
Abstract
Autophagy is a catabolic process in which an organism responds to its nutrient or metabolic emergencies. It involves the degradation of cytoplasmic proteins and organelles by forming double-membrane vesicles called “autophagosomes.” They sequester cargoes, leading them to degradation in the lysosomes. Although autophagy acts as a protective mechanism for maintaining homeostasis through cellular recycling, it is ostensibly a cause of certain cancers, but a cure for others. In other words, insufficient autophagy, due to genetic or cellular dysfunctions, can lead to tumorigenesis. However, many autophagy modulators are developed for cancer therapy. Diverse nanoparticles have been documented to induce autophagy. Also, the highly stable nanoparticles show blockage to autophagic flux. In this review, we revealed a general mechanism by which autophagy can be induced or blocked via nanoparticles as well as several studies recently performed to prove the stated fact. In addition, we have also elucidated the paradoxical roles of autophagy in cancer and how their differential role at different stages of various cancers can affect its treatment outcomes. And finally, we summarize the breakthroughs in cancer disease treatments by using metallic, polymeric, and liposomal nanoparticles as potent autophagy modulators.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
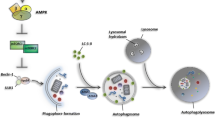
Autophagy is a catabolic process in which an organism responds to its nutrient or metabolic emergencies. It involves the degradation of cytoplasmic proteins and organelles by forming double-membrane vesicles, termed “autophagosomes,” which sequester cargoes, leading them to degradation in the lysosomes. The formation of autophagosomes is controlled by a specific set of autophagy genes called ATG genes (Fig. 1). An aberrant behavior in these one or more autophagy genes often leads to various pathological conditions, including tumorigenesis, inflammatory conditions, and neurodegeneration.

Mechanism of autophagy: Activation of growth receptor, stress, and nutrient deprivation induces AMPK signaling and inhibits mTOR signaling, which activates the ULK complex (FIP 200, ATG 13, ULK 1/2) and induces the nucleation process. ULK complex further activates PI3K class III complex 1 (Beclin 1, Vps 15, Vps 34, and ATG 14L) and initiates the development of the phagophore. The maturation, as well as the elongation of the developed phagophore, is mediated via the interaction of multiple ATGs like ATG5-ATG12, ATG16, etc. The interaction generates two "ubiquitin-like conjugation systems" which catalyze phosphatidylethanolamine (PE)-conjugated LC3 (LC3II) formation. LC 3II then binds on the surface of autophagosomes. The binding of LC3II on the surface of autophagosomes helps in the internalization of the cellular substrates within the autophagosomes. Further, the presence of exosomal sorting complex (CHMP2A, and VPS4) promotes the closure of the membrane of autophagosomes, which marked the complete maturation of the autophagosomes. The matured autophagosomes amalgamate with the lysosomes in the presence of SNARE proteins and form autolysosomes. The acidic hydrolases existing within the lysosomes provide degradation of nutrients as well as metabolites within the autophagic cargo. These degrading products further get recycled back to the cytoplasm
Autophagy and cancer association: genetic evidence
Cancer is not a single disease. It is an amalgamation of more than 100 distinct and different diseases. Although there are various types of cancer, all cancers initiate from the abnormal growth of cells [1,2,3,4,5].
Cancer is caused by the malignant transformation of a normal cell into a cancerous cell. It occurs as a result of a successive accumulation of genetic mutations and epigenetic changes that overpower infallible cellular mechanisms such as apoptosis (programmed cell death type I) or oncogene-induced senescence [6, 7].
Some first shreds of genetic evidence point out the relationship between autophagy and cancer. First, the study in 1999 [8] indicated that human breast carcinoma cell lines frequently contain allelic deletions of Beclin1, which was monoallelically deleted in human breast, ovarian, and prostate cancers and was expressed at reduced levels in those tumors. The tumor suppressor Beclin1 is part of the phosphatidylinositol 3-kinase class III (PI(3)KCIII) lipid–kinase complex that induces autophagy [9].
Later in 2006, the novel ultraviolet irradiation resistance-associated gene (UVRAG), a tumor suppressor gene, was identified, which is a positive regulator of the Beclin1–PI (3) KCIII complex and hence autophagy. UVRAG was monoallelically mutated at high frequency in human colon cancers [10, 11].
In ensuing studies, targeted mutant mouse models were used, showing that Beclin 1-/- mutant mice die early in embryogenesis (due to disruption in autophagy and not due to apoptosis). In contrast, Beclin 1 +/- mutant mice (hemizygous to Beclin1) are viable but suffer from a high incidence of spontaneous tumors [12, 13]. In 2007, the findings suggested that the complete loss of the gene encoding the UVRAG-binding protein BIF-1 resulted in tumor susceptibility in mice [14]. All these studies, taken together, strongly suggested that autophagy plays a vital role in human tumor suppression. What is important to note here is that in all the mouse models described above, there was a deletion of the autophagy-related gene in the target cells since their embryonic stage. Hence, all the tumors observed resulted from all-time autophagy impaired cells. Therefore, these experiments gave no clarity about the role of autophagy loss in an established tumor, which is a more common and likely scenario to occur. To find answers to this, robust research was initiated in this field.
Cancer autophagy-related dichotomies
The cytoprotective trait of autophagy is to lessen or moderate the influence of metabolic stress, which occurs due to nutrient and oxygen unavailability. During the growth of tumors, the vascularisation system is hampered, limiting the nutrient supply in the hypoxic regions within tumors [15]. Since autophagy is known to be activated in these regions and could be utilized to keep these tumor cells alive, it seems paradoxical how the inactivation of a process that can keep tumor cells alive benefits tumor development. The quick fix for this contradicting viewpoint is that autophagy evades stress to maintain cellular homeostasis. However, insufficient autophagy can no longer provide nutrients, prevent the accumulation of defective proteins and organelles, manage oxidative stress, and limit inflammation. These factors, along with genomic instability, lead to tumorigenesis.
Autophagy was not only found to promote cell survival in solid tumors [16] but was also found to contribute to cell death [17]. Hence, there were dichotomies and paradoxes in understanding the role of autophagy in both genesis and suppression of cancer. To elude these incongruities coupled with cancer and autophagy, the answer lies in the fact that autophagy's exact role is dependent on the context. This includes the type of tumor in consideration, the stage of tumor/neoplasia, the cellular and metabolic context in which the tumor cell lines, etc. In the following section, we discuss these disjunctions in more detail.
Autophagy in oncogenesis
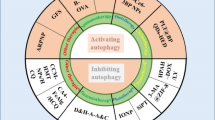
The role of autophagy in oncogenesis is contradictory. Chronic suppression of autophagy during the initial tumor stage may stimulate oncogenesis. However, once a tumor is established or is in the advanced stage of progression, autophagy might go against the body and promote tumor survival (Fig. 2). This role of autophagy depends on many factors, and its exact mechanism of switching its role is elusive.

Conflicting role of autophagy during oncogenesis: The oncosuppressive properties of autophagy include preservation of normal metabolism, degradation of oncogenic proteins, maintenance of genomic proteins, and regulation of immunosurveillance. On the other hand, autophagy speeds up the cancer progression and undergoes resistance to chemotherapy, once the cancerous transformation has occurred. The cancer-supporting properties depict the capacity of autophagy in preserving the cancer stem cells, promoting resistance in cancerous cells toward various endogenous conditions like anoikis, hypoxia, nutrient deprivation, and epithelial-to-mesenchymal transition (EMT) that could otherwise lead to cell death
Tumor suppressive or pro-death mechanism?
Autophagy is a principal mechanism in maintaining cellular homeostasis and chromosomal stability. Inhibition or lack of autophagy results in increased levels of reactive oxygen species, which leads to accumulation of DNA damage, manifesting itself as gene amplification, increased double-strand breaks, and polyploid nuclei [18, 19]. Under stress conditions, deregulation or loss of autophagy leads to increased DNA damage, which makes the cell more prone to de novo tumor formation and development. How this is achieved and the appropriate mechanism behind it is equivocal. Nonetheless, the cumulation of many corroborations outlines that autophagy acts as a tumor-suppressive or pro-death mechanism under several defined conditions.
BECN-1
The first link between autophagy and tumor suppression came from the discovery that BECN (the gene that encodes for Beclin1) is a haploinsufficient tumor suppressor gene and was found to be monoallelically deleted in approximately 50% of human breast, ovarian and prostate cancers and expressed only at low levels in brain tumors [9, 20].
ATG5 and ATG7
ATG5 and ATG7 are two other critical autophagy regulators whose deletion and subsequent aftereffects have been analyzed in experimental animal models. In contrast to the embryonic death in the case of BECN1, it was found that mice with systemic mosaic deletion of Atg5 and liver-specific Atg7 − / − mice develop benign liver adenomas [21, 22]. As Atg7 and Atg5 are essential for amino acid supply in neonates and starvation-induced bulk degradation in mice, the depletion of these genes leads to the death of both mice within 24 h of their birth. This occurs as a result of autophagy inhibition, due to which transplacental nutrient supply is suddenly interrupted, and neonates face severe starvation and ultimately death [22, 23].
ATG4C
The most widely expressed mammalian orthologue of yeast Atg4 is ATG4C (autophagin-3). Mutant mice deficient in ATG4C were generated and found fertile and viable without any abnormalities in the early neonatal period. However, tissue-specific autophagy alterations were leading to decreased autophagy in the diaphragm. To boot, animals deficient in Atg4C show an increased susceptibility to develop fibrosarcomas induced by chemical carcinogens [24]. Giving substance to this is the study [25], which showed that the expression level of ATG4C is deeply associated with the prognosis of glioma patients.
UVRAG
As mentioned earlier, in “Autophagy and cancer association: genetic evidence” section, UVRAG (a tumor suppressor gene) is found to be monoallelically mutated at high frequency in human colon cancers. In contrast, in the xenograft studies, UVRAG's expression in human HCT116 colon carcinoma cells makes them less susceptible to tumorigenicity [10, 11]. Frameshift mutations of UVRAG are also present in gastric carcinomas in addition to colorectal carcinomas with microsatellite instability and promote tumorigenesis [26, 27]. Furthermore, disruption of the association of UVRAG with centrosomes causes centrosome instability and aneuploidy, which is a hallmark of cancer [28].
BIF1
Bif-1 (also known as endophilin B1 or SH3GLB1) is another protein that joins the UVRAG–Beclin 1 complex as a potential activator of autophagy and tumor suppressor. Unlike Beclin-1-deficient mice, which were embryonically lethal, Bif-1-/- developed normally and were indistinguishable from their wild-type littermates, except for an enlarged spleen. However, these mice had an 89.7% chance of developing spontaneous tumors at an average age of 12 months. Hence, this showed that the knockout of Bif-1 significantly enhances the development of spontaneous tumors in mice [14].
Accumulation of p62
p62 (also known as sequestosome-1 or SQSTM 1) is a multi-domain protein that interacts with the autophagy machinery as a key adaptor of target cargo by linking LC3 with ubiquitin moieties on misfolded proteins. Autophagy, therefore, mediates the clearance of p62 together with ubiquitylated proteins. Hence, suppression of autophagy results in p62 accumulation and contributes to tumorigenesis [29]. Increased evidence of p62 upregulation and/or reduced degradation has been implicated in tumor formation, cancer promotion as well as in resistance to therapy. The growth of liver tumors caused by inhibition of autophagy was greatly reduced by p62 deletion [21]. Chronic p62 elevation contributes to hepatocellular carcinoma by preventing oncogene-induced senescence and death of cancer-initiating cells and enhancing their proliferation [30]. Tumor promotion activity of p62 was observed in hepatitis B virus (HBV)-associated hepatocarcinoma [31], breast [32], gastric [33], and prostate cancers [34].
Oncogene-induced senescence
Autophagy is regarded as an effector mechanism of senescence which is essential for the rapid protein remodeling required to make the efficient transition of a tumor from a proliferative to a senescent state. Furthermore, inhibition of autophagy delays the senescence phenotype. Hence, autophagy might be required in this process as a tumor suppressor [35].
Tumor growth or pro-survival mechanism?
As seen in the previous section, insufficient autophagy can promote a normal cell to a state of a malignant tumor. However, increasing evidence also suggests that the tumor in the advanced stages of tumorigenesis is strongly dependent on autophagy for survival and proliferation. Poor sensitivity of anticancer therapies showed that autophagy, rather than inducing autophagic cell death (as presumed earlier), is helping cancer cells cope with the metabolic stress and hypoxia progressing to tumor growth. Many observations suggest the need for autophagy in cancer development.
BECN-1
In contrast to the previous study, which depicted decreased beclin-1 expression in breast cancer cells compared to the normal breast cells [8], another study indicated that expression of beclin‐1 was detected in 95% of the colorectal carcinomas and 83% of the gastric carcinomas. In contrast, normal mucosal cells of both stomach and colon showed no or very weak expression of beclin‐1. This identification suggests the possibility that neo‐expression of beclin‐1 may play a role in both colorectal and gastric tumorigenesis [36].
Another study showed that monoallelic deletion of beclin-1 blocks or delays tumor formation in TSC2 + / − and ATM − / − mice, respectively [37, 38].
FIP200
Again, in contrast to monoallelic deletion of beclin-1 leading to tumor progression, the knockout of essential autophagy proteins including FIP200 (FAK family-interacting protein of 200 kDa) in various tissues did not lead to malignant tumor development in vivo. Furthermore, inhibition of autophagy by FIP200 ablation suppresses mammary tumor initiation and progression in a mouse model of breast cancer. This provided strong evidence for a pro-tumorigenesis role of autophagy in oncogene-induced tumors in vivo [39].
ATG5 and ATG7
It was observed that both the mRNA and protein levels of ATG5 and ATG7 were increased in cells overexpressing K-RasV12. K-RasV12 overexpression leads to malignant transformation in human breast epithelial cells. Hence, this provided the first evidence that autophagy is essential for oncogenic K-Ras (K-RasV12)-induced malignant cell transformation [40].
Cytoprotective traits
The cardinal role of autophagy in established tumors is to fulfill their nutrient/oxygen requirements and allow them to survive in conditions of metabolic and cellular stresses.
It was seen that normal cells die within 24 h when subjected to extreme nutrient starvation. On the other hand, liver cancer cells died within 36 h, > 50% of pancreatic cancer cells survived, even after starvation for 48 h, 1 of 3 gastric cancer cells survived > 36 h, and > 50% of the colon cancer cells survived > 36 h [41]. In solid tumors, enhanced autophagy is frequently found in the hypoxic regions contributing to cell survival [42]. Due to increased cell proliferation, cancer cells have a high demand for nutrients and oxygen. When the blood supply is insufficient, particularly during the initial steps of tumor formation or in the poorly vascularized regions of solid tumors, cancer cells encounter metabolic stress and induce autophagy to allow them to survive [43, 44].
Induction of dormancy
Dormancy is a stage in cancer progression where the cells cease dividing but survive in a quiescent state while waiting for appropriate environmental conditions to begin proliferation again [45]. It is seen that autophagy has a crucial role in inducing tumor cell dormancy under stressful conditions.
Pharmacologic or genetic inhibition of autophagy in dormant breast cancer cells results in significantly decreased cell survival and metastatic burden in mouse and human 3D in vitro and in vivo preclinical models of dormancy [46]. Another study shows that the tumor suppressor gene aplasia Ras homolog member I (ARHI; also known as DIRAS3), in an ovarian cancer xenograft mouse model, leads to acute cell death and tumor regression initially. However, subsequently, the tumoral resurgence is furnished when ARHI expression and autophagy are terminated. This showed that autophagy helps the cells to remain dormant and still survive [47, 48].
Inhibition of anoikis
Anoikis is a programmed cell death induced upon cell detachment from the extracellular matrix, thus avoiding colonizing of distant organs [49]. Inhibition of cell death by anoikis due to autophagy induction during the later stages of tumor progression, such as tumor cell dissemination and metastasis, can lead to tumor cell survival [50]. Due to anoikis inhibition, the cancer cells undergo malignant transformation and can be transferred through blood or lymphatic circulation to a distant location, undergoing metastasis [51]. It was shown that the knockout of Atg5 and Atg7 genes in the 3D culture model of MCF-10A induced pro-survival autophagy in the epithelial cells promoting cell survival during anoikis [50, 52]. Furthermore, clinical studies deduced that the under-expression and overexpression of autophagy in melanomas were associated with poor prognosis. However, the latter manifested metastases much earlier than the former [53]. Similarly, hyperactivation of autophagy in HCC (hepatocellular carcinoma) facilitates malignant tumor differentiation, which results in a more aggressive cancer cell phenotype and poor prognosis of HCC [54].
Adaptation to reprogrammed metabolism
Cancer cells are observed to rewire their metabolism and energy production networks to support their increased biosynthetic needs [55]. In 2011, Drs. Hanahan and Weinberg have recognized cancer metabolic reprogramming as one of the ten cancer hallmarks and published it in their seminal review paper [6]. In the 1920s, the legendary German biochemist Otto Warburg established the first link between tumorigenesis and metabolic reprogramming and is also known as the "Warburg effect." Normally differentiating cells rely primarily on mitochondrial oxidative phosphorylation for fulfilling their metabolic requirements and energy generation. In contrast, high levels of aerobic glycolysis activity were observed in cancer cells [56]. The hypothesis is that mitochondrial dysfunction leads to its repurposing, facilitating the uptake and incorporation of nutrients into the biomass (e.g., nucleotides, amino acids, and lipids) needed to produce a new cell [56, 57].
Shifting of the metabolic pathway from oxidative phosphorylation to aerobic glycolysis is induced and regulated by several oncogenes. In cells transfected with activated RAS and SRC oncogenes, the rate of glucose uptake was markedly increased, which is a sign of their metabolic reprogramming [58, 59]. Activation of RAS can also induce autophagy in tumor cells [60,61,62]. There is an accumulation of abnormal mitochondria and reduced oxygen consumption in Ras expressing cells. As mitochondria sustain the viability of Ras expressing cells in starvation, autophagy is required to maintain a pool of functional mitochondria necessary to support Ras-driven tumors' growth [60, 62]. Hence, inhibiting autophagy virtually abolishes the tumorigenicity of RAS and reduces glucose metabolism.
The MYC oncogene, which contributes to the genesis of many human cancers, encodes a transcription factor c-Myc, which links altered cellular metabolism to tumorigenesis [63]. Similarly, the constitutive activity of the serine/threonine kinase AKT (also known as protein kinase B (PKB)) is a common perturbation observed in malignant cells. It was observed that Akt activation did not lead to the increased proliferation of malignant cells but stimulated glucose consumption in transformed cells without affecting the rate of oxidative phosphorylation [64, 65]. All these studies suggested that activation of oncogenes, such as RAS, SRC, MYC, and AKT, seems to orchestrate the metabolic changes associated with cell transformation [66].
Survival and self-renewal of cancer cells
Since the establishment of the cancer stem cell (CSC) theory and the discovery of CSCs in individual cancer types, autophagy has been proposed to be a key mechanism in their homeostasis, dismissal, or spread [67]. Cancer stem cells (CSCs) are a small subpopulation of cells within tumors with capabilities of self-renewal, differentiation, and tumorigenicity when transplanted into an animal host [68]. Pluripotency is the key feature of CSC, which allows them to divide indefinitely and maintain the undifferentiated state [69].
From the study of breast cancer CSCs, it was found that autophagy regulates breast CSC maintenance in autophagy-dependent breast cancer cells by modulating IL6 secretion, hence maintaining the pluripotency of the CSCs [70, 71]. Furthermore, autophagy is found to be upregulated in the mammospheres [72, 73], compared to adherent cells, and both Beclin 1 and ATG4, two essential autophagy proteins, are needed for their maintenance and expansion [74].
In recent studies, autophagy's relationship with various CSCs has been identified and documented. These include breast [75, 76], pancreatic, liver [77], osteosarcoma [78], ovarian [79], and glioblastoma [80] CSCs, in which autophagy impairment negatively affects the expression of staminal markers and consequently the cell self-renewal capacity [74].
Autophagy in different stages of oncogenesis
From various studies, it has been observed that the process of autophagy always acts as a means of protector during oncogenesis, even if it serves dual roles of tumor suppressor and promoter in various stages of oncogenesis. Autophagy safeguards healthy cell homeostasis during the early stages of oncogenesis via restricting the genome instability by retarding the stem cells involved damage/repair cycle and prohibiting the formation of inflammation or tumor microenvironment. During the late stage of oncogenesis, autophagy safeguards the survival of tumor cells by providing support for metabolic demand and reducing metabolic damage. Further, autophagy increases the migratory behavior of cancer cells by initiating the anoikis resistance and dormancy [81].
Autophagy modulation: overcoming resistance to cancer therapies
As discussed previously, the bipolar nature of autophagy leads to its completely contrasting traits in various cancers. It can be upregulated or downregulated at different stages and conditions of cancers. Additionally, this behavior impacts the anticancer therapies devised for multiple cancer types. Depending on its subsequent roles, it can have either a beneficial or a detrimental influence on the patients undergoing cancer treatment therapies. Let us see how.
Drugs inhibiting autophagy
As discussed in “Drugs inducing autophagy” section, autophagy acts as a pro-survival mechanism by aiding the hypoxic and nutrient-deficient tumor cells to survive such hostile conditions. In this case, the activation of autophagy leads to tumorigenesis. Similar to this, recent studies have demarcated growing evidence of autophagy's potential for protecting cancer cells against chemotherapy by mediating their acquired resistance phenotype.
Recent studies have provided increasing shreds of evidence that autophagy is seen to upregulate in tumor cell lines on exposure to various anticancer therapies. These can include radiation therapies, chemotherapy, and targeted cancer therapies [82, 83]. Hence, pre-studies demonstrated that introducing drugs that could inhibit autophagy, along with anticancer drugs, resulted in the increased sensitization of tumor cells to anticancer therapeutic agents [84] (Fig. 3).

Induction and inhibition of autophagy cancer therapy: Cancer cells experience either apoptotic or autophagic cell death or both when treated with autophagic inducers in combination with chemotherapeutics entrapped in nanoparticles. Such an approach provides a cytotoxic process. While some cancerous cells activate autophagy as a response to alleviate the stress induced by the chemotherapeutics and provide cytoprotective autophagy which promotes cell survival. In such cases, nanoparticles are administered comprising of a combination of autophagic inhibitors and chemotherapeutics. Autophagic inhibitors sensitize the cancerous cells toward the chemotherapeutics, as a result, increase the cytotoxic effects induced by the chemotherapeutics leading to apoptosis
Many in vitro and in vivo studies have been carried out to validate this hypothesis. In human colon cancer cell lines, autophagy was seen to have a cytoprotective role against 5-fluorouracil (5-FU)-based adjuvant chemotherapy. However, autophagy inhibition by 3-methyladenine (3-MA) or small interference RNA targeting Atg7 (Atg7 siRNA) produced a synergistic effect along with 5-FU by significantly augmenting 5-FU-induced apoptosis [85]. A similar observation was noted when 5-FU was used with another autophagy inhibitor, chloroquine (CQ) [86]. In hepatocellular carcinoma (HCC) tumors, the anticancer effects of bevacizumab and sorafenib were augmented by using them in combination with chloroquine or 3-MA and CQ, respectively [86, 87]. Furthermore, in human lung cancer cell lines, EGFR tyrosine kinase inhibitors, gefitinib, and erlotinib, when solely used, were seen to acquire therapy resistance by inducing high levels of autophagy. However, cytotoxicity induced by these drugs was significantly enhanced when used in amalgamation with autophagy inhibitors like CQ and siRNAs targeting ATG5 and ATG7, which are essential autophagic components [88, 89]. Also, in CML, the drug suberoylanilide hydroxamic acid (SAHA), which is a histone deacetylase (HDAC) inhibitor, when used in combination with 3-MA, CQ, or bafilomycin A, helps overcome autophagy-aided drug resistance [90].
Multiple potential anticancer therapies and autophagy inhibitor drug combinations have been identified and documented. It is seen that different autophagy inhibitors work in association with different anticancer therapies by blocking the autophagic process at different stages. Some examples include CQ, HCQ (anti-malarial drugs), and bafilomycin A1 sensitize tumor cells to therapy by inhibiting autophagosome fusion with lysosomes and its degradation. 3-MA, LY294002, and Wortmannin lead to Class III PI3K inhibition, impeding autophagy's initiation or expansion stage. Furthermore, autophagic inhibition is also achieved by genetic silencing of autophagy regulatory genes [91,92,93,94].
Drugs inducing autophagy
As discussed in “Drugs inhibiting autophagy”, autophagy is also a self-cannibalistic process despite its clear pro-survival role and cytoprotective traits. This is due to its ability to induce cell death of cancer cells in case of excess or enhanced autophagy. Increasing evidence has been provided stating it exhibits a pro-death mechanism in cases of hampered or defective apoptosis [95,96,97,98]. This role of autophagy contributes to the efficacy of anticancer drugs by generating an autophagy-mediated cell death mechanism (Fig. 2). Anti-tumor action of cannabinoids (autophagy inducers) was enhanced in glioma cancer cells by the inhibition of mTORC1 and thereby activation of an ER stress response which promoted autophagy [99]. Similarly, the activity of gemcitabine (used in the treatment of advanced pancreatic adenocarcinoma) was augmented by adding cannabinoids. Their combined effect strongly inhibited the growth of human pancreatic tumor cells xenografted in nude mice without apparent toxic effects [100]. Furthermore, cannabinoids were also found to generate an anti-tumoral action on hepatocellular carcinoma by activating the AMPK pathway and autophagy [101]. These studies highlighted the possibility of designing new therapeutic strategies that could exploit autophagy's pro-death mechanism. In Atg5 and Atg7 knockout cultured colon cancer cells, the cytotoxicity of FK-16 (derived from anticancer peptide LL-37) was seen to be attenuated, which pointed toward the pro-death nature of FK-16-induced autophagy [102]. The cell death induced by temozolomide in the glioblastoma cell line was potentiated by the mTOR inhibitor, RAD001 [103].
All these results strengthen the connection between autophagy and anticancer sensitivity. Hence, all these results point toward autophagy induction being considered as a new hope for possibilities in targeting and designing treatments for these malignancies [104].
Targeting autophagy in cancer: nanotechnology-based approaches
We have seen how the contrasting roles of autophagy and its modulation in cancer can be exploited to understand the designing of chemotherapies and treatments for cancer patients. On the one hand, in tumors with elevated levels of autophagy, anticancer agents are administered along with autophagy inhibitors. This inhibits tumor cell survival under metabolic and chemotherapy stress. On the other hand, overstimulation of autophagy, especially in apoptosis defective cells, by the use of autophagy inducers along with cytotoxic drugs may lead to autophagy-mediated tumor cell death [104]. Hence, the importance and utility of conventional chemotherapeutic drugs could not be denied; however, they do carry a variety of side effects along with them [105]. Therefore, new and improved treatments involving the role of nanotechnology provide novel approaches to harness more possibilities in cancer treatments [106]. This section covers the various aspects and advances in the use of nanoparticle-based strategies as potent modulators of autophagy and their therapeutic implications in cancer.
Why are nanoparticles (NPs) compelling candidates compared to common autophagy-inducing drugs?
Conventional drugs pose certain obstacles to offering efficient, safe, and innovative agents to patients for fighting cancer. Various side effects are triggered by conventional drugs, which include poor specificity, uneven tissue, and organ distribution, rapid clearance of the drug, and its biodegradation [105]. The use of nanomedicine addresses these issues and provides multiple advantages faced by traditional drugs [107,108,109,110,111].
NPs have specific properties, making them versatile carriers contributing to a wide range of biomedical and pharmaceutical applications [112,113,114,115,116]. The first property includes NPs platforms which are "scaffolds" for assembling defined multi-functional structures. These provide abilities to generate a wealth of possibilities by simply incorporating slight variations in composition, surface coating, and ligand choice [117,118,119]. Secondly, they have a high surface-to-volume ratio. This property provides vital importance for optimizing drug payloads and other NP-based interactions [120,121,122]. Thirdly is its controlled shape and size. The shape of the NPs directly affects their function in vivo [123,124,125,126]. Further, the careful size modulation of these structures can provide a perfect transition from effective renal clearance (for smaller NPs) to accumulation in the body (for larger NPs), depending on the therapy requirements [127,128,129]. Lastly, the optical properties of NPs help in facilitating an image-based drug delivery hence combining diagnosis and treatment [130]. Thus, these four properties broadly answer the reason for an enthusiastic interest in this field, which tends to mark a paradigm shift in cancer treatment [131].
Elaborating on the importance of NPs concerning cancer specifically, it is found that all these properties, though useful, do lead to issues like NP toxicity.
Hence, NPs can enhance the effectiveness of standard chemotherapy treatment by acting as cytotoxics [132, 133]. NPs can be classified into four categories based on their size, shape, morphology, and chemical properties. These categories include metals, liposomes, polymers, and carbon-based NPs.
Modulation of autophagy by metallic nanoparticles
Metallic NPs can selectively overstimulate autophagy. They cause this “selective” overstimulation by creating imbalances in the cellular signaling pathways, which do not affect the non-cancerous cells. This approach provides an exciting area of exploration and carries the potential of designing therapies for various cancers.
Silver-based nanoparticles
When silver nanoparticles (AgNPs) were embedded into specific polysaccharides (EPS), then it was observed that they produced pronounced cytotoxic activity against breast cancer cell lines. This mode of action of AgNPs was elucidated by using SKBR3 breast cancer cells. In particular, AgNPs were found to increase ROS generation, supporting cell death primarily through autophagic cell death mechanism and minorly through apoptotic cell death [134] (Fig. 4). In another study for identifying the underlying molecular mechanism of AgNPs against pancreatic cancer cells, it was found that these NPs induced tumor cell death through necrosis and apoptosis, occurring with autophagy. Further, identification of elevated levels of autophagy biomarker, LC3-II, in the tumor cells as opposed to the non-tumor cells showed the selectivity of AgNPs for inducing cytotoxicity [135].

Modulation mechanism of NP in autophagic-signaling pathways: Current evidence suggests that the nanoparticles (NP) are regarded as an endosomal pathogen to the cells. So, like the cellular substrates, the NP also undergoes ubiquitination which leads to their sequestration via phagophore. NP can induce autophagy by inducing mitochondrial damage and ER stress, by altering autophagic-dependent genes and proteins, and by suppressing and downregulating Akt-mTOR signalings. NP can also inhibit autophagy by inducing lysosomal degradation, and upregulating Akt-mTOR signalings
Many studies were carried out that looked at the collective effects of an anticancer drug along with forms of AgNPs. A report depicted a synergistic effect between Cisplatin, a very widely used chemotherapeutic drug) and rGO-AgNPs (a reduced graphene oxide–silver NP nanocomposite) against human cervical cancer (HeLa) cells. This combination led to more pronounced effects on the expression of autophagy genes and the accumulation of autophagosomes and autophagolysosomes, which were associated with the generation of ROS and cell death. These findings depicted the ability of rGO-AgNPs to potentiate cisplatin-induced cytotoxicity, autophagy, and apoptosis in HeLa cells [136]. Another notable study illustrated synergistic interactions between a novel bacterium synthesized AgNPs and salinomycin against ovarian cancer cells. In comparison with their individual anticancer effects, their combination produced a dramatic inhibitory effect on cell viability and morphology. The combination of salinomycin and AgNPs produced more cytotoxicity and expression of apoptotic genes, along with significantly inducing the accumulation of autophagolysosomes in these ovarian cancer cells. This increased autophagy led to mitochondrial dysfunction, eventually leading to cell death. This proposed a relevant targeted therapeutic strategy for treatment against ovarian cancer [137]. Another study demonstrated that the anti-tumor effect of AgNPs in the B16 mouse melanoma cell model was augmented by wortmannin, a widely used inhibitor of autophagy [138].
The ability of AgNPs was not limited to potentiating the effect of chemotherapeutic drugs, but they were also found to be effective nanoradiosensitizers for the treatment of glioma. In comparison with AuNPs, AgNPs exhibited more powerful radiosensitizing abilities, leading to a higher rate of apoptotic cell death. Furthermore, as compared to AuNPs, AgNPs together with radiotherapy led to a significant upregulation of autophagy as revealed by LC3-II protein level, acridine orange (AO), and monodansylcadaverine (MDC) staining. This study suggested that the modulation of autophagy by treatment with AgNPs may improve glioblastoma therapeutic outcomes [139].
Gold-based nanoparticles
Gold nanoparticles (AuNPs) act as drug delivery scaffolds because their surfaces can act as modifiable sites. These sites can be easily modified with various substances like chemotherapeutics, oligonucleotides, and proteins. AuNPs have great utility in biomedical research due to their fascinating characteristics. These include low toxicity and immunogenicity, good biocompatibility, and excellent stability [140]. In 2011, the mechanism of action of AuNPs on lysosomes was studied. It revealed that AuNPs could be taken up into the lysosomes through endocytosis. Its subsequent accumulation could impair lysosomal degradation and induce autophagosome accumulation. Hence, they play a significant role in interrupting the autophagic pathway [141].
A study reported that 11-mercaptoundecanoic acid-modified AuNPs, when conjugated with chloroquine (GNP-Chl), led to inhibition of cancer cell growth in MCF-7 breast cancer cells. It revealed that cell death was mediated by autophagy [142].
EGFR is found to be overexpressed in 70–80% of TNBC and is a promising target for TNBC treatment [143]. A study demonstrated that near-infrared photothermal therapy (NIR-PTT) using anti-EGFR AuNPs (anti-EGFR-GNs) increases autophagic cell death in breast cancer cells. The researchers validated this idea by investigating autophagic cell death on treatment with anti-EGFR-GNs combined NIR-PTT in TNBC cells and mouse xenograft tumors. Interestingly, on the administration of 3-MA, an autophagy inhibitor, the cytotoxicity of TNBC cells was rescued, implying high levels of autophagy induction by anti-EGFR-GNs-combined NIR-PTT. Many autophagic vesicles and a significant increase in autophagy-specific markers accompanying the inhibition of the AKT-mTOR signaling pathway were responsible for inducing autophagy. Additionally, increased levels of LC3 and beclin-1 in mouse xenograft tumors further validated the result. Hence, the combination of AuNPs with NIR-PTT represents a promising strategy for effectively targeting EGFR in TNBC [144].
Rad6 protein is overexpressed in TNBC cells and is a key player in DNA damage tolerance, post-replication DNA repair mechanism, and mitochondrial stability [145, 146]. Hence, its inhibition (by SMI#9) would provide a new way to combat TNBC. A recent study showed that chemically modified SMI#9-GNP is endocytosed and releases active SMI#9, which selectively induces cytotoxicity only in cancer cells. Increased levels of autophagy and apoptosis were observed in TNBC cells after treatment with SMI#9-GNP. Also, SMI#9-GNP, when combined with Cisplatin, is seen to increase cisplatin sensitivity synergistically. These observations show the essential role of nanotechnology-based Rad6-targeting therapy for TNBCs [147].
TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) is a highly regarded anti-tumor agent, but its resistance in most cancers led to therapeutic inefficacy. So, in another study, it was found that the combination of AuNPs with TRAIL exhibited greater potency in promoting apoptosis in non-small cell lung cancer (NSCLC) cells compared with TRAIL alone. It was found that the combination was able to promote Drp1-mediated mitochondrial damage leading to apoptosis, autophagy, and mitophagy activation. As a result, the combined administration of TRAIL along with AuNPs may promise a therapeutic strategy to overcome TRAIL resistance in many cancer cells [148].
Metal oxide-based nanoparticles
Metal oxide NPs depict a versatile behavior and provide effective biomedical applications.
Zinc oxide nanoparticles
A study revealed that exposure of human ovarian cancer cells (SKOV3) to zinc oxide nanoparticles (ZnO NPs) leads to significant induction of cytotoxicity, apoptosis, and autophagy. Apoptotic features like reactive oxygen species generation and autophagic markers like upregulation of p53 and LC3 indicate that ZnO NPs upregulate apoptosis and autophagy [149, 150]. Another study elucidated the anticancer effect of ZnO NPs on CAL 27 human tongue cancer cells by identifying the role of PINK1/Parkin-mediated mitophagy in this effect. They confirmed that the CAL 27 cell viability decreased upon treatment with ZnO NPs due to reactive oxygen species generation and activation of PINK1/Parkin-mediated mitophagy [151]. The conjugation of ZnO NPs with Meso-Tetra (4-Carboxyphenyl) Porphyrin (MTCP) was seen to augment cytotoxicity in the two human breast adenocarcinoma cell lines (MCF-7 and MDA-MB-468). This was caused by the non-canonical apoptosis and autophagy induction in these tumor cell lines [152]. Hence, ZnO NPs possess interesting characteristics which could be exploited for devising effective tumor therapies.
Iron oxide nanoparticles
Iron oxide nanoparticles (IO NPs) have shown their potential in biomedicine applications, such as tumor therapy, specifically by inducing autophagy-mediated cell death of cancer cells. A study revealed that IO NPs selectively induce autophagy in cancer cells (A549) and not in normal cells (IMR-90) [153]. Due to the drug delivery and multi-imaging functions of IO NPs, they are widely used in cancer treatments. Yet, their use is limited due to their therapeutic efficacy and biological safety issues. IO NPs induce autophagosome accumulation through multiple mechanisms like lysosome impairment, mitochondrial dysfunction, and ER stress [154].
IO NPs have multi-functional properties of super-paramagnetism. Combining anti-EGFR with IO NPs suppresses lung tumor growth by abrogating G2/M cell-cycle arrest and inducing DNA damage, autophagy, and apoptosis [155]. In a different study, N-((2-hydroxy-3-trimethylammonium) propyl) chitosan chloride (HTCC)/alginate-encapsulated Fe3O4 magnetic nanoparticles (HTCC-MNPs) were developed using an ionic gelation method. These were applied to multi-drug-resistant (MDR) gastric cancer models. They observed that the HTCC–MNPs were more cytotoxic in both the SGC7901 human gastric cancer cell line and MDR variant cell line (SGC7901/ADR) than in normal gastric epithelial cell line (GES). They predicted the reason for cell death as attributed to autophagy induction due to identification of co-localization of LC3 with lysosomal marker LAMP2 and an increased LC3-II/LC3-I ratio [140, 156]. Finally, they concluded that the tumor volume was reduced via the induction of cellular autophagy and apoptosis, which was characterized by ROS generation and mitochondrial damage [156].
Some studies revealed the importance of analyzing the size and coating characteristics of the IONPs in their application. An interesting study revealed that Polyethylenimine (PEI)-coated IONPs lead to severe toxicity in both SKOV3 human ovarian cancer cells and macrophages due to multiple mechanisms, including the disruption of cell membrane integrity, ROS generation, and apoptosis. On the other hand, no apparent cytotoxicity was observed for PEGylated IONPs at even much higher concentrations. The authors observed changes in autophagosome formation when SKOV3 cells were exposed to PEGylated IONPs through TEM imaging and by detecting the level of autophagy marker LC3-II [116, 133].
Copper and cuprous oxide nanoparticles
Cuprous oxide nanoparticles (CuO NPs) or copper oxide nanoparticles (CO NPs) are also used to selectively induce cytotoxicity in tumor cells, leading to the suppression of tumor proliferation. They adopt mechanisms that include stimulating apoptosis and autophagic cell death, tumor growth and metastasis inhibition, and nephrotoxicity [150, 157,158,159]. These pharmacological effects make it a potential nanomedicine for cancer therapy.
An excellent study provided evidence of the therapeutic potential of CuO NPs and their effectiveness in comparison with cisplatin in the treatment of cervical carcinoma. According to TEM and autophagic flux, CuO NPs were found to induce autophagosome formation, which increased in a time- and concentration-dependent manner. Also, it was seen that they led to autophagy induction through the AKT/mTOR pathway by decreasing the phosphorylation of AKT and mTOR [160].
Another study revealed that CuO NPs induce autophagy in MCF3 breast cancer cells in addition to oxidative stress generation and deregulation of normal cellular activities. However, in this case, the authors hypothesized that autophagy is being adopted as a survival strategy by the MCF3 cells, thus representing its pro-survival role. Hence, the inhibition of autophagy by 3-MA led to the induction of apoptosis. Finally, it was concluded that combining CuO NPs with an autophagy inhibitor is essential for apoptosis induction in breast cancer cells [161].
Carbon-based nanoparticles
Many carbon-based nanoparticles (C-NPs) modulate autophagy to target cancer cells effectively. Carboxyl-functionalized carbon nanotubes (COOH-CNTs), a type of functionalized single-walled carbon nanotubes, induced autophagic cell death in human lung adenocarcinoma A549 cells by affecting the mTOR/AKT pathway (by the formation of autophagosomes and LC3-II upregulation). However, other functionalized CNTs (like PABS-CNT and PEG-CNT) did not replicate the same behavior, implicating the impact of surface group characteristics on autophagic modulation. More so, the administration of autophagy inhibitors like 3-MA evaded lung toxicity in mice, potentiated by COOH-CNT [162].
A study on C-NPs demonstrated that derivatized fullerene C60 induces autophagy, augmenting chemotherapeutic killing of both normal and drug-resistant cancer cells by reducing drug resistance [163]. Further, it is also noteworthy that graphene-based nanomaterials (GNM), such as graphene, graphene oxide, and graphene quantum dots also play a role in the induction of autophagy [164]. Several studies have validated this. For instance, co-treatment of graphene oxide (GO) nanomaterial with cisplatin induces significant cell death, by inducing autophagic flux and chemosensitizing colon cancer CT26 cells [165], ovarian cancer Skov-3 cells, cervical cancer HeLa cells, as well as prostate cancer (Tramp-C1) cells [166]. Additionally, GO and Chloroquine co-administration in A549 human lung carcinoma cells caused cellular toxicity by the cytotoxic block of autophagic flux, leading to necroptosis [167].
Silica-based nanoparticles
Silica-based nanoparticles (Si-NPs) possess many distinctive properties, making them a very suitable drug delivery vehicle and numerous applications in gene transfection, biosensing, and bioimaging. It has interesting characteristics like biocompatibility, tunable pore size, high surface area, and ease of modification [168,169,170] and is hence used as a potential approach in cancer therapy.
A study revealed that Si-NPs impart selective toxicity in glioblastoma cells compared to normal skin fibroblasts. On treatment with Si-NPs, the authors noticed the induction of apoptosis and necrosis in LBC3 cells, whereas the induction of necrosis in LN-18 cells. This revealed the anticancer therapeutic potential of Si-NPs for the treatment of glioblastoma [171]. The accumulation of Si-NPs in the human cervix carcinoma cells leads to lysosomal dysfunction and inhibition of autophagy-mediated protein turnover. As a result, the metabolic activity of the cancer cells is hampered [172]. In a recent study, the authors chemically synthesized genistein-PEGylated silica hybrid nanoparticles (Gen-PEG-Si-NPs) by incorporating genistein (Gen) into PEG-Si-NPs. As a result, Gen's antioxidant and anti-proliferative effects on HT29 human colon cancer cells were potentiated, which activated both cell death mechanisms (apoptosis and autophagy), in contrast to Gen, which only triggered apoptosis [173]. The study proposed using Gen-PEG-Si-NPs as an alternative treatment for colorectal cancer in the near future.
In addition to its benefits, it has also been observed that Si-NPs induce ROS and autophagy dysfunction in HCT-116 colon cancer cells, L-02, and HepG2 hepatoma cells, generating an increased concern for the careful study of Si-NPs toxic effects [174,175,176].
Modulation of autophagy by polymer nanoparticles
Polymeric NPs exhibit various advantages as compared to conventional delivery systems like improved stability, and biocompatibility, non-toxicity, biodegradability, increased accumulation at the cancer site via enhanced permeability and retention (EPR) effect, and controlled drug release [177,178,179,180]. Also, the polymeric NPs reduce the multi-drug resistance-associated with many chemotherapeutics [181].
Eudragit RS polymeric NPs were found to be cytotoxic for NR8383 rat macrophages. In continuation with this discovery, a study in 2011 demonstrated the relationship between the cytotoxicity induced by Eudragit RS in NR8383 rat macrophages and autophagy activation. TEM showed internalization of Eudragit RS NPs in the cells leading to mitochondrial damage. The authors proposed that cells adopt the mechanism of autophagy followed by cell death to get rid of the oxidative stress created due to damaged mitochondria [182].
Cationic polyamidoamine dendrimer (PAMAM) triggered autophagic cell death in A549 lung cancer cells by the deregulation of the mTOR pathway [183]. However, it was not specified whether the upregulated LC3, as observed by western blotting and microscopy, was due to the enhanced on rate or decreased off rate of autophagosomes. Hence, the blockage of autophagy cannot be kept aside [184]. Another study demonstrated that PAMAM dendrimers resulted in lysosomal impairment due to the alkalinization of lysosomes [185].
Polymer therapeutics or polymer conjugates are observed as one of the most growing first-generation polymeric nanoparticles. A study was performed where polymeric nanomicelles were prepared for the co-delivery of doxorubicin and LY294002 (autophagy inhibitor) for the effective treatment of oral squamous cell carcinoma (OSCC). In this study, the hydrophobic DOX was conjugated with pH-responsive, hydrophilic polyacylhydrazone (HPAH). The amphiphilicity of HPAH − DOX conjugates facilitated the self-assembly of nanomicelles within an aqueous solution, and the pH responsiveness of the conjugates facilitated the release and rapid entry of LY294002 and DOX to an intracellular acidic environment inducing inhibition of proliferation of cancer cells. It was observed that the preferential release of LY294002 inhibited the autophagy of tumor cells, making the cancer cells more sensitive to the subsequent liberation of DOX [186]. Similarly, N-(2-hydroxypropyl) methacrylamide (HPMA)–doxorubicin (Dox) conjugate PK1 (FCE28068), was prepared as a synthetic polymer–drug conjugate. The results showed that the cancer cells treated with FCE28068 exhibited increased levels of LC3II and Beclin 1, the proteins associated with the formation of the autophagosome. Moreover, it was observed that the process of autophagy proceeds via glutamine metabolism, and the treatment with FCE28068 showed a reduction in glutamine concentration, proving the facilitation of autophagy in MCF-7 cells [187]. In this context, polymer–peptide conjugates also play an important role in nanomedicine as cancer therapy due to their longer circulation time and improved therapeutic efficacy for targeted tumor delivery than free peptides. 14-amino acid polycationic peptide (KLAKLAK)2 (abbr. KLAK) was conjugated with self-assembled polymers (P-KLAK) which caused autophagosomes accumulation via lysosome impairment facilitating autophagy blockage and mitochondrial damage, inducing U87 cell death [188].
Modulation of autophagy by liposomal nanoparticles
Liposomal NPs also possess many distinctive properties due to their flexible surface characteristics [189,190,191]. Cationic lipids are a commonly used class of transfection agents. In 2010, a study demonstrated the induction of autophagy by dioleoyl trimethylammonium propane (DOTAP), a cationic lipid in HeLa cells of cervical cancer. Analysis of autophagy was carried out by observation under TEM and autophagic flux. The results suggested that autophagy induced by cationic lipids was primarily due to increased formation of autophagosomes and not decreased turnover. Also, the process was mTOR independent. The researchers hypothesized that the induction of autophagy might be caused by the triggering of an additional degradative pathway (i.e., autophagy) in response to DOTAP, a synthetic lipid [192]. However, the mechanism of autophagy induction by cationic lipids was not understood. So, another study in 2013 demonstrated that cationic liposomes enter cells in endosomes, activate autophagy, and generate tubulovesicular autophagosomes, which capture autophagic cargoes followed by a slower rate of lysosomal fusion [193].
Naturally, it is known that the administration of transfection agents stimulates autophagy induction, which leads to poor transfection effectiveness. Hence, another possible strategy could be the inhibition of autophagy to improve transfection efficiency. So, the study also showed that gene delivery and expression increased many folds in atg5 (-/-) cells, which were autophagy defective cells [193]. Interestingly, neutral lipid (i.e., dioleoylphosphatidylethanolamine; DOPE) was inefficient in modulating autophagy in HeLa cells [192]. This observation, however, was in stark contrast to another study also carried out in 2013. The authors showed that neutral PEGylated C6-ceramide nanoliposomes activate autophagy in liver HepG2 cells. They further illustrated the synergistic combination therapy combining nanoliposomal C6-ceramide, an autophagy inducer, and vinblastine, the autophagy maturation inhibitor, as being associated with autophagy dysfunction in hepatocarcinoma and colorectal cancer models [194].
Liposomal NPs show several advantages, especially as a modulator of autophagy for the treatment of glioma. For instance, functionalized liposomes can cross the blood–brain barrier in the CNS, which is otherwise impossible for chemotherapeutic drugs and autophagy inhibitors to penetrate. Based on this property, R6dGR peptide-functionalized liposome (R6dGR-Lip) was developed, which encapsulated ZD6474 and hydroxychloroquine (HCQ) inhibitors for the targeting of glioma cells through blockage of autophagic flux [195].
Thus, the versatile liposomal NPs have been identified as the most successful among the others in oncological settings. They can be used as carriers of chemotherapeutics and/or autophagy modulators, due to their useful biological properties including low toxicity and immunogenicity, along with the added advantages of being cheap and biodegradable [195,196,197,198].
The various nanoformulations developed in the last decade to modulate the autophagic pathways for the treatment of cancer have been summarized in Table 1.
Future perspective
At present, immunotherapy is rising as a promising treatment approach against cancer. The immune system serves an important role in cancer treatment by recognizing and destroying cancer cells during various stages of cancer development. Various studies showed that autophagy could up-modulate as well as down-modulate the immune response by releasing cytokines and regulating immune cells. Cancer immunotherapy is emerging as a new generation of anticancer therapeutics. Till now, antibody targeting therapy in synergistic with autophagy has been reported. A humanized mAb (farletuzumab) was developed against folate receptor in an ovarian cancer model, which exhibited an anticancer activity via antibody-dependent cellular cytotoxicity by assisting late-stage autophagy. Presently, therapies directed at autophagy to improve the immune responses and anticancer effects of immunotherapy have become an eventual strategy, with increased antigen presentation and higher sensitivity to cytotoxic T cells (CTLs). It was observed that autophagy exhibits a pivotal role in antigen processing for both MHC-I and MHC-II presentation. From a recent study, it was observed that the alpha-tocopheryloxyacetic acid (a-TEA) enhances MHC-I cross-presentation of cancer antigens to antigen-special CD8 + T cells by exhilarating late autophagy. Such an approach was observed as an adjuvant approach for improving cancer immunotherapy. Moreover, it was reported in recent times that DCs-based vaccines have shown promising therapeutic activity in improving cancer immunotherapy by enhancing antigen presentation. For instance, lactosylated N-Alkyl polyethylenimine coated superparamagnetic iron oxide (SPIO) nanoparticles increased the vaccine activity of DCs by inducing autophagy. Further, autophagy can enhance the therapeutic efficacy of DNA vaccines by synthetizing intracellular vaccine-encoded tumor antigens. However, some reports stated that the effects of cancer immunotherapy could be impaired by hypoxia-induced autophagy. Hypoxia-induced autophagy impairs CTLs-mediated cancer cell lysis which is associated with pSTAT3 (hypoxia-dependent phosphorylation of STAT3). Hypoxia-induced autophagy further impairs NK-mediated killing, leading to a decrease in cancer immunotherapy effect. In this context, a study was performed where 3-MA (selective PI3K inhibitor, autophagy inhibitor) was employed which showed a significant increase of IL-24-induced apoptosis in oral squamous cell carcinomas (OSCC), demonstrating the combination of autophagy inhibitors and IL-24 as a promising approach for cancer immunotherapy. In addition to this, many efforts are engrossed on how to modulate autophagy to boost both innate and adaptive immune response and bypass anticancer immune resistance in immunotherapy for cancer [232].
Conclusion
In conclusion, from the historical evidence to modern-day research, autophagy is one of the most compelling mechanisms, with its relevance in cancer. Though its exact mechanism, leading to tumorigenesis or tumor suppression, is ambivalent, empirical evidence and research show a strong correlation between autophagy modulation and tumor death. Hence, the findings summarized in this review bring into light the optimistic hope of a clinically relevant, effective complementary therapy by exploiting nanoparticle-based systems to modulate autophagy in cancer.
Data availability
Not applicable.
References
Bhattacharya S, Patel KK, Dehari D, Agrawal AK, Singh S. Melatonin and its ubiquitous anticancer effects. Springer US. Mol Cell Biochem [Internet]. 2019;462:133–55. https://doi.org/10.1007/s11010-019-03617-5.
Aqil F, Jeyabalan J, Agrawal AK, Kyakulaga AH, Munagala R, Parker L, et al. Exosomal delivery of berry anthocyanidins for the management of ovarian cancer. Food Funct Royal. 2017;8:4100–7.
Agrawal AK, Aqil F, Jeyabalan J, Spencer WA, Beck J, Gachuki BW, et al. Milk-derived exosomes for oral delivery of paclitaxel. Nanomedicine Nanotechnology. Elsevier Inc. Biol Med [Internet]. 2017;13:1627–36. https://doi.org/10.1016/j.nano.2017.03.001.
Munagala R, Aqil F, Jeyabalan J, Agrawal AK, Mudd M, Kyakulaga AH, et al. Exosomal formulation of anthocyanidins against multiple cancer types. Cancer Lett. 2018;393:94–102.
Jain S, Jain R, Das M, Agrawal AK, Thanki K, Kushwah V. Combinatorial bio-conjugation of gemcitabine and curcumin enables dual drug delivery with synergistic anticancer efficacy and reduced toxicity. RSC Adv. 2014;4:29193–201.
Hanahan D, Weinberg RA. The Hallmarks of Cancer. Cell. 2000;100:57–70.
Crighton D, Ryan KM. Splicing DNA-damage responses to tumour cell death. Biochim Biophys Acta- Rev Cancer. 2004;1705:3–15.
Aita VM, Liang XH, Murty VVVS, Pincus DL, Yu W, Cayanis E, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59:59–65.
Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin1. Nature. 1999;402:672–6.
Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol. 2006;8:688–98.
Knævelsrud H, Ahlquist T, Merok MA, Nesbakken A, Stenmark H, Lothe RA, et al. UVRAG mutations associated with microsatellite unstable colon cancer do not affect autophagy. Autophagy. 2010;6:863–70.
Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–20.
Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–82.
Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sato Y, Liang C, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol. 2007;9:1142–51.
Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64.
Yun CW, Lee SH. The roles of autophagy in cancer. Int J Mol Sci. 2018;19:1–18.
Rosenfeldt MT, Ryan KM. The multiple roles of autophagy in cancer. Carcinogenesis. 2011;32:955–63.
Karantza-Wadsworth V, Patel S, Kravchuk O, Chen G, Mathew R, Jin S, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007;21:1621–35.
Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007;21:1367–81.
Miracco C, Cosci E, Oliveri G, Luzi P, Pacenti L, Monciatti I, et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. Int J Oncol. 2007;30:429–36.
Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800.
Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol. 2005;169:425–34.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–6.
Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J Biol Chem. 2019;282:18573–83.
Wen ZP, Zeng WJ, Chen YH, Li H, Wang JY, Cheng Q, et al. Knockdown ATG4C inhibits gliomas progression and promotes temozolomide chemosensitivity by suppressing autophagic flux. J Exp Clin Cancer Res. 2019;38:1–15.
Kim MS, Jeong EG, Ahn CH, Kim SS, Lee SH, Yoo NJ. Frameshift mutation of UVRAG, an autophagy-related gene, in gastric carcinomas with microsatellite instability. Hum Pathol. 2008;39:1059–63.
He S, Zhao Z, Yang Y, O’Connell D, Zhang X, Oh S, et al. Truncating mutation in the autophagy gene UVRAG confers oncogenic properties and chemosensitivity in colorectal cancers. Nat Commun. 2015;6:1–14.
Zhao Z, Oh S, Li D, Ni D, Pirooz SD, Lee J-H, et al. A Dual Role for UVRAG in Maintaining Chromosomal Stability Independent of Autophagy. Dev Cell. 2012;22:1001–16.
Moscat J, Diaz-Meco MT. p62 at the Crossroads of Autophagy, Apoptosis, and Cancer. Cell. 2009;137:1001–4.
Taniguchi K, Yamachika S, He F, Karin M. p62/SQSTM1- Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016;590:2375–97.
Liu B, Fang M, He Z, Cui D, Jia S, Lin X, et al. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015;6:1–10.
Thomasina Barron E, Smyth PPA, McDermott EW, Tobbia IN, Higgins NJO. Quantitative cytochemistry of glucose-6-phosphate dehydrogenase in benign and malignant breast tumours. Eur J Cancer Clin Oncol. 1991;27:985–9.
Chen Z, Wang J, Yuan W, Chen Z, Wu S, Chen J, et al. Overexpression of G6PD is associated with poor clinical outcome in gastric cancer. Tumor Biol. 2012;33:95–101.
Zampella EJ, Ii TGP. Glucose-6-Phosphate Dehydrogenase : A Possible Clinical lndica tor for Prosta tic Carcinoma. Cancer. 1982;49:384–7.
Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803.
Ahn CH, Jeong EG, Lee JW, Kim MS, Kim SH, Kim SS, et al. Expression of beclin-1, an autophagy-related protein, in gastric and colorectal cancers. APMIS. 2007;115:1344–9.
Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci USA. 2011;108:12455–60.
Valentin-Vega YA, Kastan MB. A new role for ATM: Regulating mitochondrial function and mitophagy. Autophagy. 2012;8:840–1.
Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–27.
Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924–32.
Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H. Remarkable tolerance of tumor cells to nutrient deprivation: Possible new biochemical target for cancer therapy. Cancer Res. 2000;60:6201–7.
Mathew R, Karantza-Wadsworth V, White E. Role of Autophagy in Cancer. Nat Rev Cancer. 2007;7:961–7.
Harris AL. Hypoxia - A key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47.
Lorin S, Hamaï A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Elsevier Ltd. Semin Cancer Biol [Internet]. 2013;23:361–79. https://doi.org/10.1016/j.semcancer.2013.06.007.
Endo H, Inoue M. Dormancy in cancer. Cancer Sci. 2019;110:474–80.
Vera-Ramirez L, Vodnala SK, Nini R, Hunter KW, Green JE. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Springer US. Nat Commun [Internet]. 2018;9. https://doi.org/10.1038/s41467-018-04070-6.
Lu Z, Luo RZ, Lu Y, Zhang X, Yu Q, Khare S, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J Clin Invest. 2008;118:3917–29.
Amaravadi RK. Autophagy-induced tumor dormancy in ovarian cancer. J Clin Invest. 2008;118:3837–40.
Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Elsevier B.V. Biochim Biophys Acta [Internet]. 2013;1833:3481–98. https://doi.org/10.1016/j.bbamcr.2013.06.026.
Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of Autophagy during Extracellular Matrix Detachment Promotes Cell Survival. Mol Biol Cell. 2008;19:797–806.
Kenific C, Thorburn A, Debnath J. Autophagy and Metastasis: Another double-edged sword. Curr Opin Cell Biol. 2010;22:241–5.
Yang J, Zheng Z, Yan X, Li X, Liu Z, Ma Z. Integration of autophagy and anoikis resistance in solid tumors. Anat Rec. 2013;296:1501–8.
Giatromanolaki AN, St Charitoudis G, Bechrakis NE, Kozobolis VP, Koukourakis MI, Foerster MH, et al. Autophagy patterns and prognosis in uveal melanomas. Mod Pathol Nature Publishing Group. 2011;24:1036–45.
Ding Z Bin, Shi YH, Zhou J, Qiu SJ, Xu Y, Dai Z, et al. Association of autophagy defect with a malignant phenotype and poor prognosis of hepatocellular carcinoma. Cancer Res. 2008;68:9167–75.
Jang M, Kim SS, Lee J. Cancer cell metabolism: Implications for therapeutic targets. Nature Publishing Group. Exp Mol Med. 2013;45:1–8.
Heiden MGV, Cantley LC, Thompson CB. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science. 2009;324:1029–33.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The Biology of Cancer: Metabolic Reprogramming Fuels Cell Growth and Proliferation. Cell Metab. 2008;7:11–20.
Eng CH, Abraham RT. The autophagy conundrum in cancer: Influence of tumorigenic metabolic reprogramming. Nature Publishing Group. Oncogene [Internet]. 2011;30:4687–96. https://doi.org/10.1038/onc.2011.220.
Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72.
Guo JY, Chen HY, Mathew R, Fan J, Strohecker AM, Karsli-Uzunbas G, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–70.
Lock R, Roy S, Kenific CM, Su JS, Salas E, Ronen SM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol Biol Cell. 2011;22:165–78.
Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–29.
Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res. 2009;15:6479–83.
Manning BD, Cantley LC. AKT/PKB Signaling: Navigating Downstream. Cell. 2007;129:1261–74.
Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64:3892–9.
Shortt J, Johnstone RW. Oncogenes in cell survival and cell death. Cold Spring Harb Perspect Biol. 2012;4:1–10.
Nazio F, Bordi M, Cianfanelli V, Locatelli F, Cecconi F. Autophagy and cancer stem cells: molecular mechanisms and therapeutic applications. Springer US. Cell Death Differ [Internet]. 2019;26:690–702. https://doi.org/10.1038/s41418-019-0292-y.
Yu Z, Pestell TG, Lisanti MP, Pestell RG. Cancer Stem Cells. Int J Biochem Cell Biol. 2012;44:2144–51.
Lobo NA, Shimono Y, Qian D, Clarke MF. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–99.
Maycotte P, Jones KL, Goodall ML, Thorburn J, Thorburn A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol Cancer Res. 2015;13:651–8.
Han Y, Fan S, Qin T, Yang J, Sun Y, Lu Y, et al. Role of autophagy in breast cancer and breast cancer stem cells (Review). Int J Oncol. 2018;52:1057–70.
Gong C, Song E, Codogno P, Mehrpour M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy. 2012;8:1853–5.
Wolf J, Dewi DL, Fredebohm J, Müller-Decker K, Flechtenmacher C, Hoheisel JD, et al. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013;15:1–13.
Chen X, He Y, Lu F. Autophagy in stem cell biology: A perspective on stem cell self-renewal and differentiation. Hindawi. Stem Cells Int. 2018;2018.
Gong C, Bauvy C, Tonelli G, Yue W, Deloménie C, Nicolas V, et al. Beclin 1 and autophagy are required for the tumorigenicity of breast cancer stem-like/progenitor cells. Oncogene. 2013;32:2261–72.
Chaterjee M, van Golen KL. Breast Cancer Stem Cells Survive Periods of Farnesyl-Transferase Inhibitor-Induced Dormancy by Undergoing Autophagy. Bone Marrow Res. 2011;2011:1–7.
Song Y jiao, Zhang S shan, Guo X ling, Sun K, Han Z peng, Li R, et al. Autophagy contributes to the survival of CD133+ liver cancer stem cells in the hypoxic and nutrient-deprived tumor microenvironment. Elsevier Ireland Ltd. Cancer Lett [Internet]. 2013;339:70–81. https://doi.org/10.1016/j.canlet.2013.07.021.
Zhang D, Zhao Q, Sun H, Yin L, Wu J, Xu J, et al. Defective autophagy leads to the suppression of stem-like features of CD271+ osteosarcoma cells. J Biomed Sci [Internet]. 2016;23:1–12. https://doi.org/10.1186/s12929-016-0297-5.
Peng Q, Qin J, Zhang Y, Cheng X, Wang X, Lu W, et al. Autophagy maintains the stemness of ovarian cancer stem cells by FOXA2. J Exp Clin Cancer Res. 2017;36:1–12.
Buccarelli M, Marconi M, Pacioni S, De Pasqualis I, D’Alessandris QG, Martini M, et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Springer US. Cell Death Dis [Internet]. 2018;9:1–17. https://doi.org/10.1038/s41419-018-0864-7.
Sun K, Deng W, Zhang S, Cai N, Jiao S, Song J, et al. Paradoxical roles of autophagy in different stages of tumorigenesis: Protector for normal or cancer cells. Cell Biosci [Internet]. Cell & Bioscience; 2013;3:1.
Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z, et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy. 2012;8:1798–810.
Hu YL, Jahangiri A, DeLay M, Aghi MK. Tumor cell autophagy as an adaptive response mediating resistance to treatments such as antiangiogenic therapy. Cancer Res. 2012;72:4294–9.
Sotelo J, Briceño E, López-González MA. Adding Chloroquine to Conventional Treatment for Glioblastoma Multiforme. Ann Intern Med. 2006;144:337.
Li J, Hou N, Faried A, Tsutsumi S, Kuwano H. Inhibition of autophagy augments 5-fluorouracil chemotherapy in human colon cancer in vitro and in vivo model. Elsevier Ltd. Eur J Cancer [Internet]. 2010;46:1900–9. https://doi.org/10.1016/j.ejca.2010.02.021.
Sasaki K, Tsuno NH, Sunami E, Kawai K, Hongo K, Hiyoshi M, et al. Resistance of colon cancer to 5-fluorouracil may be overcome by combination with chloroquine, an in vivo study. Anticancer Drugs. 2012;23:675–82.
Guo XL, Li D, Sun K, Wang J, Liu Y, Song JR, et al. Inhibition of autophagy enhances anticancer effects of bevacizumab in hepatocarcinoma. J Mol Med. 2013;91:473–83.
Shi YH, Ding Z Bin, Zhou J, Hui B, Shi GM, Ke AW, et al. Targeting autophagy enhances sorafenib lethality for hepatocellular carcinoma via ER stress-related apoptosis. Autophagy. 2011;7:1159–72.
Han W, Pan H, Chen Y, Sun J, Wang Y, Li J, et al. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS One. 2011;6:1–8.
Carew JS, Nawrocki ST, Kahue CN, Zhang H, Yang C, Chung L, et al. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHAto overcome Bcr-Abl-mediated drug resistance. Blood. 2007;110:313–22.
Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4:1–12.
Shimgu T, Fujiwara K, Bogler O, Akiyama Y, Meritake K, Shinojima N, et al. Inhibition of autophagy at a late stage enhances imatinib-induced cytotoxicity In human malignant glioma cells. Int J Cancer. 2009;124:1060–71.
Liu F, Liu D, Yang Y, Zhao S. Effect of autophagy inhibition on chemotherapy-induced apoptosis in A549 lung cancer cells. Oncol Lett. 2013;5:1261–5.
Cao X, Liu B, Cao W, Zhang W, Zhang F, Zhao H, et al. Autophagy inhibition enhances apigenin-induced apoptosis in human breast cancer cells. Chinese J Cancer Res. 2013;25:212–22.
Mujumdar N, Saluja AK. Autophagy in pancreatic cancer: An emerging mechanism of cell death. Autophagy. 2010;6:997–8.
Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, et al. Role of Bcl-2 family proteins in a non-apoptopic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8.
Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, et al. Regulation of an ATG7–beclin 1 Program of Autophagic Cell Death by Caspase-8. Science. 2004;304:1500–2.
Xiong H yan, Guo X ling, Bu X xin, Zhang S shan, Ma N nan, Song J rui, et al. Autophagic cell death induced by 5-FU in Bax or PUMA deficient human colon cancer cell. Elsevier Ireland Ltd. Cancer Lett [Internet]. 2010;288:68–74. https://doi.org/10.1016/j.canlet.2009.06.039.
Salazar M, Carracedo A, Salanueva ÍJ, Hernández-Tiedra S, Lorente M, Egia A, et al. Cannabinoid action induces autophagy-mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest. 2009;119:1359–72.
Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT, et al. Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism. Nature Publishing Group. Cell Death Dis [Internet]. 2011;2:1–12. https://doi.org/10.1038/cddis.2011.36.
Vara D, Salazar M, Olea-Herrero N, Guzmán M, Velasco G, Díaz-Laviada I. Anti-tumoral action of cannabinoids on hepatocellular carcinoma: Role of AMPK-dependent activation of autophagy. Cell Death Differ. 2011;18:1099–111.
Ren SX, Shen J, Cheng ASL, Lu L, Chan RLY, Li ZJ, et al. FK-16 Derived from the Anticancer Peptide LL-37 Induces Caspase-Independent Apoptosis and Autophagic Cell Death in Colon Cancer Cells. PLoS ONE. 2013;8:1–9.
Josset E, Burckel H, Noël G, Bischoff P. The mTOR inhibitor RAD001 potentiates autophagic cell death induced by temozolomide in a glioblastoma cell line. Anticancer Res. 2013;33:1845–51.
Cirone M, Gilardini Montani MS, Granato M, Garufi A, Faggioni A, D’Orazi G. Autophagy manipulation as a strategy for efficient anticancer therapies: Possible consequences. J Exp Clin Cancer Res. 2019;38:1–7.
Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: Bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5:1909–17.
Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. Elsevier B.V. J Control Release [Internet]. 2015;200:138–57. https://doi.org/10.1016/j.jconrel.2014.12.030.
Gade S, Patel KK, Gupta C, Anjum MM, Deepika D, Agrawal AK, et al. An Ex Vivo Evaluation of Moxifloxacin Nanostructured Lipid Carrier Enriched In Situ Gel for Transcorneal Permeation on Goat Cornea. Elsevier Ltd. J Pharm Sci [Internet]. 2019;108:2905–16. https://doi.org/10.1016/j.xphs.2019.04.005.
Patel KK, Gade S, Anjum MM, Singh SK, Maiti P, Agrawal AK, et al. Effect of penetration enhancers and amorphization on transdermal permeation flux of raloxifene-encapsulated solid lipid nanoparticlean ex vivo study on human skin. Springer International Publishing. Appl Nanosci [Internet]. 2019;9:1383–94. https://doi.org/10.1007/s13204-019-01004-6.
Aqil F, Munagala R, Jeyabalan J, Agrawal AK, Kyakulaga AH, Wilcher SA, et al. Milk exosomes - Natural nanoparticles for siRNA delivery. Elsevier. Cancer Lett [Internet]. 2019;449:186–95. https://doi.org/10.1016/j.canlet.2019.02.011.
Aqil F, Munagala R, Jeyabalan J, Agrawal AK, Gupta R. Exosomes for the Enhanced Tissue Bioavailability and Efficacy of Curcumin. AAPS J The AAPS Journal. 2017;19:1691–702.
Carvajal-Vidal P, González-Pizarro R, Araya C, Espina M, Halbaut L, Gómez de Aranda I, et al. Nanostructured lipid carriers loaded with Halobetasol propionate for topical treatment of inflammation: Development, characterization, biopharmaceutical behavior and therapeutic efficacy of gel dosage forms. Elsevier B.V. Int J Pharm [Internet]. 2020;585:119480. https://doi.org/10.1016/j.ijpharm.2020.119480.
Patel KK, Agrawal AK, Anjum MM, Tripathi M, Pandey N, Bhattacharya S, et al. DNase-I functionalization of ciprofloxacin-loaded chitosan nanoparticles overcomes the biofilm-mediated resistance of Pseudomonas aeruginosa. Springer International Publishing. Appl Nanosci [Internet]. 2020;10:563–75. https://doi.org/10.1007/s13204-019-01129-8.
Patel KK, Surekha DB, Tripathi M, Anjum MM, Muthu MS, Tilak R, et al. Antibiofilm Potential of Silver Sulfadiazine-Loaded Nanoparticle Formulations: A Study on the Effect of DNase-I on Microbial Biofilm and Wound Healing Activity. Mol Pharm. 2019;16:3916–25.
Harde H, Siddhapura K, Agrawal AK, Jain S. Divalent toxoids loaded stable chitosan-glucomannan nanoassemblies for efficient systemic, mucosal and cellular immunostimulatory response following oral administration. Elsevier B.V. Int J Pharm [Internet]. 2015;487:292–304. https://doi.org/10.1016/j.ijpharm.2015.04.042.
Harde H, Agrawal AK, Jain S. Trilateral, “3P” mechanics of stabilized layersomes technology for efficient oral immunization. J Biomed Nanotechnol. 2015;11:363–81.
Agrawal AK, Das M, Jain S. In situ gel systems as “smart” carriers for sustained ocular drug delivery. Expert Opin Drug Deliv. 2012;9:383–402.
Shenhar R, Rotello VM. Nanoparticles: Scaffolds and building blocks. Acc Chem Res. 2003;36:549–61.
Saha K, Bajaj A, Duncan B, Rotello VM. Beauty is skin deep: A surface monolayer perspective on nanoparticle interactions with cells and bio-macromolecules. Small. 2011;7:1903–18.
Kushwah V, Katiyar SS, Agrawal AK, Gupta RC, Jain S. Co-delivery of docetaxel and gemcitabine using PEGylated self-assembled stealth nanoparticles for improved breast cancer therapy. Elsevier Inc. Nanomed Nanotechnol: Biol Med [Internet]. 2018;14:1629–41. https://doi.org/10.1016/j.nano.2018.04.009.
Kushwah V, Katiyar SS, Dora CP, Kumar Agrawal A, Lamprou DA, Gupta RC, et al. Co-delivery of docetaxel and gemcitabine by anacardic acid modified self-assembled albumin nanoparticles for effective breast cancer management. Acta Biomater [Internet]. 2018;73:424–36. https://doi.org/10.1016/j.actbio.2018.03.057.
Singh S, Kushwah V, Agrawal AK, Jain S. Insulin- and quercetin-loaded liquid crystalline nanoparticles: Implications on oral bioavailability, antidiabetic and antioxidant efficacy. Nanomedicine. 2018;13:521–37.
Kushwah V, Agrawal AK, Dora CP, Mallinson D, Lamprou DA, Gupta RC, et al. Novel Gemcitabine Conjugated Albumin Nanoparticles: a Potential Strategy to Enhance Drug Efficacy in Pancreatic Cancer Treatment. Pharm Res. 2017;34:2295–311.
Albanese A, Sykes EA, Chan WCW. Rough around the edges: The inflammatory response of microglial cells to spiky nanoparticles. ACS Nano. 2010;4:2490–3.
Harde H, Siddhapura K, Agrawal AK, Jain S. Development of dual toxoid-loaded layersomes for complete immunostimulatory response following peroral administration. Nanomedicine. 2015;10:1077–91.
Jain S, Harde H, Indulkar A, Agrawal AK. Improved stability and immunological potential of tetanus toxoid containing surface engineered bilosomes following oral administration. Elsevier B.V. Nanomed Nanotechnol: Biol Med [Internet]. 2014;10:431–40. https://doi.org/10.1016/j.nano.2013.08.012.
Jain S, Kumar S, Agrawal AK, Thanki K, Banerjee UC. Enhanced transfection efficiency and reduced cytotoxicity of novel lipid-polymer hybrid nanoplexes. Mol Pharm. 2013;10:2416–25.
Chithrani BD, Ghazani AA, Chan WCW. Determining the size and shape dependence of gold nanoparticle uptake into mammalian cells. Nano Lett. 2006;6:662–8.
Gratton SEA, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, et al. The effect of particle design on cellular internalization pathways. Proc Natl Acad Sci USA. 2008;105:11613–8.
Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B, et al. Renal clearance of nanoparticles. Nat Biotechnol [Internet]. 2007;25:1165–70. https://doi.org/10.1038/nbt1340.
Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064–79.
Doane TL, Burda C. The unique role of nanoparticles in nanomedicine: Imaging, drug delivery and therapy. Chem Soc Rev. 2012;41:2885–911.
Jain S, Garg T, Kushwah V, Thanki K, Agrawal AK, Dora CP. α-Tocopherol as functional excipient for resveratrol and coenzyme Q10-loaded SNEDDS for improved bioavailability and prophylaxis of breast cancer. Taylor & Francis. J Drug Target. 2017;25:554–65.
Aqil F, Kausar H, Agrawal AK, Jeyabalan J, Kyakulaga AH, Munagala R, et al. Exosomal formulation enhances therapeutic response of celastrol against lung cancer. Elsevier B.V. Exp Mol Pathol [Internet]. 2016;101:12–21. https://doi.org/10.1016/j.yexmp.2016.05.013.
Buttacavoli M, Albanese NN, Di Cara G, Alduina R, Faleri C, Gallo M, et al. Anticancer activity of biogenerated silver nanoparticles: An integrated proteomic investigation. Oncotarget. 2018;9:9685–705.
Zielinska E, Zauszkiewicz-Pawlak A, Wojcik M, Inkielewicz-Stepniak I. Silver nanoparticles of different sizes induce a mixed type of programmed cell death in human pancreatic ductal adenocarcinoma. Oncotarget. 2018;9:4675–97.
Yuan YG, Gurunathan S. Combination of graphene oxide-silver nanoparticle nanocomposites and cisplatin enhances apoptosis and autophagy in human cervical cancer cells. Int J Nanomedicine. 2017;12:6537–58.
Zhang XF, Gurunathan S. Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: An effective anticancer therapy. Int J Nanomedicine. 2016;11:3655–75.
Lin J, Huang Z, Wu H, Zhou W, Jin P, Wei P, et al. Inhibition of autophagy enhances the anticancer activity of silver nanoparticles. Autophagy. 2014;10:2006–20.
Liu P, Jin H, Guo Z, Ma J, Zhao J, Li D, et al. Silver nanoparticles outperform gold nanoparticles in radiosensitizing U251 cells in vitro and in an intracranial mouse model of glioma. Int J Nanomedicine [Internet]. 2016;11:5003–14. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5055115/pdf/ijn-11-5003.pdf.
Cordani M, Somoza Á. Targeting autophagy using metallic nanoparticles: a promising strategy for cancer treatment. Springer International Publishing. Cell Mol Life Sci [Internet]. 2019;76:1215–42. https://doi.org/10.1007/s00018-018-2973-y.
Ma X, Wu Y, Jin S, Tian Y, Zhang X, Zhao Y, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5:8629–39.
Joshi P, Chakraborti S, Ramirez-Vick JE, Ansari ZA, Shanker V, Chakrabarti P, et al. The anticancer activity of chloroquine-gold nanoparticles against MCF-7 breast cancer cells. Elsevier B.V. Colloids Surf. B [Internet]. 2012;95:195–200. https://doi.org/10.1016/j.colsurfb.2012.02.039.
Crown J, O’Shaughnessy J, Gullo G. Emerging targeted therapies in triple-negative breast cancer. Elsevier Masson SAS. Ann Oncol [Internet]. 2012;23:vi56–65. https://doi.org/10.1093/annonc/mds196.
Zhang M, Kim HS, Jin T, Moon WK. Near-infrared photothermal therapy using EGFR-targeted gold nanoparticles increases autophagic cell death in breast cancer. Elsevier B.V. J Photochem Photobiol B Biol [Internet]. 2017;170:58–64. https://doi.org/10.1016/j.jphotobiol.2017.03.025.
Koken MHM, Smit EME, Jaspers-Dekker I, Oostra BA, Hagemeuer A, Bootsma D, et al. Localization of two human homologs, HHR6A and HHR6B, of the yeast DNA repair gene RAD6 to chromosomes Xq24-q25 and 5q23-q31. Genomics. 1992;12:447–53.
Koken MHM, Reynolds P, Jaspers-Dekker I, Prakash L, Prakash S, Bootsma D, et al. Structural and functional conservation of two human homologs of the yeast DNA repair gene RAD6. Proc Natl Acad Sci USA. 1991;88:8865–9.
Haynes B, Zhang Y, Liu F, Li J, Petit S, Kothayer H, et al. Gold nanoparticle conjugated Rad6 inhibitor induces cell death in triple negative breast cancer cells by inducing mitochondrial dysfunction and PARP-1 hyperactivation: Synthesis and characterization. Nanomedicine. 2016;12:745–57.
Ke S, Zhou T, Yang P, Wang Y, Zhang P, Chen K, et al. Gold nanoparticles enhance TRAIL sensitivity through Drp1-mediated apoptotic and autophagic mitochondrial fission in NSCLC cells. Int J Nanomedicine. 2017;12:2531–51.
Feng Q, Liu Y, Huang J, Chen K, Huang J, Xiao K. Uptake, distribution, clearance, and toxicity of iron oxide nanoparticles with different sizes and coatings. Springer US. Sci Rep [Internet]. 2018;8:1–13. https://doi.org/10.1038/s41598-018-19628-z.
Wang Y, Zi XY, Su J, Zhang HX, Zhang XR, Zhu HY, et al. Cuprous oxide nanoparticles selectively induce apoptosis of tumor cells. Int J Nanomedicine. 2012;7:2641–52.
Wang J, Gao S, Wang S, Xu Z, Wei L. Zinc oxide nanoparticles induce toxicity in CAL 27 oral cancer cell lines by activating PINK1/Parkin-mediated mitophagy. Int J Nanomedicine. 2018;13:3441–50.
Mozdoori N, Safarian S, Sheibani N. Augmentation of the cytotoxic effects of zinc oxide nanoparticles by MTCP conjugation: Non-canonical apoptosis and autophagy induction in human adenocarcinoma breast cancer cell lines. Mater Sci Eng C. 2017;78:949–59.
Khan MI, Mohammad A, Patil G, Naqvi SAH, Chauhan LKS, Ahmad I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Elsevier Ltd. Biomaterials [Internet]. 2012;33:1477–88. https://doi.org/10.1016/j.biomaterials.2011.10.080.
Zhang X, Zhang H, Liang X, Zhang J, Tao W, Zhu X, et al. Iron oxide nanoparticles induce autophagosome accumulation through multiple mechanisms: Lysosome impairment, mitochondrial damage, and ER stress. Mol Pharm. 2016;13:2578–87.
Kuroda S, Tam J, Roth JA, Sokolov K, Ramesh R. EGFR-targeted plasmonic magnetic nanoparticles suppress lung tumor growth by abrogating G2/M cell-cycle arrest and inducing DNA damage. Int J Nanomedicine. 2014;9:3825–39.
Li X, Feng J, Zhang R, Wang J, Su T, Tian Z, et al. Quaternized chitosan/alginate-Fe3O4 magnetic nanoparticles enhance the chemosensitization of multidrug-resistant gastric carcinoma by regulating cell autophagy activity in mice. J Biomed Nanotechnol. 2016;12:948–61.
Wang Y, Yang F, Zhang HX, Zi XY, Pan XH, Chen F, et al. Cuprous oxide nanoparticles inhibit the growth and metastasis of melanoma by targeting mitochondria. Cell Death Dis. 2013;4:1–10.
Sun T, Yan Y, Zhao Y, Guo F, Jiang C. Copper oxide nanoparticles induce autophagic cell death in a549 cells. PLoS One. 2012;7:1–7.
Abudayyak M, Guzel EE, Özhan G. Copper (II) Oxide Nanoparticles Induced Nephrotoxicity In Vitro Conditions. Appl Vitr Toxicol. 2016;2:157–64.
Xia L, Wang Y, Chen Y, Yan J, Hao F, Su X, et al. Cuprous oxide nanoparticles inhibit the growth of cervical carcinoma by inducing autophagy. Oncotarget. 2017;8:61083–92.
Laha D, Pramanik A, Maity J, Mukherjee A, Pramanik P, Laskar A, et al. Interplay between autophagy and apoptosis mediated by copper oxide nanoparticles in human breast cancer cells MCF7. Elsevier B.V. Biochim Biophys Acta- Gen Subj [Internet]. 2014;1840:1–9. https://doi.org/10.1016/j.bbagen.2013.08.011.
Liu HL, Zhang YL, Yang N, Zhang YX, Liu XQ, Li CG, et al. A functionalized single-walled carbon nanotubeinduced autophagic cell death in human lung cells through Akt-TSC2-mTOR signaling. Cell Death Dis. 2011;2:1–7.
Wei P, Zhang L, Lu Y, Man N, Wen L. C60(Nd) nanoparticles enhance chemotherapeutic susceptibility of cancer cells by modulation of autophagy. Nanotechnology. 2010;21.
Ristic B, Harhaji-Trajkovic L, Bosnjak M, Dakic I, Mijatovic S, Trajkovic V. Modulation of cancer cell autophagic responses by graphene-based nanomaterials: Molecular mechanisms and therapeutic implications. Cancers (Basel). 2021;13:1–21.
Chen GY, Meng C Le, Lin KC, Tuan HY, Yang HJ, Chen CL, et al. Graphene oxide as a chemosensitizer: Diverted autophagic flux, enhanced nuclear import, elevated necrosis and improved antitumor effects. Elsevier Ltd. Biomaterials [Internet]. 2015;40:12–22. https://doi.org/10.1016/j.biomaterials.2014.11.034.
Lin KC, Lin MW, Hsu MN, Yu-Chen G, Chao YC, Tuan HY, et al. Graphene oxide sensitizes cancer cells to chemotherapeutics by inducing early autophagy events, promoting nuclear trafficking and necrosis. Theranostics. 2018;8:2477–87.
Arya BD, Mittal S, Joshi P, Pandey AK, Jaime E. Graphene oxide – chloroquine nanoconjugate induce necroptotic death in A549 cancer cells through autophagy modulation. Nanomedicine. 2018;13:2261–82.
Roggers R, Kanvinde S, Boonsith S, Oupický D. The practicality of mesoporous silica nanoparticles as drug delivery devices and progress toward this goal. AAPS PharmSciTech. 2014;15:1163–71.
Napierska D, Thomassen LCJ, Rabolli V, Lison D, Gonzalez L, Kirsch-Volders M, et al. Size-dependent cytotoxicity of monodisperse silica nanoparticles in human endothelial cells. Small. 2009;5:846–53.
Thomassen LCJ, Aerts A, Rabolli V, Lison D, Gonzalez L, Kirsch-Volders M, et al. Synthesis and characterization of stable monodisperse silica nanoparticle sols for in vitro cytotoxicity testing. Langmuir. 2010;26:328–35.
Krętowski R, Kusaczuk M, Naumowicz M, Kotyńska J, Szynaka B, Cechowska-Pasko M. The effects of silica nanoparticles on apoptosis and autophagy of glioblastoma cell lines. Nanomaterials. 2017;7:1–22.
Schütz I, Lopez-Hernandez T, Gao Q, Puchkov D, JaBerlinbs S, Nordmeyer D, et al. Lysosomal dysfunction caused by cellular accumulation of silica nanoparticles. J Biol Chem. 2016;291:14170–84.
Pool H, Campos-Vega R, Herrera-Hernández MG, García-Solis P, García-Gasca T, Sánchez IC, et al. Development of genistein-PEGylated silica hybrid nanomaterials with enhanced antioxidant and antiproliferative properties on HT29 human colon cancer cells. Am J Transl Res. 2018;10:2306–23.
Wang J, Yu Y, Lu K, Yang M, Li Y, Zhou X, et al. Silica nanoparticles induce autophagy dysfunction via lysosomal impairment and inhibition of autophagosome degradation in hepatocytes. Int J Nanomedicine. 2017;12:809–25.
Yu Y, Duan J, Yu Y, Li Y, Liu X, Zhou X, et al. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. Elsevier B.V. J Hazard Mater [Internet]. 2014;270:176–86. https://doi.org/10.1016/j.jhazmat.2014.01.028.
Wei F, Wang Y, Luo Z, Li Y, Duan Y. New findings of silica nanoparticles induced ER autophagy in human colon cancer cell. Sci Rep. 2017;7:1–11.
Patel KK, Tripathi M, Pandey N, Agrawal AK, Gade S, Anjum MM, et al. Alginate lyase immobilized chitosan nanoparticles of ciprofloxacin for the improved antimicrobial activity against the biofilm associated mucoid P. aeruginosa infection in cystic fibrosis. Elsevier. Int J Pharm [Internet]. 2019;563:30–42. https://doi.org/10.1016/j.ijpharm.2019.03.051.
Jain S, Kumar S, Agrawal AK, Thanki K, Banerjee UC. Hyaluronic acid-PEI-cyclodextrin polyplexes: Implications for in vitro and in vivo transfection efficiency and toxicity. RSC Adv. 2015;5:41144–54.
Harde H, Agrawal AK, Jain S. Tetanus toxoids loaded glucomannosylated chitosan based nanohoming vaccine adjuvant with improved oral stability and immunostimulatory response. Pharm Res. 2015;32:122–34.
Agrawal AK, Urimi D, Jain S. Multifunctional Polymeric Nano-Carriers. 2015.
Jain S, Spandana G, Agrawal AK, Kushwah V, Thanki K. Enhanced Antitumor Efficacy and Reduced Toxicity of Docetaxel Loaded Estradiol Functionalized Stealth Polymeric Nanoparticles. Mol Pharm. 2015;12:3871–84.
Eidi H, Joubert O, Némos C, Grandemange S, Mograbi B, Foliguet B, et al. Drug delivery by polymeric nanoparticles induces autophagy in macrophages. Elsevier B.V. Int J Pharm [Internet]. 2012;422:495–503. https://doi.org/10.1016/j.ijpharm.2011.11.020.
Li C, Liu H, Sun Y, Wang H, Guo F, Rao S, et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol. 2010;1:37–45.
Peynshaert K, Manshian BB, Joris F, Braeckmans K, De Smedt SC, Demeester J, et al. Exploiting intrinsic nanoparticle toxicity: The pros and cons of nanoparticle-induced autophagy in biomedical research. Chem Rev. 2014;114:7581–609.
Thomas TP, Majoros I, Kotlyar A, Mullen D, Holl MMB, Baker Jr JR. Cationic Poly(amidoamine) Dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations. Biomacromolecules [Internet]. 2009;10:3207–14. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3624763/pdf/nihms412728.pdf.
Saiyin W, Wang D, Li L, Zhu L, Liu B, Sheng L, et al. Sequential release of autophagy inhibitor and chemotherapeutic drug with polymeric delivery system for oral squamous cell carcinoma therapy. Mol Pharm. 2014;11:1662–75.
Armiñán A, Palomino-Schätzlein M, Deladriere C, Arroyo-Crespo JJ, Vicente-Ruiz S, Vicent MJ, et al. Metabolomics facilitates the discrimination of the specific anti-cancer effects of free- and polymer-conjugated doxorubicin in breast cancer models. Biomaterials. 2018;162:144–53.
Qiao ZY, Lai WJ, Lin YX, Li D, Nan XH, Wang Y, et al. Polymer-KLAK Peptide Conjugates Induce Cancer Cell Death through Synergistic Effects of Mitochondria Damage and Autophagy Blockage. Bioconjug Chem. 2017;28:1709–21.
Kushwah V, Jain DK, Agrawal AK, Jain S. Improved antitumor efficacy and reduced toxicity of docetaxel using anacardic acid functionalized stealth liposomes. Elsevier B.V. Colloids Surfaces B Biointerfaces [Internet]. 2018;172:213–23. https://doi.org/10.1016/j.colsurfb.2018.08.047.
Agrawal AK, Harde H, Thanki K, Jain S. Improved stability and antidiabetic potential of insulin containing folic acid functionalized polymer stabilized multilayered liposomes following oral administration. Biomacromol. 2014;15:350–60.
Jain S, Patil SR, Swarnakar NK, Agrawal AK. Oral delivery of doxorubicin using novel polyelectrolyte-stabilized liposomes (Layersomes). Mol Pharm. 2012;9:2626–35.
Man N, Chen Y, Zheng F, Zhou W, Wen LP. Induction of genuine autophagy by cationic lipids in mammalian cells. Autophagy. 2010;6:449–54.
Roberts R, Al-Jamal WT, Whelband M, Thomas P, Jefferson M, Van Den Bossche J, et al. Autophagy and formation of tubulovesicular autophagosomes provide a barrier against nonviral gene delivery. Autophagy. 2013;9:667–82.
Adiseshaiah PP, Clogston JD, McLeland CB, Rodriguez J, Potter TM, Neun BW, et al. Synergistic Combination Therapy with Nanoliposomal C6- Ceramide and Vinblastine is Associated with Autophagy Dysfunction in Hepatocarcinoma and Colorectal Cancer Models. Cancer Lett. 2013;337:254–65.
Condello M, Mancini G, Meschini S. The Exploitation of Liposomes in the Inhibition of Autophagy to Defeat Drug Resistance. Front Pharmacol. 2020;11:1–15.
Belfiore L, Saunders DN, Ranson M, Thurecht KJ, Storm G, Vine KL. Towards clinical translation of ligand-functionalized liposomes in targeted cancer therapy: Challenges and opportunities. Elsevier B.V. J Control Release [Internet]. 2018;277:1–13. https://doi.org/10.1016/j.jconrel.2018.02.040.
Gilabert-Oriol R, Ryan GM, Leung AWY, Firmino NS, Bennewith KL, Bally MB. Liposomal formulations to modulate the tumour microenvironment and antitumour immune response. Int J Mol Sci 2018.
Lamichhane N, Udayakumar TS, D’Souza WD, Simone CB, Raghavan SR, Polf J, et al. Liposomes: Clinical applications and potential for image-guided drug delivery. Molecules. 2018;23:1–17.
Jeong JK, Gurunathan S, Kang MH, Han JW, Das J, Choi YJ, et al. Hypoxia-mediated autophagic flux inhibits silver nanoparticle-triggered apoptosis in human lung cancer cells. Nature Publishing Group. Sci Rep [Internet]. 2016;6:1–13. https://doi.org/10.1038/srep21688.
Lin J, Liu Y, Wu H, Huang Z, Ma J, Guo C, et al. Key Role of TFEB Nucleus Translocation for Silver Nanoparticle-Induced Cytoprotective Autophagy. Small. 2018;14:1–10.
Fageria L, Pareek V, Dilip RV, Bhargava A, Pasha SS, Laskar IR, et al. Biosynthesized Protein-Capped Silver Nanoparticles Induce ROS-Dependent Proapoptotic Signals and Prosurvival Autophagy in Cancer Cells. ACS Omega. 2017;2:1489–504.
Faedmaleki F, Shirazi FH, Salarian AA, Ashtiani HA, Rastegar H. Toxicity effect of silver nanoparticles on mice liver primary cell culture and HepG2 cell line. Iran J Pharm Res. 2014;13:235–42.
Chen Y, Yang T, Chen S, Qi S, Zhang Z, Xu Y. Silver nanoparticles regulate autophagy through lysosome injury and cell hypoxia in prostate cancer cells. J Biochem Mol Toxicol. 2020;34:1–9.
Al-kawmani AA, Alanazi KM, Farah MA, Ali MA, Hailan WAQ, Al-Hemaid FMA. Apoptosis-inducing potential of biosynthesized silver nanoparticles in breast cancer cells. J King Saud Univ Sci [Internet]. 2020;32:2480–8. https://doi.org/10.1016/j.jksus.2020.04.002.
Ren KW, Li YH, Wu G, Ren JZ, Lu H Bin, Li ZM, et al. Quercetin nanoparticles display antitumor activity via proliferation inhibition and apoptosis induction in liver cancer cells. Int J Oncol. 2017;50:1299–311.
Bhowmik T, Gomes A. NKCT1 (purified Naja kaouthia protein toxin) conjugated gold nanoparticles induced Akt/mTOR inactivation mediated autophagic and caspase 3 activated apoptotic cell death in leukemic cell. Elsevier Ltd. Toxicon [Internet]. 2016;121:86–97. https://doi.org/10.1016/j.toxicon.2016.08.004.
Kubota T, Kuroda S, Kanaya N, Morihiro T, Aoyama K, Kakiuchi Y, et al. HER2-targeted gold nanoparticles potentially overcome resistance to trastuzumab in gastric cancer. Elsevier Inc. Nanomed Nanotechnol: Biol Med [Internet]. 2018;14:1919–29. https://doi.org/10.1016/j.nano.2018.05.019.
Lou M, Zhang L na, Ji P gang, Feng F qiang, Liu J hui, Yang C, et al. Quercetin nanoparticles induced autophagy and apoptosis through AKT/ERK/Caspase-3 signaling pathway in human neuroglioma cells: In vitro and in vivo. Elsevier Masson SAS. Biomed Pharmacother [Internet]. 2016;84:1–9. https://doi.org/10.1016/j.biopha.2016.08.055.
Luo C lin, Liu Y qiong, Wang P, Song C hua, Wang K juan, Dai L ping, et al. The effect of quercetin nanoparticle on cervical cancer progression by inducing apoptosis, autophagy and anti-proliferation via JAK2 suppression. Elsevier Masson SAS. Biomed Pharmacother [Internet]. 2016;82:595–605. https://doi.org/10.1016/j.biopha.2016.05.029.
Ruan S, Xie R, Qin L, Yu M, Xiao W, Hu C, et al. Aggregable Nanoparticles-Enabled Chemotherapy and Autophagy Inhibition Combined with Anti-PD-L1 Antibody for Improved Glioma Treatment. Nano Lett. 2019;19:8318–32.
Bai DP, Zhang XF, Zhang GL, Huang YF, Gurunathan S. Zinc oxide nanoparticles induce apoptosis and autophagy in human ovarian cancer cells. Int J Nanomedicine. 2017;12:6521–35.
Farasat M, Niazvand F, Khorsandi L. Zinc oxide nanoparticles induce necroptosis and inhibit autophagy in MCF-7 human breast cancer cells. Biologia (Bratisl). 2020;75:161–74.
Ren X, Chen Y, Peng H, Fang X, Zhang X, Chen Q, et al. Blocking Autophagic Flux Enhances Iron Oxide Nanoparticle Photothermal Therapeutic Efficiency in Cancer Treatment. ACS Appl Mater Interfaces. 2018;10:27701–11.
Huang D, Zhou H, Gao J. Nanoparticles modulate autophagic effect in a dispersity-dependent manner. Nature Publishing Group. Sci Rep. 2015;5:1–10.
Xie Y, Jiang J, Tang Q, Zou H, Zhao X, Liu H, et al. Iron Oxide Nanoparticles as Autophagy Intervention Agents Suppress Hepatoma Growth by Enhancing Tumoricidal Autophagy. Adv Sci. 2020;1903323:1–13.
Lin YR, Chan CH, Lee HT, Cheng SJ, Yang JW, Chang SJ, et al. Remote magnetic control of autophagy in mouse b-lymphoma cells with iron oxide nanoparticles. Nanomaterials. 2019;9:1–13.
Du S, Li J, Du C, Huang Z, Chen G, Yan W. Overendocytosis of superparamagnetic iron oxide particles increases apoptosis and triggers autophagic cell death in human osteosarcoma cell under a spinning magnetic field. Oncotarget. 2017;8:9410–24.
Levada K, Pshenichnikov S, Omelyanchik A, Rodionova V, Nikitin A, Savchenko A, et al. Progressive lysosomal membrane permeabilization induced by iron oxide nanoparticles drives hepatic cell autophagy and apoptosis. Springer Singapore. Nano Converg [Internet]. 2020;7:1–17. https://doi.org/10.1186/s40580-020-00228-5.
Shi M, Cheng L, Zhang Z, Liu Z, Mao X. Ferroferric oxide nanoparticles induce prosurvival autophagy in human blood cells by modulating the Beclin 1/Bcl-2/VPs34 complex. Int J Nanomedicine. 2015;10:207–16.
Jiang YW, Gao G, Jia HR, Zhang X, Zhao J, Ma N, et al. Copper oxide nanoparticles induce enhanced radiosensitizing effect via destructive autophagy. American Chemical Society. ACS Biomater Sci Eng. 2019;5:1569–79.
Xiong Q, Liu A, Ren Q, Xue Y, Yu X, Ying Y, et al. Cuprous oxide nanoparticles trigger reactive oxygen species-induced apoptosis through activation of erk-dependent autophagy in bladder cancer. Springer US. Cell Death Dis [Internet]. 2020;11:1–13. https://doi.org/10.1038/s41419-020-2554-5.
Li X, Xu H, Li C, Qiao G, Farooqi AA, Gedanken A, et al. Zinc-doped copper oxide nanocomposites inhibit the growth of pancreatic cancer by inducing autophagy through AMPK/mTOR pathway. Front Pharmacol. 2019;10:1–11.
Nowak JS, Mehn D, Nativo P, García CP, Gioria S, Ojea-Jiménez I, et al. Silica nanoparticle uptake induces survival mechanism in A549 cells by the activation of autophagy but not apoptosis. Toxicol Lett. 2014;224:84–92.
Li Y, Cho MH, Lee SS, Lee DE, Cheong H, Choi Y. Hydroxychloroquine-loaded hollow mesoporous silica nanoparticles for enhanced autophagy inhibition and radiation therapy. Elsevier B.V. J Control Release [Internet]. 2020;325:100–10. https://doi.org/10.1016/j.jconrel.2020.06.025.
Chen T, Cen D, Ren Z, Wang Y, Cai X, Huang J, et al. Bismuth embedded silica nanoparticles loaded with autophagy suppressant to promote photothermal therapy. Elsevier. Biomaterials [Internet]. 2019;221:1–9. https://doi.org/10.1016/j.biomaterials.2019.119419.
Zhang X, Dong Y, Zeng X, Liang X, Li X, Tao W, et al. The effect of autophagy inhibitors on drug delivery using biodegradable polymer nanoparticles in cancer treatment. Elsevier Ltd. Biomaterials [Internet]. 2014;35:1932–43. https://doi.org/10.1016/j.biomaterials.2013.10.034.
Zhang X, Yang Y, Liang X, Zeng X, Liu Z, Tao W, et al. Enhancing therapeutic effects of docetaxel-loaded dendritic copolymer nanoparticles by co-treatment with autophagy inhibitor on breast cancer. Theranostics. 2014;4:1085–95.
Wang FZ, Xing L, Tang ZH, Lu JJ, Cui PF, Qiao JB, et al. Codelivery of Doxorubicin and shAkt1 by Poly(ethylenimine)-Glycyrrhetinic Acid Nanoparticles to Induce Autophagy-Mediated Liver Cancer Combination Therapy. Mol Pharm. 2016;13:1298–307.
Feng Y, Gao Y, Wang D, Xu Z, Sun W, Ren P. Autophagy Inhibitor (LY294002) and 5-fluorouracil (5-FU) Combination-Based Nanoliposome for Enhanced Efficacy Against Esophageal Squamous Cell Carcinoma. Nanoscale Res Lett. 2018;13:1–9.
Zhang J, Zhu S, Tan Q, Cheng D, Dai Q, Yang Z, et al. Combination therapy with ropivacaine-loaded liposomes and nutrient deprivation for simultaneous cancer therapy and cancer pain relief. Theranostics. 2020;10:4885–99.
Chang CM, Lan KL, Huang WS, Lee YJ, Lee TW, Chang CH, et al. 188Re-liposome can induce mitochondrial autophagy and reverse drug resistance for ovarian cancer: From bench evidence to preliminary clinical proof-of-concept. Int J Mol Sci. 2017;18:1–15.
Jiang GM, Tan Y, Wang H, Peng L, Chen HT, Meng XJ, et al. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol Cancer. 2019;18:1–22.
Acknowledgements
Authors are grateful to the Indian Institute of Technology (Banaras Hindu University), Varanasi, for providing infrastructure facilities. Dulla Naveen Kumar is thankful to SERB for providing financial assistance in terms of JRF in one of the SERB sponsored projects (SRG/2019/000150).
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Shloka Negi was involved in literature search and writing of article; Aiswarya Chaudhuri sketched images and structured the article; Dulla Naveen Kumar formatted the tables; Deepa Dehari compiled the data; Sanjay Singh corrected and restructured the article; Ashish Kumar Agrawal took part in literature search, refinement, and improvement of article.
Corresponding author
Ethics declarations
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Conflict of interests
The authors declare that they have no conflicts of interest with the contents of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Negi, S., Chaudhuri, A., Kumar, D.N. et al. Nanotherapeutics in autophagy: a paradigm shift in cancer treatment. Drug Deliv. and Transl. Res. 12, 2589–2612 (2022). https://doi.org/10.1007/s13346-022-01125-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-022-01125-6