
Abstract
The present investigation demonstrates the development of crosslinked β-cyclodextrin nanoparticles (β-CD NPs) for enhancing the therapeutic efficacy of docetaxel (DTX) against breast cancer. Initially, a partial inclusion complex between β-CD and polypropylene glycol (PPG) was formed to induce self-assembly. This was followed by crosslinking of β-CDs using epichlorohydrin (EPI) and removal (by solubilization) of PPG to yield uniform β-CD NPs. The formed particles were used for loading DTX to form DTX β-CD NPs. The resultant DTX β-CD NPs exhibited particle size of 223.36 ± 17.73 nm with polydispersity index (PDI) of 0.13 ± 0.09 and showed entrapment efficiency of 54.53 ± 2%. Increased cell uptake (~5-fold), cytotoxicity (~3.3-fold), and apoptosis were observed in MDA-MB-231 cells when treated with DTX β-CD NPs in comparison to free DTX. Moreover, pharmacokinetic evaluation of DTX β-CD NPs revealed ~2 and ~5-fold increase in AUC0-∞ and mean residence time (MRT) of DTX when compared to Docepar®. Further, the anti-tumor activity using DMBA-induced cancer model showed that DTX β-CD NPs were capable of reducing the tumor volume to ~40%, whereas Docepar® was able to reduce tumor volume till ~80%. Finally, the toxicity evaluation of DTX β-CD NPs revealed no short-term nephrotoxicity and was confirmed by estimating the levels of biomarkers and histopathology of the organs. Thus, the proposed formulation strategy can yield uniformly formed β-CD NPs which can be effectively utilized for improving the therapeutic efficacy of DTX.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Till today, cancer is still considered to be the most lethal type of non-communicable disease for humans [1]. However, this has propelled several research experts and clinicians to explore new avenues to improve the quality of life and prolong the life expectancy of cancer patients. Mainly, radiotherapy, chemotherapy, and surgical resection are the go to strategies for managing cancers [2,3,4]. The surgical removal of tumors is a promising approach if resection is done effectively followed by stringent chemotherapy. The traditional chemo- and radio-therapy has shown promising results in managing cancer but has revealed many undesired consequences [5,6,7]. Additionally, in some cases, drugs with potent anticancer effect exhibit poor water solubility, instability, and low biocompatibility which add up to the difficulties [8, 9]. One such drug is DTX, which is a second-generation taxoid cytotoxic agent. Over the past decade, DTX has been considered as an effective anticancer drug against a variety of cancers including breast, ovarian, and non-small cell lung carcinoma [8]. But due to its poor water solubility and systemic toxicity, its clinical utility has been restricted [10, 11]. Currently, the marketed intravenous formulations of DTX (Taxotere®) comprises of polysorbate 80 (non-ionic surfactant) and ethanol for solubilizing DTX. But due to the use of these components, incidences of hypersensitivity reactions in patients were observed. Hence, to eliminate the use of polysorbate 80 and ethanol-based intravenous injection, several formulations have been explored, which include chitosan nanoparticles [12], lipid-based systems [13], and polymer-based micelles [14]. One of such alternative technologies is the administration of DTX in cyclodextrin (CD) nanoparticulate systems.
CDs are cyclic oligosaccharides formed by 6, 7, or 8 glucose units by α-1,4 glycosidic bonds, which are called α, β, γ-cyclodextrin, respectively. Further, they can act as supra-molecular hosts for therapeutic molecules to form inclusion complex which can further act as a building block for a suitable delivery system [15]. CDs are extensively explored because of their superior biocompatibility, aqueous solubility, and facile modification with desired ligands. Further, CDs can be polymerized to impart self-assembly and to strengthen the drug interactions by offering tuneable structures [16]. The most widely used CDs are the β-CDs, due to their favorable hydrophobic inclusion cavity. However, β-CDs exhibit solubility and toxicity issues which restrict their intravenous administration as it is without modifications [17, 18]. Hence, modified or crosslinked (using hydrophilic crosslinkers) versions of β-CDs have been developed and explored. One such crosslinker is epichlorohydrin (EPI) which is an organochlorine compound and is been widely explored in the field of drug delivery. For example, the therapeutic efficacy of drugs like glipizide [19] and triclosan [20] was improved by forming an inclusion complex with β-CD followed by crosslinking with EPI. Similarly in a report published by Gidwani et al. revealed that EPI crosslinked CDs can show efficient inclusion capacity with bendamustine compare to other hydrophilic derivatives of β-CDs [21].
However, when a crosslinker is used directly in a CD-drug inclusion complex solution for crosslinking, it renders particles with high polydispersity index (PDI). This is due to the fact that the number of CD-drug complexes present in the vicinity of each other to crosslink is never uniform. Hence, to achieve a mono-disperse system, it is important to create uniformly aggregated CD-drug inclusion complexes which can be effectively crosslinked. To form uniform CD aggregates, polyrotaxanes can be used. Polyrotaxanes are a type of inclusion complex which are formed through selective threading of CD’s into linear polymer chains like poly(ethylene glycol) (PEG) and PPG [22, 23]. Condensation/cross-linking of these inclusion complexes gives novel nanostructures including CD tubes and CD nanocapsules which are widely explored for drug delivery [24, 25]. A similar approach was attempted by Zhu et al. to develop EPI cross-linked α-CD nanoparticles for effective delivery of cisplatin [26]. Initially, an inclusion complex of α-CD and PEG in aqueous system was prepared to achieve uniform aggregates followed by crosslinking with EPI and drug loading. Further, they explored the role of PEG in the formulation and its implication on particle size and PDI uniformity.
Hence, based on this concept, we developed DTX-loaded EPI cross-linked β-CD nanoparticles via partial inclusion complex (DTX β-CD NPs). Initially, PPG was used to form a partial inclusion complex with β-CDs, so as to self-assemble the β-CDs into uniform aggregates (β-CD PPG). The free hydroxyl groups of the β-CD PPG aggregates were crosslinked using EPI. Due to the self-assembled β-CD PPG aggregates, the crosslinking of β-CD was observed in a controlled manner and formation of larger non-uniform aggregates was avoided. Once the crosslinking was done, the threaded PPG chains from the aggregate were removed by solubilizing the PPG in a suitable medium [27, 28]. Finally, the blank EPI crosslinked β-CD nanoparticles were loaded with DTX to form DTX β-CD NPs. These formed nanoparticles were then extensively characterized and systematically evaluated for their anti-cancer effect at in vitro and in vivo levels.
Materials and methods
Materials
DTX was received as a gift sample from Fresenius Kabi, India. β-Cyclodextrin was procured from Gangawal Chemicals, India. Polypropylene glycols of different grades (400, 725, and 1000) were procured from LOBA Cheme, India. EPI was acquired from HiMedia, India. Triton™ X-100, Tween® 80, coumarin-6 (C-6), DMBA, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were acquired from Sigma. Fetal bovine serum (FBS) was procured from Invitrogen™, Life Technologies, Thermo Fisher Scientific Inc. (USA). Commercial kits for estimation of aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine level were procured from Accurex Biomedical Pvt. Ltd., Mumbai, India. Acetonitrile (HPLC grade) was procured from Merck Specialities Pvt. Ltd. (Mumbai, India). Purified water (Millipore, Billerica, MA, USA) filtered from 0.45 μm hydrophilic PVDF filters (Millipore Millex-HV) was used in all experiments. All other reagents were of analytical grade.
Preparation and optimization of DTX β-CD NPs
Blank β-CD NPs were prepared by following the reported protocol with slight modifications [26]. Different critical parameters like PPG grades, ratio of β-CD:EPI, and drug loading were optimized to attain desired size, PDI, and drug content. Briefly, β-CD (1 mM) was dissolved in 10 mL of 0.3% w/v PPG solution and was kept for stirring (until dissolved) at 600 RPM. EPI was added to the above solution, and pH was adjusted to 11.0 with dilute NaOH to provide alkaline condition. After magnetic stirring for 12 h, the solution was neutralized with dilute HCl and was subjected to dialysis (molecular weight 12 kDa) against water for 24 h to remove unreacted β-CD and PPG. Finally, CHCl3 (5 to 7 mL) was added and the reaction mixture was centrifuged to remove unreacted PPG [29]. These blank β-CD NPs were characterized by FTIR for confirmation of crosslinking. Further, DTX was dissolved in ethanol and was loaded in to the blank β-CD NPs by nanoprecipitation method by using syringe injector at the rate of 1 mL/min. This mixture was kept for 24 h under stirring for complex formation followed by centrifugation at 10,000 RPM for 5 min to separate the uncomplexed drug. The supernatant was collected and lyophilized. For optimizing the lyophilization process, several cryoprotectants (mannitol, trehalose, sucrose, dextrose, and inulin) were used. The critical attributes (size, PDI, and % entrapment efficiency) were evaluated before and after lyophilization with different cryoprotectants. Also, the ability of reconstitution in 0.5 mL through vortexing was evaluated to select the best suitable cryoprotectant (optimization data for lyophilization is presented in supplementary file).
Characterization of DTX β-CD NPs
Physicochemical characterization
Particle size and PDI of DTX β-CD NPs were measured by photon correlation spectroscopy using Zetasizer Nano ZS 90 (Malvern Instruments, UK). Each of the samples was diluted suitably using HPLC grade water before analysis.
Morphological characterization
The morphological characteristics of DTX β-CD NPs were examined by scanning electron microscopy (SEM) (S-3400N, Hitachi High- Tech Co., Japan). Protocol followed for sample preparation and analysis was same as reported in our previous report [30].
Complexation studies
The physical interaction of DTX and β-CD was characterized by FTIR analysis in the range of 4000 cm−1 and 400 cm−1 (Perkin-Elmer Inc., USA, with spectrum™ software). Samples were mixed with KBr powder and were pressed using press sheet to form 1 mm pellets for analysis.
Crystallinity analysis
Crystallinity of drug was studied by PXRD (D8 Advanced Diffractometer, Bruker AXS GmbH. Germany) using Ni-filtered Cu Kα (wavelength 1.5406 Å) radiation and equipped with EVA software. The data was recorded over the scanning range of 4° to 40° 2θ scan rate of one second and at a step size of 0.01. Alignment calibration was done using the Alumina powder.
Quantification of DTX
The percentage of DTX in nanoparticles was determined by using validated HPLC analytical method (CBM-20 A pump, SIL-20AC Auto sampler, RF-10AXL with PDA detector) as reported in our previous report. DTX β-CD NPs were dispersed in water and were centrifuged at 10,000 RPM for 5 min to separate the uncomplexed drug. The supernatant was collected and diluted with methanol and analyzed at 230 nm.
Storage stability studies
Freeze-dried formulation was evaluated for storage stability at 4 ± 2 °C and 25 ± 2 °C temperatures for 3 months. After storage, the samples were evaluated for change in critical quality attributes like particle size, PDI, and %EE.
In vitro hemolysis
In vitro hemolysis was performed to evaluate the hemolytic potential of β-CD NPs in comparison to β-CDs. This is because when β-CDs are exposed to red blood cells (RBCs), they tend to cause hemolysis. So, can crosslinking the β-CDs with hydrophilic crosslinker (EPI) assist in reducing the hemolysis was the main intention of this study. The methodology employed to evaluate the % hemolysis was similar to the methods illustrated in literature [31, 32]. Briefly, the heparinized blood from female Swiss Albino mice was collected in microcentrifuge tubes and was subjected to centrifugation at 10,000 RPM for 10 min to separate the RBCs. The supernatant was discarded and RBCs were re-dispersed with isotonic PBS (pH 7.4) so as to form 1% RBC suspension. About 900 μL of 1% RBC suspension was mixed with 100 μL of β-CD NPs (stock 10 µg/µL) and β-CD (stock 10 µg/µL), respectively. RBCs mixed with PBS and Triton™ X-100 were considered as negative and positive control, respectively. The samples were incubated at 37 °C for 2 h and centrifuged at 10,000 RPM for 5 min. The supernatants were then separated and allowed to stand at room temperature for 30 min to oxidize hemoglobin (Hb). The absorbance of oxygenated hemoglobin’s (Oxy-Hb) was measured spectrophotometrically at 540 nm, and the % hemolysis was calculated by using the following equation:
where Abssample and Abs100 denote the absorbance shown by the sample and the control, respectively. The RBCs settled at the bottom were treated with 300 μL of glutaraldehyde solution (0.5% v/v in PBS) and were incubated at 4 °C for 1 h. The RBC suspension was centrifuged again at 3500 RPM for 5 min. The supernatant was discarded and the RBC pellet was washed twice with deionized water. The re-dispersed RBCs were then visualized under SEM (Hitachi, S-3400N, Japan) to study the morphological changes.
In vitro drug release
In vitro drug release of DTX β-CD NPs was carried out using dialysis membrane method (molecular weight cutoff 12,000 Da) [33, 34]. The release media was phosphate buffer (pH 7.4) with 0.1% Tween® 80. Free drug and formulation (equivalent to 1 mg of DTX in 1 mL for each sample) were filled in dialysis bags and were suspended in 25 mL of release media. The assembly was maintained at a temperature of 37 ± 0.5 °C in shaking water bath at 100 RPM. The aliquot samples (1 mL) were collected at pre-determined time intervals (0.5, 1, 2, 4, 6, 8, 12, and 24 h) and were immediately replaced with fresh media pre-incubated at same temperature. The DTX concentration within the collected aliquot was quantified using validated HPLC analytical method.
Cell culture studies
MDA-MB-231 cells (epithelial, human breast cancer cells) were procured from NCCS, Pune, India. The cells were cultured at temperature of 37 °C under 5% CO2 atmosphere. The Eagle’s minimum essential medium (MEM, Sigma) media supplemented with Earle’s salts, non-essential amino acids, L-glutamine, 100 U/mL penicillin, 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, sodium bicarbonate, and sodium pyruvate (PAA Laboratories GmbH, Austria) was used as growth and nurturing media.
Cell uptake study
For qualitative and quantitative cell uptake studies, MDA-MB-231 cells were seeded at a density of 5 × 104 cells/well in 6-well tissue culture plate (Costars, Corning Inc., NY, USA) and were allowed to adhere for 24 h. Following the incubation, the media was replaced with fresh media (without FBS) containing the respective treatment group and were incubated for 3 h.
Qualitative cell uptake of coumarin-6-loaded β-CD NPs
For qualitative cell uptake, coumarin-6 (C-6) (fluorescent dye) was used in lieu of DTX to prepare β-CD NPs in similar fashion as mentioned in the “Preparation and optimization of DTX β-CD NPs” section, to trace the uptake within the cells. Once the cells were adhered, media was replaced with fresh media containing free C-6 and C-6-loaded β-CD NPs (equivalent to 1 μg/mL of C-6), respectively. The treatment groups were incubated with cells for 3 h. The media was removed and cells were washed thoroughly using Dulbecco’s phosphate-buffered saline (DPBS) to remove any un-internalized components and cellular debris. Further, the cells were fixed using glutaraldehyde (2.5% v/v; sigma, USA), washed, and were then observed using confocal laser scanning microscope (CLSM) (Olympus FV1000) under green channel [35].
Quantitative cell uptake of DTX-loaded β-CD NPs (DTX β-CD NPs)
For quantitative cell uptake, the media of adhered cells were replaced with fresh media containing DTX and DTX β-CD NPs (equivalent to 10 and 15 µg/mL of DTX). The treatment groups were incubated with cells for 0.5, 1, 2, and 3 h. The cells were washed thoroughly using DPBS to remove any uninternalized drug or formulation from the surface. The cells were lysed using Triton™ X-100 (0.2% v/v) and were diluted using methanol to extract the internalized DTX. The obtained cell lysate was centrifuged at 21,000 RPM for 10 min at 4 °C to separate the cellular debris and supernatant was separated. The supernatant was then analyzed for DTX content using validated HPLC method [36].
Cell cytotoxicity assay
The cell cytotoxicity evaluation was performed using standard MTT assay as described in our previous reports [30, 36, 37]. Briefly, cells were seeded at a density of 104 cells/well in 96-well flat-bottom tissue culture plates and were incubated for 24 h for adhesion. The media was removed and replaced with fresh media without FBS containing the treatments (free DTX, DTX β-CD NPs, and blank β-CD NPs). Based on % EE, the DTX β-CD NP treatment group was tested at different concentration levels which were equivalent to 0.1, 1, 5, and 10 μg/mL of free DTX. Similarly, blank β-CD NPs were also evaluated at same mentioned concentrations. Each treatment groups were incubated with cells for 24, 48, and 72 h, respectively. After incubation, 20 µL of MTT solution (5 mg/mL) was added in each well and incubated for 3 h. Media from all wells were carefully removed and were replaced with 100 µL of DMSO to dissolve the formed formazan crystals. The optical density of each well was analyzed at 550 nm using a Bio-Tek ELISA plate reader. The % cell viability was calculated using Eq. (2);
Apoptosis
The ability of developed formulation to induce apoptosis in breast cancer cells was determined using standard phosphatidylserine externalization assay as reported previously [11, 35, 36]. Briefly, cells were seeded at the density of 5 × 104 cells/well in 6-well tissue culture plate and were allowed to adhere overnight. The media was then replaced with fresh medium without FBS containing free DTX and DTX β-CD NPs at a dose equivalent to 10 μg/mL of DTX and were incubated for 6 h. After the incubation, cells were washed thoroughly with DPBS and were stained using Annexin V-Cy3.18 conjugate (AnnCy3) and 6-carboxyfluorescein diacetate (6-CFDA) as per the manufacturer’s protocol (Annexin V-Cy3™ Apoptosis Detection Kit, Sigma, USA). The cells were then observed under red and green fluorescent channel, respectively, using CLSM. The cells which were stained green, red, and yellow (overlay of green and red) indicated the live, necrotic, and apoptotic cell populations, respectively. For quantitative estimation, the apoptotic index (ratio of the red and green fluorescence intensity) was measured by estimating the red and green fluorescence intensity using ImageJ software (US National Institutes of Health, USA).
In vivo studies
Pharmacokinetic study
In vivo pharmacokinetics of developed formulation was performed using female Sprague–Dawley (SD) rats based on our previous protocols [14, 30, 38]. The rats were procured from central animal facility; NIPER and protocols were approved by the Institutional Animals Ethics Committee. The rats were housed and maintained (at temperature of 25 ± 2 °C; humidity 55 ± 5%; 12 h light/dark cycles) with food and water ad libitum. The rats were divided in to two groups (n = 5). In the first group, dose of 2 mg/kg marketed formulation of DTX (Docepar® a generic version of Taxotere®) was injected intravenously through tail vein while the second group received DTX β-CD NPs in equivalent dose intravenously. Blood samples were collected through tail vein into heparinized tubes and were centrifuged for 5 min at 3500 RPM. Paclitaxel was used as an internal standard in bioanalytical method [39], and concentration of DTX in plasma was estimated using validated bio-analytical HPLC method. The various pharmacokinetic parameters like total area under curve (AUC 0-α) and terminal phase half-life (t1/2) were analyzed using Kinetica Software (Version 5.0, Thermo scientific).
In vivo antitumor efficacy studies
The anticancer potential of developed formulation was evaluated using DMBA-induced breast cancer model as described in our previous reports [30, 40]. Briefly, DMBA at a dose of 45 mg/kg solubilized in soya bean oil was administered orally at a frequency of once a week for three consecutive weeks in ~2-month-old female Sprague–Dawley rats (180–210 g). Measurable volumes of tumors were observed after 10 weeks of DMBA administration. Tumor-bearing animals were randomly divided into 3 groups (n = 5). The first two groups of animals received a single dose of Docepar® and DTX β-CD NPs via intravenous injection (dose equivalent to 2 mg/kg of free DTX). The third group was kept as a control group and was injected with saline intravenously. The tumor volume was measured as prospective of tumor width (W; in mm) and length (L; in mm) with an electronic digital caliper on every alternate day by using the Eq. (3):
Further, the % change in tumor volume was estimated by evaluating the percentage tumor growth or reduction with respect to the tumor volume observed during the initial day of the treatment.
Toxicity evaluation
The toxicity profile of the developed formulations was evaluated using healthy female Swiss Albino mice (25–30 g). Animals were randomly divided in to three groups (n = 5). The first two groups were intravenously injected through tail vein with Docepar® (marketed formulation) and DTX β-CD NPs at a dose equivalent to 5 mg/kg of DTX. The third group was injected with saline and was considered as control. After 7 days of injection, the mice were humanly sacrificed and blood samples were collected through cardiac puncture in microcentrifuge tubes preloaded with heparin (40 IU/mL of blood). The collected samples were centrifuged at 3500 RPM for 10 min, which separated the RBCs from plasma. The separated plasma was subjected to analyze the levels of aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), and creatinine. The separated RBCs were dispersed using glutaraldehyde solution (0.5% v/v in saline) and were incubated at 4 °C for 1 h. The formed RBC suspensions were again centrifuged at 3500 RPM for 10 min, and the settled RBCs were washed thrice using isotonic saline solution. Finally, the re-suspended RBCs were visualized under SEM (Hitachi, S-3400N, Japan) to identify any morphological changes. Further, for histological evaluation, vital organs like liver, kidney, and spleen were isolated and were outsourced to pathology lab (Nimbus Path Lab, Mohali, India) for tissue slicing and hematoxylin and eosin staining.
Statistical analysis
The data for all in vitro and in vivo results was expressed in terms of mean ± standard deviation (SD). GraphPad Prism® (version 5.01) was used to perform the statistical analysis utilizing one-way ANOVA followed by a Bonferroni multiple comparison tests.
Results and discussion
Preparation and optimization of DTX β-CD NPs
The preparation of DTX β-CD NPs involves the formation of partial inclusion complexes of β-CD and PPG as illustrated in Fig. 1a. It has been reported that when PPG with molecular weight (MW) of 400–1000 Da is used, it can form partial inclusion complex with β-CDs [28, 41]. Further, if the MW of PPG is increased beyond 1000 Da, chance of acquiring the desired results shortens. Hence, we selected PPG with MW of 400 Da to form the partial inclusion complex (Table 1). The formed partial inclusion complex possessed an ability to self-assemble in aqueous media. Wherein, the hydrophobic part of the β-CD constitutes to form the core and incompletely threaded PPG chains forms the corona. The presence of PPG in the corona, enabled them to interact (hydrogen bonds) with water molecules which stabilized the β-CDs and prevented their larger aggregation [26, 28]. Finally, the self-assembled β-CDs were crosslinked using EPI. EPI is a bifunctional agent containing an epoxide and chloroalkyl moiety which can easily interact and crosslink the β-CDs. To understand the role of PPG as a stabilizer (by forming partial inclusion complex), we tried to crosslink the β-CDs (under similar conditions and concentration) in absence of PPG. Large aggregates of β-CDs with high PDI were formed (Fig. 1b (ii)). Moreover, we observed that the molar ratio of EPI with respect to β-CDs critically affects the size and PDI of the aggregates and same is reported in the literature as well [42, 43]. But considering that EPI may exhibit some undesired in vivo effects, we selected β-CD/EPI molar ratio of 1:8 to be optimum.

a Describes the steps involved in formulating DTX β-CD NPs and also in brief illustrates its further processing. b Displays the SEM images of (i) crosslinked β-CD NPs formed using PPG and (ii) crosslinked β-CD NPs formed without using PPG
Once, the partial inclusion complexes of β-CD and PPG were crosslinked with EPI at 1:8 ratio, the PPG was removed by solubilizing it in CHCl3 to yield blank β-CD NPs with size of 211.56 ± 14.45 nm and PDI of 0.228 ± 0.031. Drug loading into blank NPs was optimized based on particle size, PDI and %EE (Table 2). Thus, with 5% of drug loading, highest % EE (54.533 ± 2.004) and size of 223.366 ± 17.730 nm (PDI of 0.13 ± 0.09) was observed and hence was considered ahead. Further, the formed DTX β-CD NPs were lyophilized using different cryoprotectants (refer supplementary file). However, lyophilization using mannitol revealed cake like elegancy and exhibited insignificant changes in particle size, PDI and %EE upon re-dispersion. Also, easy re-dispersion was observed with mannitol within 20 s. Whereas, other cryoprotectants rendered significant changes in these critical attributes due to the formation of collapsed network formation (Table S1).
Shape and morphology of DTX β-CD NPs
Morphology of DTX β-CD NPs through SEM revealed that the particles were spherical and uniform as shown in Fig. 1b. However, size evaluated using SEM was not matching with the size values evaluated through zeta-sizer. This is due to the fact that the zeta-sizer is based on DLS principle which considers the hydrodynamic radius of the particles, whereas SEM provides the actual surface scanned image.
Complexation studies
The complexation studies were performed using FTIR and PXRD analysis.
FTIR analysis
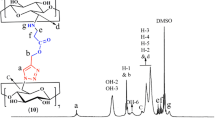
Figure 2a depicts the FTIR spectra of β-CD and cross linked blank β-CD NPs. Blank crosslinked β-CD NPs showed C–O–C stretching band at 1157 cm−1, CH2-Cl wagging band at 1250 cm−1, and C–Cl stretching band at 755 cm−1, which confirms the crosslinking of β-CD with EPI. Figure S1 depicts the FTIR spectra of β-CD, β-CD-PPG physical mixture, PPG and β-CD-PPG complex. The characteristics peaks of β-CDs (1000–1300 cm−1 due to stretching of ether bond) shows significant shift and characteristic peak of PPG at 3461 (O–H stretching), 2971–2931 (-C-H stretching) appeared in case of complex, while these peaks were absent in case of physical mixture. Figure 2b depicts the FTIR spectra of DTX, blank nanoparticles, physical mixture and DTX β-CD NPs. Interaction between DTX and β-CD NPs was confirmed from peak broadening and characteristic peak disappearance of DTX (peaks at 1713, 1093, 1369 cm−1) and β-CD (peaks at 1602.49, 1497.33, 1453.05, and 1419.03 cm−1). The bands at 1024.18 (C = O stretching), 1071.16 (C-N stretching), 1167.97 (C–O–C linkage) and 2908.58 cm−1 (C-H stretching) showed a significant shift and displayed a characteristic DTX peak at 1713.36 cm−1 with comparative changes in frequency for DTX β-CD NPs. This confirmed that there is an interaction between DTX and β-CDs. These peaks were absent in case of physical mixture suggesting superposition of both DTX and β-CD.

FTIR spectra of a β-CD and EPI crosslinked β-CD, b β-CD-PPG complex and DTX, blank β-CD NPs, physical mixture, and DTX β-CD NPs
PXRD analysis
PXRD analysis was used to evaluate the crystallinity state of drug in the formulation. Characteristic diffraction peaks at 3.24°, 7.09°, 7.81°, 9.93°, and 10.75° 2θ values of DTX were observed in case of physical mixture while these peaks were absent in case of DTX β-CD NPs. This indicated that the crystalline nature of DTX was converted into amorphous form when complexed with β-CD [44] (Fig. 3).

PXRD spectra of DTX, blank β-CD NPs, physical mixture and DTX β-CD NPs
Storage stability studies
Storage stability studies were performed for 3 months at 4 ± 2 °C and 25 ± 2 °C temperatures. Supplementary Table S2 represents the results of storage stability. It was observed that there is no significant change (p > 0.05) in the size and PDI of the developed formulation till 3 months which indicates that the developed system is stable.
In vitro hemolysis
The in vitro hemolysis study revealed that β-CD exhibited significantly (p ≤ 0.001) higher % hemolysis (25.22 ± 3.08) in comparison to β-CDs NPs (5.95 ± 0.55). However, insignificant change in % hemolysis was observed for β-CDs NPs when compared to control (Fig. S2). This indicates that β-CD NPs are more compatible than the un-modified β-CDs which is true and same is reported in literature [45]. Further, the SEM images reveal higher RBC membrane deformation in case of β-CDs than the β-CD NPs (Fig. S2). Probable reason for these observations can be the tendency of β-CDs to abstract cholesterol from the RBC plasma membrane which can lead to membrane destabilization and lysis [45,46,47]. However, crosslinking β-CDs to form matrix type NPs can restrict the hydrophobic region to the core part of the particle which can hinder the cholesterol abstraction by great extent. Also, EPI being hydrophilic in nature it can impart hydrophilicity in the periphery of the NPs which can further hinder the cholesterol abstraction from RBCs plasma membrane [42]. Thus, due to such toxic nature of the β-CDs (alone; non-crosslinked), they were not considered ahead for further investigation.
In vitro drug release
The in vitro cumulative release profiles of DTX and DTX β-CD NPs in PBS (pH 7.4) containing 0.1% w/v of Tween® 80 is shown in Fig. 4. The free DTX showed an obvious rapid release of ~95% within 8 h. Whereas, DTX β-CD NPs displayed a biphasic release pattern in which initially a burst release (~20%) was observed within first 2 h, followed by sustained release (~60%) up to 24 h. The probable reason for such biphasic release could be due to the physical adsorption of the drug on NPs which exhibited the initial burst release and the following prolonged release resulted from the drug diffusion through the NPs.

In vitro release of free DTX and DTX β-CD NPs in phosphate buffer pH (7.4) (entire experiment was performed in triplicate, n = 3)
Cell culture studies
Cell uptake
Figure 5a displays the CLSM images of MDA-MB-231 cells when incubated with C-6-loaded β-CD NPs and free C-6 for 3 h. Qualitatively, higher green fluorescence intensity can be observed in cells incubated with C-6 loaded β-CD NPs compared to the cell incubated with free C-6 dye. Furthermore, the quantitative cell uptake demonstrated significantly (p < 0.05) higher levels of DTX in cells treated with DTX β-CD NPs as compared to free DTX-treated cells as shown in Fig. 5b. However, the DTX titter within the cells after 2 h of incubation showed insignificant (p > 0.05) increase. This directed that under set conditions (cell density and treatment concentrations of NPs), maximum uptake within the cells was accomplished within 2 h and after that, a plateau/saturation phase is achieved. Overall, the DTX β-CD NPs exhibited ~5-fold higher uptake in comparison to free DTX. This indicated that the formed NPs were capable of overthrowing the issue of intracellular entry faced with DTX. This hinted that DTX β-CD NPs can effectively improve the therapeutic efficacy of the conventional DTX.

a Represents the CLSM images of the qualitative cell uptake of free C-6 and C-6-loaded β-CD NPs (C-6 β-CD NPs) in MDA-MB-231 cells. b Represents the quantitative uptake of free DTX and DTX β-CD NPs in MDA-MB-123 cells when incubated for specified duration (0.5, 1, 2, and 3 h) and concentrations (equivalent to 10 and 15 µg/mL of DTX) (values are represented as mean ± SD, nsp > 0.05, ** p < 0.01, and ***p < 0.001)
In vitro cytotoxicity
Figure 6 shows the % cell viability of MDA-MB-231 cells when treated with blank β-CD NPs, DTX β-CD NPs, and free DTX at different time points. The cytotoxicity of DTX β-CD NPs was significantly higher (p < 0.01) than that of free drug, and it was further assessed by IC50 values. The IC50 value of DTX was reduced from 5.78 to 1.75 μg/mL when treated with DTX β-CD NPs, i.e., ~3.3-fold reduction was observed. Such higher cytotoxicity was attributed because of the obvious reason that the formed NPs were able to increase the intracellular DTX titter as compared to free DTX. However, blank β-CD NPs showed > 85% cell viability at all concentrations and time points indicating that the resultant toxicity was only due to the loaded drug and not due to the carrier.

Cell cytotoxicity of various formulations after a 24 h, b 48 h, and c 72 h (values represented as mean ± SD, where ***p ≤ 0.001 and *p ≤ 0.05)
Apoptotic assay
The apoptotic potential of the formulation was evaluated using standard phosphatidylserine (PS) externalization assay. In normal cells, PS is located in the inner leaflet of the plasma membrane. During apoptosis, the distribution is randomized which results in appearance of PS on the outer leaflet of the membrane. Here, we have used AnnCy3 dye which can effectively bind with the exposed PS on the cell membrane. Along with AnnCy3, 6-CFDA was also used which permeates into the cells and gets hydrolyzed by the esterases present within the live cells to form fluorescent compound called 6-carboxyfluorescein, indicating that cell is alive. This combination (AnnCy3 and 6-CFDA) enables in differentiating early apoptotic (AnnCy3+ve/6-CFDA+ve), necrotic (AnnCy3+ve/6-CFDA−ve), and live cells (AnnCy3−ve/6-CFDA+ve) [48]. Similarly, Fig. 7 reveals the CLSM images of AnnCy3 and 6-CFDA stained cells after treating with free DTX and DTX β-CD NPs. The apoptotic index of free DTX and DTX β-CD NPs was found to be 0.57 and 0.93, respectively. The apoptotic results were found to be in accordance with cell uptake and cell cytotoxicity results which validates the finding that DTX β-CD NPs are more potent than the free DTX.

Showcases the CLSM images of double-stained (AnnCy3 and 6-CFDA dye) MDA-MB-231 cells, upon treatment with a free DTX and b DTX β-CD NPs. In both panels, the subpanels depict the CLSM images of cells under (a) 6-CFDA channel (green cells indicating the live population), (b) AnnCy3 channel (red cells indicating necrotic population), (c) overlay of (b) and (c) (yellow cells indicating apoptotic population), and (d) differential interference contrast image of the representative cells. The apoptotic index was measured by calculating the ratio of fluorescence intensity observed under red channel to that of the fluorescence intensity observed under green channel. The fluorescence intensities of the images were measured using ImageJ software, US National Institutes of Health
In vivo pharmacokinetics
The plasma concentration profiles of DTX after single I.V. dose of Docepar® and DTX β-CD NPs is shown in Fig. 8, and the critical pharmacokinetic parameters are illustrated in Table 3. The Docepar® showed a rapid decline in plasma concentration within 2 h of administration when compared to DTX β-CD NPs. A significant (p < 0.001) increase in AUC0-∞ (13,405.84 ± 569.52 ng.h/mL; ~2-fold) of DTX in animals injected with DTX β-CD NPs was observed in comparison to AUC0-∞ in animals injected with Docepar® (6034.47 ± 239.25 ng h/L). Half-life of DTX (10.59 ± 0.85 h in case of Docepar®) was also significantly (p < 0.05) increased with DTX β-CD NPs (31.25 ± 2.36 h). Mean residence time (MRT) of DTX for DTX β-CD NPs was found to be ~5-fold higher than Docepar®. The probable reason for such improved pharmacokinetic parameters can be the increased encapsulation of DTX within β-CD NPs due to their tuneable structure [49], and their ability to prolong the DTX release.

Evaluating the plasma concentration profile of DTX with respect to time in animals treated with Docepar® and DTX β-CD NPs (at a dose equivalent to 2 mg/kg of DTX) (values represented as mean ± SD; n = 5)
In vivo antitumor efficacy
Antitumor efficacy of NPs was explored using DMBA induced breast cancer model. The developed formulation showed significantly (p < 0.01) better effect by reducing tumor volume compared to the marketed formulation. Figure 9a, b illustrate the % change in tumor volume and % tumor burden in animals treated with saline (control), Docepar® and DTX β-CD NPs. The Docepar® and DTX β-CD NPs were able to reduce the tumor volume to ~80 and ~40% with respect to tumor volume calculated during the start of the treatment. A representative image of the isolated tumors from each treatment group is shown in Fig. 9(B’). This indicates the efficacy of DTX β-CD NPs in reducing the tumor volume as compare to its marketed counterpart. Further, prolonged release of DTX, increased intracellular entry and enhanced permeation and retention effect by the DTX β-CD NPs could be the probable reasons for such superior results.

a Illustrates the % change in tumor volume in the animals injected with mentioned treatments. b Illustrates the % tumor burden in treated animals (values represented as mean ± SD; n = 5 and *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001). (B’) Illustrates a representative digital photograph of isolated tumors from each treatment group
In vivo toxicity
The in vivo suitability of CD supra-molecular drug carriers and their detailed safety profiles are often unclear [50]. Hence, efforts need be made to ensure that the supra-molecular complexes are not only safe at in vitro level but also compatible at in vivo level as well. This is probably the first attempt where β-CDs based nanoparticles are formulated for intravenous administration to investigate their in vivo safety profiles. The in vivo toxicity assessment revealed that animals treated with Docepar® had significantly (p < 0.001) increased levels of ALT, AST, BUN, and creatinine in comparison to control group indicating potential renal impairment. However, DTX β-CD NPs showed insignificant (p > 0.05) changes in the levels of BUN and creatinine but significant (p < 0.05) increase in ALT and AST was observed in comparison to control. But the levels of ALT and AST in animals treated with DTX β-CD NPs were still lower than the levels observed for Docepar®-treated animals. This indicates that the DTX β-CD NPs renders lower chance of exhibiting hepatic stress than its marketed counterpart (Fig. 10a). Further, the histopathology examinations of organs isolated from animals treated with Docepar® revealed toxicity to kidney (necrotic tubules), liver (hepatocyte damage), and spleen (splenocyte damage). Whereas, in case of DTX β-CD NP-treated animals, organs did not exhibit any short-term nephro- and hepato-toxicity when administered intravenously (Fig. 10b). This might be due to the hydrophilicity granted by EPI to the NPs, which allowed rapid clearance and lower chance of aggregation and precipitation in kidney [51]. Similarly, hemolysis study revealed that there were no noticeable changes in the shape and morphology of the RBCs isolated from the animals treated with DTX β-CD NPs (Fig. 10c). Whereas Docepar®-treated animals revealed alteration in RBC morphology compared to control, which indicates hemo-incompatibility. So, we hypothesized that there is a scope to explore EPI crosslinked β-CDs for intravenous application as an alternative to other hydrophilic derivatives of β-CDs.

Represents the data obtained from the animals after 7 days from treatment with DTX β-CD NPs and Docepar® at their respective toxic doses. a Presents the observed plasma levels of AST, ALT, BUN, and creatinine in animals injected with respective treatments (values represented as mean ± SD; n = 5 and nsp > 0.05, *p < 0.05, and **p < 0.01 with respect to control). b Represents the histopathology of liver, spleen, and kidney isolated from the animals. Finally, c represents SEM images of RBC isolated from (i) control, (ii) DTX β-CD NPs, and (iii) Docepar®-treated animals
Conclusion
It can be concluded that our strategy of self-assembling the β-CD by forming partial inclusion complexes with PPG followed by crosslinking of β-CDs with EPI and removal of PPG by solubilization to form uniform nanoparticles was successful. However, when direct crosslinking with EPI without the formation of inclusion complex was tried, it led to form large aggregates with high PDI. Further, the uniformly formed DTX β-CD NPs showed desired drug loading and entrapment with improved (~5-fold higher than free drug) cellular internalization. Also, the DTX β-CD NPs were capable of improving the potency of the DTX by inducing apoptosis when tested at in vitro level using MDA-MB-231 cells. Similarly, the in vivo investigation also suggested that DTX β-CD NPs can exhibit better pharmacokinetic and dynamic results when compared to Docepar® (marketed counterpart). Interestingly, DTX β-CD NPs revealed no potential nephro- and hemo-toxicity. This actually makes us wonder that can this system be a potential substitute for hydrophilic (non-toxic) derivatives of β-CDs. To answer that, further exploratory investigation is required.
Availability of data and materials
Data will be available on request to authors.
Change history
19 February 2024
A Correction to this paper has been published: https://doi.org/10.1007/s13346-024-01545-6
References
Organization WH. Global status report on noncommunicable diseases 2014. World Health Organization. 2014.
Coburn N, Cosby R, Klein L, Knight G, Malthaner R, Mamazza J, et al. Staging and surgical approaches in gastric cancer: a systematic review. Cancer Treat Rev. 2018;63:104–15.
Guo X, Fang W, Li Z, Yu Z, Rong T, Fu J, et al. Adjuvant radiotherapy, chemotherapy or surgery alone for high-risk histological node negative esophageal squamous cell carcinoma: protocol for a multicenter prospective randomized controlled trial: adjuvant therapy for esophageal cancer. Thorac Cancer. 2018;9:1801–6.
Malavia N, Kuche K, Ghadi R, Jain S. A bird’s eye view of the advanced approaches and strategies for overshadowing triple negative breast cancer. J Control Release. 2021;330:72–100.
Zhu Y, Wang L, Yang Z, Wang J, Li W, Zhou J, et al. Hematopoietic effects of paeoniflorin and albiflorin on radiotherapy-induced myelosuppression mice. Evid Based Complement Alternat Med. 2016;2016:1–8.
Nguyen NP, Sallah S, Karlsson U, Antoine JE. Combined chemotherapy and radiation therapy for head and neck malignancies: quality of life issues. Cancer. 2002;94:1131–41.
Desale K, Kuche K, Jain S. Cell-penetrating peptides (CPPs): an overview of applications for improving the potential of nanotherapeutics. Biomater Sci. 2021;10.1039.D0BM01755H.
Zhang N, Zhang. How nanotechnology can enhance docetaxel therapy. IJN. 2013;2927.
Gupta A, Sharma R, Kuche K, Jain S. Exploring the therapeutic potential of the bioinspired reconstituted high density lipoprotein nanostructures. Int J Pharm. 2021;596:120272.
Ghadi R, Dand N. BCS class IV drugs: highly notorious candidates for formulation development. J Control Release. 2017;248:71–95.
Jain S, Spandana G, Agrawal AK, Kushwah V, Thanki K. Enhanced antitumor efficacy and reduced toxicity of docetaxel loaded estradiol functionalized stealth polymeric nanoparticles. Mol Pharm. 2015;12:3871–84.
Hwang H-Y, Kim I-S, Kwon IC, Kim Y-H. Tumor targetability and antitumor effect of docetaxel-loaded hydrophobically modified glycol chitosan nanoparticles. J Control Release. 2008;128:23–31.
Immordino ML, Brusa P, Arpicco S, Stella B, Dosio F, Cattel L. Preparation, characterization, cytotoxicity and pharmacokinetics of liposomes containing docetaxel. J Control Release. 2003;91:417–29.
Lee S-W, Yun M-H, Jeong SW, In C-H, Kim J-Y, Seo M-H, et al. Development of docetaxel-loaded intravenous formulation, Nanoxel-PMTM using polymer-based delivery system. J Control Release. 2011;155:262–71.
Zhang D, Lv P, Zhou C, Zhao Y, Liao X, Yang B. Cyclodextrin-based delivery systems for cancer treatment. Mater Sci Eng C. 2019;96:872–86.
Lakkakula JR, Maçedo Krause RW. A vision for cyclodextrin nanoparticles in drug delivery systems and pharmaceutical applications. Nanomedicine. 2014;9:877–94.
Li P, Song J, Ni X, Guo Q, Wen H, Zhou Q, et al. Comparison in toxicity and solubilizing capacity of hydroxypropyl-β-cyclodextrin with different degree of substitution. Int J Pharm. 2016;513:347–56.
Brewster ME, Simpkins JW, Hora MS, Stern WC, Bodor N. The potential use of cyclodextrins in parenteral formulations. PDA Journal of Pharmaceutical Science and Technology. Parenteral Drug Association (PDA). 1989;43:231–40.
Nie S, Zhang S, Pan W, Liu Y. In vitro and in vivo studies on the complexes of glipizide with water-soluble β-cyclodextrin–epichlorohydrin polymers. Drug Dev Ind Pharm. 2011;37:606–12.
Jug M, Kosalec I, Maestrelli F, Mura P. Analysis of triclosan inclusion complexes with β-cyclodextrin and its water-soluble polymeric derivative. J Pharm Biomed Anal. 2011;54:1030–9.
Gidwani B, Vyas A. Formulation, characterization and evaluation of cyclodextrin-complexed bendamustine-encapsulated PLGA nanospheres for sustained delivery in cancer treatment. Pharm Dev Technol. 2016;21:161–71.
Wenz G, Han B-H, Müller A. Cyclodextrin rotaxanes and polyrotaxanes. Chem Rev American Chemical Society. 2006;106:782–817.
Huang F, Gibson HW. Polypseudorotaxanes and polyrotaxanes. Prog Polym Sci. 2005;30:982–1018.
Omrani Z, Dadkhah TA. New cyclodextrin-based supramolecular nanocapsule for codelivery of curcumin and gallic acid. Polym Bull. 2020;77:2003–19.
Wu Y-L, Li J. Synthesis of supramolecular nanocapsules based on threading of multiple cyclodextrins over polymers on gold nanoparticles. Angew Chem Int Ed. 2009;48:3842–5.
Zhu W, Zhang K, Chen Y, Xi F. Simple, clean preparation method for cross-linked α-cyclodextrin nanoparticles via inclusion complexation. Langmuir. 2013;29:5939–43.
Blackadder DA, Le Poidevin GJ. Dissolution of polypropylene in organic solvents: 1. Partial dissolution Polymer. 1976;17:387–94.
Harada A, Kamachi M. Complex formation between cyclodextrin and poly(propylene glycol). J Chem Soc: Chem Commun; 1990. p. 1322.
Mognetti B, Barberis A, Marino S, Berta G, Francia S, Trotta F, et al. In vitro enhancement of anticancer activity of paclitaxel by a Cremophor free cyclodextrin-based nanosponge formulation. J Incl Phenom Macrocycl Chem. 2012;74:201–10.
Desale JP, Swami R, Kushwah V, Katiyar SS, Jain S. Chemosensitizer and docetaxel-loaded albumin nanoparticle: overcoming drug resistance and improving therapeutic efficacy. Nanomedicine. 2018;13:2759–76.
Mandal H, Katiyar SS, Swami R, Kushwah V, Katare PB, Kumar Meka A, et al. ε-Poly-l-Lysine/plasmid DNA nanoplexes for efficient gene delivery in vivo. Int J Pharm. 2018;542:142–52.
Jain S, Kumar S, Agrawal AK, Thanki K, Banerjee UC. Hyaluronic acid–PEI–cyclodextrin polyplexes: implications for in vitro and in vivo transfection efficiency and toxicity. RSC Adv. The Royal Society of Chemistry. 2015;5:41144–54.
Jain S, Bhankur N, Swarnakar NK, Thanki K. Phytantriol based “stealth” lyotropic liquid crystalline nanoparticles for improved antitumor efficacy and reduced toxicity of docetaxel. Pharm Res. Springer. 2015;32:3282–92.
Jain S, Deore SV, Ghadi R, Chaudhari D, Kuche K, Katiyar SS. Tumor microenvironment responsive VEGF-antibody functionalized pH sensitive liposomes of docetaxel for augmented breast cancer therapy. Mater Sci Eng C. 2021;121:111832.
Bathara M, Date T, Chaudhari D, Ghadi R, Kuche K, Jain S. Exploring the promising potential of high permeation vesicle-mediated localized transdermal delivery of docetaxel in breast cancer to overcome the limitations of systemic chemotherapy. Mol Pharm. 2020;17:2473–86.
Kushwah V, Jain DK, Agrawal AK, Jain S. Improved antitumor efficacy and reduced toxicity of docetaxel using anacardic acid functionalized stealth liposomes. Colloids Surf B. 2018;172:213–23.
Kaushik L, Srivastava S, Panjeta A, Chaudhari D, Ghadi R, Kuche K, et al. Exploration of docetaxel palmitate and its solid lipid nanoparticles as a novel option for alleviating the rising concern of multi-drug resistance. Int J Pharm. 2020;578:119088.
Kushwah V, Katiyar SS, Agrawal AK, Gupta RC, Jain S. Co-delivery of docetaxel and gemcitabine using PEGylated self-assembled stealth nanoparticles for improved breast cancer therapy. Nanomed Nanotechnol Biol Med. 2018;14:1629–41.
Swami R, Singh I, Jeengar MK, Naidu VGM, Khan W, Sistla R. Adenosine conjugated lipidic nanoparticles for enhanced tumor targeting. Int J Pharm. 2015;486:287–96.
Jain AK, Swarnakar NK, Godugu C, Singh RP, Jain S. The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials. 2011;32:503–15.
Fox TG, Flory PJ. Second‐order transition temperatures and related properties of polystyrene. I. Influence of molecular weight. J Appl Phys. 1950;21:581–91.
Renard E, Deratani A, Volet G, Sebille B. Preparation and characterization of water soluble high molecular weight β-cyclodextrin-epichlorohydrin polymers. Eur Polymer J. 1997;33:49–57.
Renard E, Sebille B, Barnathan G, Deratani A. Polycondensation of cyclodextrins with epichlorohydrin. Influence of reaction conditions on the polymer structure. Macromol Symp. 1997;122:229–34.
Youm I, Yang X, Murowchick JB, Youan B-BC. Encapsulation of docetaxel in oily core polyester nanocapsules intended for breast cancer therapy. Nanoscale Res Lett. 2011;6:630.
Irie T, Otagiri M, Sunada M, Uekama K, Ohtani Y, Yamada Y, et al. Cyclodextrin-induced hemolysis and shape changes of human erythrocytes in vitro. J Pharmacobiodyn. 1982;5:741–4.
Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim Biophys Acta Biomembr. 2007;1768:1311–24.
Kilsdonk EPC, Yancey PG, Stoudt GW, Bangerter FW, Johnson WJ, Phillips MC, et al. Cellular cholesterol efflux mediated by cyclodextrins. J Biol Chem. 1995;270:17250–6.
Farinacci M. Improved apoptosis detection in ovine neutrophils by annexin V and carboxyfluorescein diacetate staining. Cytotechnology. 2007;54:149–55.
Gidwani B, Vyas A. Synthesis, characterization and application of epichlorohydrin-β-cyclodextrin polymer. Colloids Surf B. 2014;114:130–7.
Tsoucaris G. Current challenges on large supramolecular assemblies. Springer Science & Business Media. 2012.
Szeman J, Ueda H, Szejtli J, Fenyvesi E, Machida Y, Nagai T. Complexation of several drugs with water-soluble cyclodextrin polymer. Chem Pharm Bull. 1987;35:282–8.
Acknowledgements
The authors acknowledge Fresenius Kabi for providing docetaxel as a gift sample. Authors are also thankful to Mr. Rahul Mahajan, NIPER, S.A.S. Nagar for his technical assistance.
Author information
Authors and Affiliations
Contributions
SJ, MD, and BN conceived this research and designed experiments. SJ and MD participated in designing and interpretation of the data. MD and BN performed experiments and analysis. KK wrote the paper and participated in the revisions of it. DC and SK significantly contributed in performing the in vitro and in vivo studies.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All the procedures performed in this study involving animals were in accordance with the ethical standards of NIPER and protocols were approved by the Institutional Animals Ethics Committee.
Consent for publication
All authors have read and approved the submitted manuscript.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jain, S., Desai, M.R., Nallamothu, B. et al. Partial inclusion complex assisted crosslinked β-cyclodextrin nanoparticles for improving therapeutic potential of docetaxel against breast cancer. Drug Deliv. and Transl. Res. 12, 562–576 (2022). https://doi.org/10.1007/s13346-021-00956-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-021-00956-z