budesonide decreased with the drug content of the PMAB nanoconjugates, no statistical difference in cytokines released was observed between the three nanoconjugates. The absence of difference is explained by the fact that budesonide is efficient at relatively low doses below 10 µg/mL. It has been noticed that concentrations as low as 1 nM was effective enough to inhibit the release of TNF-α, or even as low as 10 pM to inhibit IL-6 release [61]. These results highlight the potential of PMAB nanoconjugates to release intact and active budesonide effectively.
Microparticles formulation
Nanoconjugates PMAB 1:4 were selected to produce nano-embedded microparticles for their ability to release budesonide faster than more hydrophobic nanoconjugates and for their rapid cellular uptake. L-leucine was chosen as an excipient because it can generate microparticles with good properties of deep lung deposition [33, 62,63,64,65]. Moreover, as an anti-adherent excipient [66], it should also facilitate the disaggregation of nanoconjugates after contact with an aqueous solution.
NEMs were obtained by spray-drying an L-leucine solution mixed with PMAB 1:4 at different concentrations. As shown in Table 4, blank microparticles and NEMs were successfully spray-dried with a pretty high yield (> 60%). These results are consistent with previous studies showing the higher anti-adherent power of L-leucine compared to other excipients such as chitosan, DPPC, hyaluronic acid, lactose, mannitol, or maltodextrin [65, 67,68,69,70].
Introducing nanoconjugates at different concentrations to the particle formulation does not significantly modify the geometric diameter (D50), with values of around 3 µm for blank microparticles and NEMs (Table 4). This was expected as the nanoconjugates presented similar diameters, not modifying drastically the diffusion coefficient. These results are in agreement with those of Gómez-Gaete et al. [69], where increasing the incorporation of dexamethasone nanoparticles into trojan particles did not lead to a noticeable change in size (p > 0.05, from 6.7 to 6.4 µm with 0 and 33% of nanoparticles, respectively). Similarly, Stocke et al. [71] did not observe a size change when spray-drying 5 to 20 wt% iron oxide nanoparticles into microparticles.
The morphology of the particles was then observed by SEM (Fig. 5). Whatever the formulation, spherical hollow microparticles can be observed with or without nanoconjugates, consistent with previous findings using L-leucine as an excipient [62,63,64]. More importantly, nanoconjugates can be perfectly observed in the NEM shell, showing the successful incorporation of nanoconjugates in different contents. The sizes of small spherical objects visible on the surface of the hollow microparticles are consistent with those of the nanoconjugates measured before spray-drying (Table 3). The blank microparticles have a rough surface but are exempt from small spherical objects such as those observed on the NEMs.

Scanning electron microscopy (SEM) images of blank and nanoconjugate-embedded hollow microparticles obtained by spray-drying
Aerodynamic properties
To determine more precisely the aerosolization behavior of airborne particles and better predict their deposition in lungs [72], the aerodynamic properties were investigated using a multistage liquid impinger (MSLI). As shown in Table 5, the addition of nanoconjugates in the formulation did not significantly modify the aerodynamic properties of the microparticles compared to the blank microparticles. The MMADs of microparticles were quite similar between blank microparticles and NEMs. In addition, no observable differences were reported in the NEMs group, regardless of the concentration of the nanoconjugates. All presented MMADs are close to the 1-—5 µm range; thus, they are in the target size range for an optimal deposition in the peripheral lung [73,74,75,76].
Powder for all the formulations was expelled from the capsules with a high EF of around 99% thanks to the anti-adherent properties of L-leucine (Table 5). Concerning the FPF and AF, nanoconjugates do not modify the particles' distribution in the different stages. They are found mainly in the lowest stage of the MSLI (around 83% for FPF and 69% for AF), representing the optimum distribution to achieve efficient delivery into the deep lungs. These results are consistent with previous observations where microparticles formulated with leucine also exhibited high FPF of up to 74% [33, 65]. Therefore, we could expect that after their administration through inhalation, most of the powder would directly be available in the alveoli to distribute the nanoconjugates near the target alveolar macrophages, with reduced loss of the microparticles in the higher airways.
Microparticle redispersion behavior
The possibility of redispersing nanoconjugates from NEMs was investigated by dynamic light scattering after incubation with an aqueous solution simulating the lung fluid (SLF). As observed in Table 6, NEMs spray-dried with nanoconjugates, whatever their concentration, showed good redispersion properties, allowing favorable deagglomeration of microparticles. Sizes and PDI measured after redispersion were slightly larger than the original measurements (109–117 nm vs. 93 nm), but no visible agglomerates were present.
Observation of NC 1:4 NEMs by TEM after SLF incubation clearly shows individual nanoconjugates with a size distribution consistent with the measurements of nanoconjugates before spray-drying (Fig. S7). Along with the SEM images (Fig. 5), this demonstrates that the spray-drying process preserves the primary structure of the PMAB 1:4 nanoconjugates and that they can be restored nearly to their initial state after redispersion in an aqueous solution. NEMs have an optimal structure for the deposition of nanoconjugates in the alveolar region of the lungs while maintaining their initial characteristics to improve the delivery of corticosteroids to alveolar macrophages.
In vivo lung distribution
To assess the in vivo behavior, NEMs formulated with 10% NC 1:4 were administered to healthy rats at 60 µg of budesonide. After administration, there were no fatalities, and none of the animals showed distress after the treatments. Thus, the tested dose of 220 µg.kg−1 is safe even though this dose is much higher than the human dose (1.4 to 5.7 µg.kg−1), but it remains in the usual range for budesonide tested on rats (0.2 to 1 mg.kg−1) [77,78,79,80].
As shown in Fig. 6, after inhalation, plasma pharmacokinetics obtained showed low and stable budesonide concentrations over time with a Cmax at 6.3 ± 6.2 ng.mL−1 after 30 min. Compared to free budesonide [81,82,83], this behavior reflects the low absorption of budenoside contained in NEMs to the systemic compartment. During chronic treatment with inhaled corticosteroids, this low systemic uptake should limit systemic side effects [8, 84].

Mean total budesonide concentration in plasma, lung tissue, ELF (epithelial lining fluid) and alveolar cells versus time profiles after intratracheal administration of NEMs (10% NC 1:4) containing 60 µg of budesonide. Data represents the mean ± SD (n = 5)
In lung tissue, budesonide concentration was high after inhalation and then decreased before rising to the maximum level at 6 h (Fig. 6). Similar behavior was observed in the ELF and in the alveolar cells collected from the BAL fluid, but in this latter case, the maximum level was reached after 3 h (Fig. 6). After 6 h, the concentration decreased in all tissues. Once deposited in the lungs, the microparticle structure should get disrupted and release nanoconjugates, which are further internalized by alveolar macrophages. Following the uptake of nanoconjugates, budesonide will be released through hydrolysis of the ester bond between the drug and the polymer due to the presence of esterases in lysosomes [85, 86]. As shown in Fig. 6, this behavior is supported by the increase in budesonide concentration in alveolar cells up to 3 h and then decreases, whereas in lung tissue and ELF the peak is reached at 6 h. The first part could correspond to the internalization of nanoconjugates by alveolar macrophages followed by hydrolysis of PMAB. This will induce a release of budesonide into ELF and a distribution in lung tissue, explaining the increase of concentration at 6 h in these two areas. The decrease after 6 h corresponds to the elimination phase of the drug.
Conclusion
In this study, we assessed the potential of a budesonide-poly(malic acid) polymeric conjugate to self-assemble into nanoconjugates capable of being administered through inhalation. The present work demonstrates that controllable and high drug-loading conjugates could be synthesized. They can then form stable nanoconjugates by nanoprecipitation in water, modulating the budesonide release rate. In vitro, macrophages rapidly internalize nanoconjugates under 24 h and do not exhibit cytotoxicity until relatively high concentrations. Moreover, they are as efficient as free budesonide to reduce pro-inflammatory cytokines.
Nanoconjugate-embedded microparticles with good aerosolization efficiency were produced by spray-drying the nanoconjugate PMAB 1:4 suspension with a leucine solution. With a large respirable fraction, NEMs would efficiently deposit the drug-bearing nanoconjugates in the peripheral lung. They were easily suspendable in simulated lung fluid and were used for in vivo experiments on healthy rats. Pulmonary administration of budesonide showed a sustained release as the drug could be detected in the lungs, epithelial lining fluid, and alveolar cells up to 48 h after administration. Moreover, its systemic levels remained low compared to the lungs, suggesting limited side effects.
Our study has several limitations. First, we could only investigate the nanoconjugates' internalization by endocytosis. However, we have not yet demonstrated that lysosomes effectively take up nanoconjugates and the drug being released. Further studies with lysosomal markers will be necessary. In addition, in vivo studies were carried out on healthy animals, whereas the therapeutic efficacy remains to be explored using an LPS-induced acute lung injury model.
Overall, NEMs containing nanoconjugates constitute an excellent approach to improving the deposition rate of a drug in the alveolar region. They represent a promising formulation for pulmonary administration, allowing the administration of low doses of the drug with reduced systemic exposure that would undoubtedly lead to reducing side effects.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
References
Tashkin DP, Strange C. Inhaled corticosteroids for chronic obstructive pulmonary disease: What is their role in therapy? Int J COPD. 2018;13:2587–601.
Agusti A, Fabbri LM, Singh D, Vestbo J, Celli B, Franssen FME, et al. Inhaled corticosteroids in COPD: Friend or foe? Eur Respir J [Internet]. 2018;52:1801219. Available from: https://doi.org/10.1183/13993003.01219-2018.
Thomson NC. The Role of Smoking in Asthma and Chronic Obstructive Pulmonary Disease Overlap. Immunol Allergy Clin North Am [Internet]. 2022;42:615–30. Available from: https://linkinghub.elsevier.com/retrieve/pii/S088985612200011X.
Barnes PJ. Alveolar macrophages as orchestrators of COPD. COPD [Internet]. 2004;1:59–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16997739.
Barnes PJ. Cellular and molecular mechanisms of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35:71–86.
Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38:497–515.
2023 GOLD Report - Global Initiative for Chronic Obstructive Lung Disease - GOLD [Internet]. Available from: https://goldcopd.org/2023-gold-report-2/.
Oray M, Abu Samra K, Ebrahimiadib N, Meese H, Foster CS. Long-term side effects of glucocorticoids. Expert Opin Drug Saf [Internet]. 2016;15:457–65. Available from: https://doi.org/10.1517/14740338.2016.1140743.
Lipworth BJ. Systemic adverse effects of inhaled corticosteroid therapy: A systematic review and meta-analysis. Arch Intern Med. 1999;159:941–55.
Pandey R, Sharma A, Zahoor A, Sharma S, Khuller GK, Prasad B. Poly (DL-lactide-co-glycolide) nanoparticle-based inhalable sustained drug delivery system for experimental tuberculosis. J Antimicrob Chemother. 2003;52:981–6.
De Boer AH, Gjaltema D, Hagedoorn P, Frijlink HW. Can “extrafine” dry powder aerosols improve lung deposition? Eur J Pharm Biopharm. 2015;96:143–51.
Amani A, York P, Chrystyn H, Clark BJ. Evaluation of a nanoemulsion-based formulation for respiratory delivery of budesonide by nebulizers. AAPS PharmSciTech. 2010;11:1147–51.
Britland S, Finter W, Chrystyn H, Eagland D, Abdelrahim ME. Droplet aerodynamics, cellular uptake, and efficacy of a nebulizable corticosteroid nanosuspension are superior to a micronized dosage form. Biotechnol Prog. 2012;28:1152–9.
Zhang Y, Zhang J. Preparation of budesonide nanosuspensions for pulmonary delivery: Characterization in vitro release and in vivo lung distribution studies. Artif Cells, Nanomedicine Biotechnol. 2016;44:285–9.
Heyder J, Gebhart J, Rudolf G, Schiller CF, Stahlhofen W. Deposition of particles in the human respiratory tract in the size range 0005–15 μm. J Aerosol Sci. 1986;17:811–25.
Chiraz JM, Andrieu V, Elaissari A, Fessi H. Beclomethasone-loaded lipidic nanocarriers for pulmonary drug delivery: Preparation, characterization and In Vitro drug release. J Nanosci Nanotechnol. 2011;11:1841–51.
Midhun BT, Shalumon KT, Manzoor K, Jayakumar R, Nair SV, Deepthy M. Preparation of budesonide-loaded polycaprolactone nanobeads by electrospraying for controlled drug release. J Biomater Sci Polym Ed. 2011;22:2431–44.
Amini MA, Faramarzi MA, Gilani K, Moazeni E, Esmaeilzadeh-Gharehdaghi E, Amani A. Production, characterisation, and in vitro nebulisation performance of budesonide-loaded PLA nanoparticles. J Microencapsul. 2014;31:422–9.
Prosperi D, Colombo M, Zanoni I, Granucci F. Drug nanocarriers to treat autoimmunity and chronic inflammatory diseases. Semin Immunol. 2017;34:61–7.
Lühder F, Reichardt H. Novel Drug Delivery Systems Tailored for Improved Administration of Glucocorticoids. Int J Mol Sci [Internet]. 2017/08/25. 2017;18:1836. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28837059. 2017 Aug 25.
Gauthier A, Fisch A, Seuwen K, Baumgarten B, Ruffner H, Aebi A, et al. Glucocorticoid-loaded liposomes induce a pro-resolution phenotype in human primary macrophages to support chronic wound healing. Biomaterials. 2018;178:481–95.
Gómez-Gaete C, Tsapis N, Besnard M, Bochot A, Fattal E. Encapsulation of dexamethasone into biodegradable polymeric nanoparticles. Int J Pharm. 2007;331.
Beloqui A, Cococ R, Alhouayek M, Solińis MÁ, Rod́riguez-Gáscon A, Muccioli GG, et al. Budesonide-loaded nanostructured lipid carriers reduce inflammation in murine DSS-induced colitis. Int J Pharm. 2013;454:775–83.
Balzus B, Sahle FF, Hönzke S, Gerecke C, Schumacher F, Hedtrich S, et al. Formulation and ex vivo evaluation of polymeric nanoparticles for controlled delivery of corticosteroids to the skin and the corneal epithelium. Eur J Pharm Biopharm. 2017;115:122–30.
O Abioye A, Tangyie Chi G, T Kola-Mustapha A, Ruparelia K, Beresford K, Arroo R. Polymer-Drug Nanoconjugate An Innovative Nanomedicine Challenges and Recent Advancements in Rational Formulation Design for Effective Delivery of Poorly Soluble Drugs. Pharm Nanotechnol. 2016;4:38–79.
Lorscheider M, Tsapis N, ur-Rehman M, Gaudin F, Stolfa I, Abreu S, et al. Dexamethasone palmitate nanoparticles: An efficient treatment for rheumatoid arthritis. J Control Release [Internet]. 2019;296:179–89. Available from: https://www.sciencedirect.com/science/article/pii/S0168365919300331?dgcid=raven_sd_aip_email.
Pang X, Jiang Y, Xiao Q, Leung AW, Hua H, Xu C. PH-responsive polymer-drug conjugates: Design and progress. J Control Release. 2016;222:116–29.
Shen S, Wu Y, Liu Y, Wu D. High drug-loading nanomedicines : progress, current status, and prospects. Int J Nanomedicine. 2017;12:4085–109.
Ljubimova JY, Fujita M, Khazenzon NM, Lee BS, Wachsmann-Hogiu S, Farkas DL, et al. Nanoconjugate based on polymalic acid for tumor targeting. Chem Biol Interact. 2008;171:195–203.
Pinto Carneiro S, Moine L, Tessier B, Nicolas V, dos Santos O, Fattal E. Pyrazinoic acid-Poly(malic acid) biodegradable nanoconjugate for efficient intracellular delivery. Precis Nanomedicine. 2019;2:303–17.
Tsapis N, Bennett D, Jackson B, Weitz DA, Edwards DA. Trojan particles: Large porous carriers of nanoparticles for drug delivery. Proc Natl Acad Sci U S A. 2002;99:12001–5.
Anton N, Jakhmola A, Vandamme TF. Trojan microparticles for drug delivery Pharmaceutics. 2012;4:1–25.
Kunda NK, Alfagih IM, Miyaji EN, Figueiredo DB, Gonçalves VM, Ferreira DM, et al. Pulmonary dry powder vaccine of pneumococcal antigen loaded nanoparticles. Int J Pharm. 2015;495:903–12.
Ruge CA, Bohr A, Beck-Broichsitter M, Nicolas V, Tsapis N, Fattal E. Disintegration of nano-embedded microparticles after deposition on mucus: A mechanistic study. Colloids Surfaces B Biointerfaces. 2016;139:219–27.
Wang Y, Beck-Broichsitter M, Yang M, Rantanen J, Bohr A. Investigation of nanocarriers and excipients for preparation of nanoembedded microparticles. Int J Pharm. 2017;526:300–8.
Zhang L, Yang L, Zhang X, Jiaqi L, Fan L, Beck-Broichsitter M, et al. Sustained therapeutic efficacy of budesonide-loaded chitosan swellable microparticles after lung delivery: Influence of in vitro release, treatment interval and dose. J Control Release. 2018;283:163–74.
Nguyen T, Francis MB. Practical synthetic route to functionalized rhodamine dyes. Org Lett. 2003;5:3245–8.
Su T, Peng X, Cao J, Chang J, Liu R, Gu Z, et al. Functionalization of biodegradable hyperbranched poly(α, β-malic acid) as a nanocarrier platform for anticancer drug delivery. RSC Adv. 2015;5:13157–65.
Fessi H, Puisieux F, Devissaguet JP, Ammoury N, Benita S. Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm. 1989;55:R1-4.
Martinez Barbosa ME, Cammas S, Appel M, Ponchel G. Investigation of the degradation mechanisms of poly(malic acid) esters in vitro and their related cytotoxicities on J774 macrophages. Biomacromol. 2004;5:137–43.
Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63.
O’Malley B. European Pharmacopoeia. 9th ed. Br Med J Strasbourg. 1971.
Marques MRC, Loebenberg R, Almukainzi M. Simulated biological fluids with possible application in dissolution testing. Dissolution Technol. 2011;18:15–28.
Marchand S, Gobin P, Brillault J, Baptista S, Adier C, Olivier JC, et al. Aerosol therapy with colistin methanesulfonate: A biopharmaceutical issue illustrated in rats. Antimicrob Agents Chemother. 2010;54:3702–7.
Kiem S, Schentag JJ. Interpretation of antibiotic concentration ratios measured in epithelial lining fluid. Antimicrob Agents Chemother. 2008;52:24–36.
Politou A, Temussi PA. Revisiting a dogma: The effect of volume exclusion in molecular crowding. Curr Opin Struct Biol. 2015;30:1–6.
Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99:306–14.
Yang W, Peters JI, Williams RO. Inhaled nanoparticles-A current review. Int J Pharm. 2008;356:239–47.
Borm PJA, Kreyling W. Toxicological hazards of inhaled nanoparticles - Potential implications for drug delivery. J Nanosci Nanotechnol. 2004;4:521–31.
Fidler IJ, Raz A, Fogler WE, Kirsh R, Bugelski P, Poste G. Design of Liposomes to Improve Delivery of Macrophage-augmenting Agents to Alveolar Macrophages. Cancer Res. 1980;40:4460–6.
Leonard F, Srinivasan S, Liu X, Collnot EM, Ferrari M, Lehr CM, et al. Design and in vitro characterization of multistage silicon-PLGA budesonide particles for inflammatory bowel disease. Eur J Pharm Biopharm. 2020;151:61–72.
Matter B, Ghaffari A, Bourne D, Wang Y, Choi S, Kompella UB. Dexamethasone Degradation in Aqueous Medium and Implications for Correction of In Vitro Release from Sustained Release Delivery Systems. AAPS PharmSciTech. 2019;20:1–11.
Miller-Larsson A, Mattsson H, Hjertberg E, Dahlbäck M, Tunek A, Brattsand R. Reversible fatty acid conjugation of budesonide: Novel mechanism for prolonged retention of topically applied steroid in airway tissue. Drug Metab Dispos. 1998;26:623–30.
Yeo Y, Park K. Control of encapsulation efficiency and initial burst in polymeric microparticle systems. Arch Pharm Res. 2004;27:1–12.
Ahsan F, Rivas IP, Khan MA, Torres Suárez AI. Targeting to macrophages: Role of physicochemical properties of particulate carriers - Liposomes and microspheres - On the phagocytosis by macrophages. J Control Release. 2002;79:29–40.
Barnes PJ. Molecular mechanisms of steroid action in asthma. J Allergy Clin Immunol. 1996;97:159–68.
Vert M, Lenz RW. Preparation and properties of poly- beta -malic acid: a functional polyester of potential biomedical importance. Am Chem Soc Div Polym Chem Prepr. 1979;20:608–11.
Loyer P, Cammas-Marion S. Natural and synthetic poly(malic acid)-based derivates: A family of versatile biopolymers for the design of drug nanocarriers. J Drug Target. 2014;22:556–75.
Simón-Vázquez R, Tsapis N, Lorscheider M, Rodríguez A, Calleja P, Mousnier L, et al. Improving dexamethasone drug loading and efficacy in treating arthritis through a lipophilic prodrug entrapped into PLGA-PEG nanoparticles. Drug Deliv Transl Res [Internet]. 2022 [cited 2022 Apr 19];12:1270–84. Available from: https://springerlink.bibliotecabuap.elogim.com/article/10.1007/s13346-021-01112-3. [cited 2022 Apr 19].
Chan ED, Riches DWH. IFN-γ + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38mapk in a mouse macrophage cell line. Am J Physiol - Cell Physiol. 2001;280:C441–50.
Furman S, Nissim-Bardugo E, Zeeli S, Weitman M, Nudelman A, Finkin-Groner E, et al. Synthesis and in vitro evaluation of anti-inflammatory activity of ester and amine derivatives of indoline in RAW 264.7 and peritoneal macrophages. Bioorganic Med Chem Lett. 2014;24:2283–7.
Feng AL, Boraey MA, Gwin MA, Finlay PR, Kuehl PJ, Vehring R. Mechanistic models facilitate efficient development of leucine containing microparticles for pulmonary drug delivery. Int J Pharm. 2011;409:156–63.
Lucas P, Anderson K, Potter UJ, Staniforth JN. Enhancement of small particle size dry powder aerosol formulations using an ultra low density additive. Pharm Res. 1999;16:1643–7.
Najafabadi AR, Gilani K, Barghi M, Rafiee-Tehrani M. The effect of vehicle on physical properties and aerosolisation behaviour of disodium cromoglycate microparticles spray dried alone or with L-leucine. Int J Pharm. 2004;285:97–108.
Rabbani NR, Seville PC. The influence of formulation components on the aerosolisation properties of spray-dried powders. J Control Release. 2005;110:130–40.
Staniforth JN. Powders comprising anti-adherant materials for use in dry powder inhalers [Internet]. United States; 2002. Available from: http://www.google.com/patents/US6475523.
Huang YC, Yeh MK, Cheng SN, Chiang CH. The characteristics of betamethasone-loaded chitosan microparticles by spray-drying method. J Microencapsul. 2003;20:459–72.
Pourshahab PS, Gilani K, Moazeni E, Eslahi H, Fazeli MR, Jamalifar H. Preparation and characterization of spray dried inhalable powders containing chitosan nanoparticles for pulmonary delivery of isoniazid. J Microencapsul. 2011;28:605–13.
Gómez-Gaete C, Fattal E, Silva L, Besnard M, Tsapis N. Dexamethasone acetate encapsulation into Trojan particles. J Control Release. 2008;128:41–9.
Vehring R. Pharmaceutical particle engineering via spray drying. Pharm Res. 2008;25:999–1022.
Stocke NA, Meenach SA, Arnold SM, Mansour HM, Hilt JZ. Formulation and characterization of inhalable magnetic nanocomposite microparticles (MnMs) for targeted pulmonary delivery via spray drying. Int J Pharm. 2015;479:320–8.
Chen W, Fryrear DW. Aerodynamic and geometric diameters of airborne particles. J Sediment Res. 2001;71:365–71.
Gonda I. Study of the Effects of Polydispersity of Aerosols on Regional Deposition in the Respiratory Tract. J Pharm Pharmacol. 1981;33:52P.
The Task Group on Lung Dynamics. Deposition and retention models for internal dosimetry of the human respiratory tract: Task group on lung dynamics. Health Phys. 1966;12:173–207.
Zeng XM. Particulate Interactions in Dry Powder Formulations for Inhalation. Part. Interact. Dry Powder Formul Inhal New York: Taylor & Francis. 2001.
Patton JS, Byron PR. Inhaling medicines: Delivering drugs to the body through the lungs. Nat Rev Drug Discov. 2007;6(1):67–74. https://doi.org/10.1038/nrd2153.
Cui N, Friend DR, Fedorak RN. A budesonide prodrug accelerates treatment of colitis in rats. Gut. 1994;35:1439–46.
Jansson AH, Eriksson C, Wang X. Effects of budesonide and N-acetylcysteine on acute lung hyperinflation, inflammation and injury in rats. Vascul Pharmacol. 2005;43:101–11.
Rauf A, Bhatnagar A, Sisodia SS, Khar RK, Ahmad FJ. Lungs deposition and pharmacokinetic study of submicron budesonide particles in Wistar rats intended for immediate effect in asthma. EXCLI J. 2017;16:236–44.
Fu TT, Zhao Y, Yang FF, Wen H, Liu CY, Liao YH. Ciclesonide and budesonide suspensions for nebulization delivery: An in vivo inhalation biopharmaceutics investigation. Int J Pharm. 2018;549:21–30.
Liu T, Han M, Tian F, Cun D, Rantanen J, Yang M. Budesonide nanocrystal-loaded hyaluronic acid microparticles for inhalation: In vitro and in vivo evaluation. Carbohydr Polym. 2018;181:1143–52.
Chanoine F, Grenot C, Heidmann P, Junien JL. Pharmacokinetics of butixocort 21-propionate, budesonide, and beclomethasone dipropionate in the rat after intratracheal, intravenous, and oral treatments. Drug Metab Dispos. 1991;19(2):546–53.
Boobis AR. Comparative physicochemical and pharmacokinetic profiles of inhaled beclomethasone dipropionate and budesonide. Respir Med. 1998;92:2–6.
Dahl R. Systemic side effects of inhaled corticosteroids in patients with asthma. Respir Med. 2006;100(8):1307–17.
Dannenberg AM, Burstone MS, Walter PC, Kinsley JW. A histochemical study of phagocytic and enzymatic functions of rabbit mononuclear and polymorphonuclear exudate cells and alveolar macrophages. I. Survey and quantitation of enzymes, and states of cellular activation. J Cell Biol. 1963;17:465–86.
Laskin DL, Malaviya R, Laskin JD. Pulmonary Macrophages. Comp Biol Norm Lung Second Ed. 2015;7:629–49.
Acknowledgements
The authors would like to thank S. Denis for cell culture, C. Cailleau for animal experiment, V. Nicolas (MIPSIT, Université Paris-Saclay) for confocal microscopy, A. Solgadi (MIPSIT, Université Paris-Saclay) for HPLC-MS, Natalie Hue (ICSN, CNRS) for UPLCMS and R. Pires Brazuna (ICPME, CNRS) for scanning electron microscopy. The present work has benefited from the core facilities of Imagerie‐Gif, (http://www.i2bc.paris-saclay.fr/), member of IBiSA (http://www.ibisa.net), supported by “France‐BioImaging” (ANR‐10‐INBS‐04‐01), and the Labex “Saclay Plant Science” (ANR‐11‐IDEX‐0003‐02) with the help of C. Boulogne and C. Gillet.
Funding
The authors declare that no funds, grants, or other support were received during the preparation of this manuscript. The authors have no relevant financial or non-financial interests to disclose.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study's conception and design. Barbara Tessier and Arnaud Peramo performed material preparation, data collection, and analysis. Barbara Tessier wrote the manuscript's first draft, and all authors commented on previous versions. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval
This study was performed in line with the principles of the Declaration of Helsinki. Animal experiments were carried out following the Guide for Care and Use of Laboratory Animals as recommended by the European Parliament and Council (Directive 2010/63/EU, September 22, 2010). The protocol for the present experiment (APAFIS#14993–2018050714402162 v1) was approved by the local ethics committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tessier, B., Moine, L., Peramo, A. et al. Poly(malic acid)-budesonide nanoconjugates embedded in microparticles for lung administration. Drug Deliv. and Transl. Res. 14, 2062–2078 (2024). https://doi.org/10.1007/s13346-024-01571-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-024-01571-4