
Abstract
Amphotericin B (AmB) exhibits potential antileishmanial activity, with only a little rate of recurrence. However, low bioavailability and severe nephrotoxicity are among the major shortcomings of AmB-based therapy. Various AmB nanoformulations have been developed, which to an extent, have reduced its toxicity and increased the drug efficacy. To further reduce the nonspecific tissue distribution and the cost of the treatment, the current AmB-based formulations require additional improvements. Combination of natural bioenhancers with AmB is expected to further increase its bioavailability. Therefore, we developed a nanoformulation of AmB and piperine (Pip), a plant alkaloid, known to enhance the bioavailability of various drugs, by entrapping them in guar gum, a macrophage targeting polymer. Owing to the ease of oral delivery, these nanoparticles (NPs) were coated with eudragit to make them suitable for oral administration. The formulated eudragit-coated AmB and Pip-loaded NPs (Eu-HDGG-AmB-Pip-NPs) exhibited controlled release of the loaded therapeutic agents and protected the drug from acidic pH. These NPs exhibited effective suppression of growth of both promastigotes and amastigotes of Leishmania donovani parasite under in vitro. In vivo evaluation of these NPs for therapeutic efficacy in golden hamster-L. donovani model demonstrated enhanced drug bioavailability, non-nephrotoxic nature, and potential antileishmanial activity with up to 96% inhibition of the parasite.

Graphical abstract
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Visceral leishmaniasis (VL) is a chronic protozoan infection caused by parasite of genus Leishmania [1, 2]. The parasite resides and grows in macrophages of reticuloendothelial system [2]. VL is associated with severe immune dysfunction and high mortality if left untreated [1, 2]. Pentavalent antimonials (PA) were the mainstays for VL treatment due to their high efficacy and cost effectiveness [3]. However, toxicity to various organs and emergence of drug resistance limit their use. Miltefosine and sitamaquine are among the other effective antileishmanial agents, but emergence of drug-resistant strains, teratogenicity, abortificient activity, methhemoglobinemia, and nephrotoxicity [4, 5] are among the reported side effects.
The polyene antibiotic, amphotericin B (AmB), has demonstrated excellent cure rate with no reported resistance and a little rate of recurrence [6]; however, low bioavailability [7] and nephrotoxicity are the major disadvantages of AmB treatment [8]. Current AmB-based formulations are also having several disadvantages such as nonspecific tissue toxicity, stability issue, nephrotoxicity, and high cost [9,10,11,12] necessitating the development of alternative treatment strategies. Nanotechnology-based drug delivery systems offer promising approach to reduce the side effects and increase the treatment efficacy of AmB [7, 9].
The combination of natural compounds, which are known to increase the absorption of therapeutic agents, has gained immense interest for improving the bioavailability of poorly bioavailable drugs [13]. Piperine, the pungent constituent of Piper nigrum, has been extensively evaluated for its bioavailability enhancing effect [14]. Therefore, we decided to combine the bioenhancing effect of Pip with antileishmanial activity of AmB by entrapping both of them in a single nanoformulation.
Guar gum is a natural polysaccharide known to target mannose-like receptors on macrophages [15]. Various guar gum-based nanoparticles have been reported to deliver the therapeutic agents to macrophages [16,17,18]. Moreover, guar gum is recognized to activate macrophages by induction of pro-inflammatory polarization, which may further increase the treatment efficacy of the delivered agents via activating host immune defense [19,20,21]. Therefore, we selected guar gum as a carrier to deliver the therapeutic agents to the macrophages to achieve further enhancement in treatment efficacy by site specific delivery. Considering the convenience of oral route, we planned to formulate enteric coated nanoparticles [22] and selected a pH responsive polymer, eudragit L30D to protect the nanoformulation from the acidic pH of GI tracts.
Eventually, we prepared eudragit L30D-coated AmB and Pip-loaded guar gum (HDGG) nanoparticles (Eu-HDGG-AmB-NPs) and evaluated their in vitro and in vivo antileishmanial activity. The therapeutic efficacy of Eu-HDGG-AmB-NPs was evaluated in vitro on promastigotes and amastigotes of Leishmania donovani (L. donovani) and in vivo in golden hamster model of L. donovani.
Materials and methods
Materials
AmB and dialysis membrane were purchased from Sigma-Aldrich, USA. Eudragit L30D was purchased from Evonik, Germany. All other chemicals and organic solvents either of analytical or HPLC grade were procured from Sigma-Aldrich, USA. Guar gum was obtained from M/s. Sunita Hydrocolloids Pvt. Ltd. (SHPL), Jodhpur, India, and purified using standard methods. Distilled water from a three-stage Milli-pore Milli-Q plus 185 purification system (Bedford, MA, USA) was used in all experiments.1H-NMR was recorded on a Bruker Avance 400 MHz instrument (Bruker, Germany) using appropriate solvents and the chemical shifts are denoted in ppm. Multiplicities of NMR signals are designated as s (singlet), d (doublet), t (triplet), and m (multiplet). Size measurements of the projected nanoparticles were carried out on Zetasizer Nano-ZS (Malvern Instruments, UK).
Parasite
Leishmania donovani promastigotes (WHO designation MHOM/IN/80/Dd8), originally obtained as a gift from the (late) Prof. P. C. C. Garnham and routinely maintained at the institute in golden hamsters, were used in the present study. Promastigotes were grown in medium 199 (Sigma) supplemented with 10% heat-inactivated fetal calf serum (GBL) and 1% penicillin (50 U/ml) and streptomycin (50 mg/mL) solution (Sigma) at 24 °C (Debrabant et al.,1995).
Synthesis of nanoparticles
Synthesis of 6-O-(3-hexadecyloxy-2-hydroxypropyl)-guar gum (HDGG) (1)
Guar gum (500 mg, 1.56 mmol) was dissolved in 50 mL of distilled water and an aqueous sodium hydroxide solution (1 M, 20 mL) was added to it and the solution was stirred at 25 °C for 15 min. Then 3-(hexadecyloxy)-1-chloropropan-2-ol [23] [183 mg, 0.54 mmol, for 35–37% substitution (attempted)], dissolved in tetrahydrofuran (THF) (5.0 mL), was added into the reaction mixture. The resulting solution was heated at 50 °C for 24 h. The reaction mixture was concentrated on a rotary evaporator and the aqueous phase was washed with diethyl ether (300 mL). The aqueous phase was then concentrated on a rotary evaporator and subjected to dialysis (MWCO 12kD) against double distilled water for 6 h. The dialyzed solution was then lyophilized to obtain 6-O-(3-hexadecyloxy-2-hydroxypropyl)-guar gum (HDGG), as a light yellow solid. The product obtained in ~ 67% yield was then characterized by 1H NMR spectroscopy.
1H NMR (D2O) δ: 4.3-3.6 (guar gum protons), 2.3-1.4 (m, 33H, cetyl protons).
Preparation of amphotericin B (AmB) entrapped HDGG nanoparticles (HDGG-AmB-NPs) (2)
HDGG (1) (50 mg) was dissolved in double distilled water (20 mL) and to this, AmB (5 mg) dissolved in dimethylsulphoxide (DMSO) (4 mL), was added over a period of 20 min. The yellow colored solution was stirred gently at 300×g for 16 h in dark at 25 °C. Subsequently, the resulting solution was concentrated and dialyzed (MWCO 12kD) against double distilled water for 24 h at 25 °C with the water being changed at least 4 times to remove unwanted materials. It was found that the time (24 h) was sufficient to release unbound AmB and DMSO in a controlled experiment. The dialyzed solution was lyophilized (without any cryoprotectant) in a speed vac for 24 h to obtain a yellow solid, HDGG-AmB-NPs in ~ 92% yield.
Similarly, blank HDGG (without drug) nanoparticles (HDGG-NPs) (4) (~ 77% yield) were also prepared.
For the preparation of AmB and Pip entrapped HDGG nanoparticles (HDGG-AmB-Pip-NPs) (3), HDGG (1) (50 mg) was dissolved in double distilled water (50 mL) and to this, AmB (5 mg) dissolved in dimethylsulphoxide (DMSO) (4 mL), was added over a period of 20 min. After that, Pip (5 mg) dissolved in DMSO (5 mL) was added over a period of 20 min to the reaction mixture. The ratio of two drugs was kept in 1:1 ratio by following reported literature [24] with the intension of maximum drug loading. The light yellow colored solution was stirred gently at 300 x g for 16 h in dark at 25 °C. Afterward, the resulting solution was concentrated and dialyzed (MWCO 12kD) against double distilled water for 24 h at 25 °C with the water being changed at least 4–6 times to remove unwanted materials. The dialyzed solution was lyophilized in a speed vac for 24 h to obtain a light yellow solid, HDGG-AmB-Pip-NPs (3) in ~ 54% yield.
All the NP formulations were characterized by DLS.
Preparation of eudragit-coated HDGG-AmB-NP (Eu-HDGG-AmB NPs) (5)
HDGG (500 mg) was dissolved in double distilled water (300 mL) and to this, a solution of AmB (50 mg), dissolved in DMSO (1 mL), was added over a period of 20 min. After that, eudragit L30D (30% aqueous solution) (100 μL) was added drop wise to the solution. The light yellow colored solution was stirred gently at 300×g for 16 h in dark at 25 °C. Subsequently, the resulting solution was concentrated and dialyzed against double distilled water for 24 h at 25 °C with the water being changed at least 4 times to remove unwanted materials. The dialyzed solution was lyophilized in a speed vac for 30 h to obtain light yellow solid, Eu-HDGG-AmB-NPs (480 mg) in ~ 87% yield.
Similarly, eudragit-coated HDGG-NPs (Eu-HDGG-NPs) (7) were also prepared with ~ 82% yield.
To prepare eudragit-coated HDGG-AmB-Pip-NPs (Eu-HDGG-AmB-Pip-NPs) (6), HDGG (500 mg) was dissolved in double distilled water (300 mL) and to this, a solution of AmB (50 mg), dissolved in DMSO (10 mL), was added over a period of 20 min. Subsequently, Pip (50 mg) dissolved in DMSO (10 mL) was added over a period of 20 min to the reaction mixture. After that, eudragit L30D (30% aqueous solution) (100 μL) was added drop wise to the solution. The light yellow colored solution was stirred gently at 300×g for 16 h in dark at 25 °C. Subsequently, the resulting solution was concentrated and dialyzed against double distilled water for 24 h at 25 °C with the water being changed at least 4–6 times to remove unwanted materials. The dialyzed solution was lyophilized in a speed vac for 30 h to obtain light yellow solid, Eu-HDGG-AmB-NPs in ~ 80% yield.
Characterization of the NPs
Percent yield
After attaining the constant weight, yield (%) of NPs was calculated by using the formula:
Particle size measurement
The mean particle size and polydispersity index (PDI) of all NPs were determined by dynamic light scattering (DLS) technique employing a nominal 5 mW He-Ne laser operating at 633 nm wavelengths. All freeze-dried NPs were dispersed in aqueous buffer and the size was measured. The measurements were carried out at 25 °C with the following settings: 10 measurements per sample; refractive index of HDGG 1.23, viscosity of water, 0.89 cP. The particle size was measured in triplicate.
Drug loading and entrapment efficiency (%EE) determination
The drug loading and encapsulation efficiency were determined by analyzing the NPs spectrophotometrically using Lambda Bio 20 UV/VIS Spectrophotometer (Perkin Elmer, USA). The amount of drug present in the nanoparticles was estimated as follows: a known amount of NPs (5 mg, dry powder) was dispersed in 1 mL dimethyl sulphoxide (DMSO) by stirring the sample vigorously and the absorbance of the solution was measured at 406 nm and 341 nm for AmB and Pip, respectively. The amount of drug present was calculated from a previously drawn calibration curve of concentration vs. absorbance with different known concentrations of drug and piperin. The entrapment efficiency (%EE) was calculated using the formulas as given below. All the measurements were performed in triplicate.
Evaluation of drug release from the NPs
To determine the release profile of drug loaded HDGG-NPs, a known quantity of the particles (~ 5 mg) was dispersed in 1 mL of 50% ethanol in PBS solution (pH 7.4), and kept in the dialysis tube, which was suspended in 20 mL of 50% ethanol in PBS (pH 7.4) solution in a glass vial and the solution was stirred at 240×g at 37 °C. At pre-determined intervals of time, samples were collected (ca. 200 μL) from the glass vial followed by spectroscopic analysis at 406 nm and 341 nm for AmB and Pip, respectively, using UV/VIS Spectrophotometer. The same amount of fresh buffer was added to the glass vial and the release study was continued. The quantity of the released drug was then calculated using a previously drawn standard curve of the pure drug in 50% ethanol in PBS. The release study of eudragit-coated nanoparticles was carried out at acidic pH (1.5) [0.1 N HCl buffer solution of pH 1.5] to obtain the release pattern in acidic medium.
In vitro antileishmanial activity assay
Antipromastigote assay
The L. donovani promastigotes (MHOM/IN/Dd8/80), originally obtained from Imperial college, London, and transfected with fire fly luciferase gene, as described earlier [25], were maintained in medium 199 (Sigma) supplemented with 10% fetal calf serum (Gibco) and gentamycin (40 μg/mL) solution (Sigma). Exponentially growing transgenic promastigotes (1 × 106 promastigotes/100 μL/well) were seeded in 96-well flat bottom tissue culture plates (Cellstar) and allowed to grow for 72 h at 24 °C, in the presence of 1–100 μg/mL of free AmB and AmB loaded NPs (stock prepared in 100% DMSO/water, initial concentration, 1 mg/mL followed by dilution in media) in duplicate for each concentration. After 72 h of incubation, 50 μL of the parasite suspension was transferred in a black 96-well plate (Nunc) followed by addition of 50 μL Steady Glo reagent (Promega), incubated for 1 min with mild shaking, and read on luminometer (Berthhold). The inhibition of parasite growth was determined by comparison of the luciferase activity of drug-treated parasites with that of untreated control parasites by the following formula:
where N is the average relative luminescence unit (RLU) of control wells and n is the average RLU of treated wells. The experiment was repeated three times.
Anti-amastigote assay
Macrophage cells (J744) were harvested from exponentially growing culture. The cells were diluted to 1 × 106/mL in RPMI medium plus 10% FCS (Sigma, USA) and layered in 16 well chamber slides (Nunc) under a final volume of 100 μL/well and allowed to adhere for 24 h at 37 °C in a 5% CO2–95% air mixture. Adherent macrophages were infected with stationary phase L. donovani promastigotes at ratio 1:10 for 24 h at 37 °C in a 5% CO2–95% air mixture. After incubation, non-phagocytozed parasites were removed by washing and infected cultures were incubated further at 37 °C in a 5% CO2–95% air mixture in the presence of free AmB and its NPs for 72 h. Free AmB solution prepared in DMSO and AmB-loaded NPs solution prepared in water were then diluted in complete RPMI medium and added at twofold dilutions up to 7 points in complete medium starting from 200 ng/mL concentrations. Finally, slides were fixed with 100% methanol and stained with 20% Giemsa stain for 45 min on day 6 [26]. The number of amastigotes per 500 cell nuclei was counted in each well and the parasitic burden was expressed in terms of the number of amastigote per 100 cell nuclei. Drug activity (percent inhibition) was determined by comparing amastigotes count in treated and untreated macrophages by the general formula:
where N is the average number of amastigotes per 100 cell nuclei of untreated well and n is the average number of amastigotes per 100 cell nuclei of treated well.
Evaluation of in vivo antileishmanial efficacy in L. donovani-hamster model
The modified method of Bhatnagar et al. [27] was used for in vivo screening. Golden hamsters (Inbred strain) of either sex weighing 40–45 g were infected intra-cardialy with 1 × 107 amastigotes per animal. Pre-treatment spleen biopsy in all the animals was carried out to assess the degree of infection on day 20 of infection. The animals with + 1 infection (5–15 amastigotes/100 spleen cell nuclei) were included in the chemotherapeutic trials. The infected animals were randomized into several groups on the basis of their parasitic burdens. Six animals were used for each test sample and untreated controls. Drug treatment by intraperitoneal (i.p.) route or oral route was initiated after 2 days of biopsy and continued for 5 consecutive days. Post-treatment biopsies were done on day 7th and 28th day of the last drug administration and amastigote counts were assessed by Giemsa staining. Intensity of infection in both, treated and untreated animals, as also the initial count in treated animals was compared and the efficacy was expressed in terms of parasite burden (number of amastigotes per 500 macrophage cell nuclei and percentage inhibition (PI) using the following formula:
where PI is the percent inhibition of amastigotes multiplication, ANAT is the actual number of amastigotes in treated animals, INAT is the initial number of amastigotes in treated animals, and TIUC is the times increase of parasites in untreated control animals.
For the in vivo evaluation, aqueous solution of the AmB was prepared by suspending the accurately weighed sample in a standard suspension vehicle of 10% Tween-80/Ethanol (70:30) in ddH2O. The final volume contains 10% of the vehicle for inoculation by the intraperitoneal or oral route. All AmB loaded NPs were dispersed in water.
Pharmacokinetics and biodistribution
Pharmacokinetic (PK) and biodistribution study of nanoformulation of AmB in hamsters was done according to Gershkovich et al. [29]. AmB NPs 6 were dispersed in MilliQ water with the aid of continuous stirring for 30 min at a magnetic hot plate and was dosed (2.5 mg/kg of Eu-HDGG-AmB-Pip-NPs) orally by gavage to hamsters (105 to 140 g, male N = 20) for 5 days in a volume of 10 mL/kg. On the 5th day after dosing, four animals were sacrificed at 0.5, 2, 4, 8, and 24 h post-administration and liver, kidney, spleen, brain, and blood samples were harvested. Blood samples were put on ice immediately after collection and then centrifuged to obtain plasma sample (12,000 rpm, 5 min) within 15 min of sampling and stored at − 20 °C until analysis. The tissue samples were blotted with absorbent paper to remove surface blood and stored at − 20 °C until analysis. Concentration of AmB in both plasma and tissue homogenate samples was then determined by LC-MS/MS. Pharmacokinetics of free AmB was also conducted in hamsters at 2.5 mg/kg dose for comparison with NPs.
UPLC-MS/MS analysis of plasma and tissue samples
Calibration standards of AmB were prepared by spiking the appropriate aliquots of working standard solutions into pooled blank hamster plasma and tissue samples. For analysis, 100 μL of plasma or tissue samples were precipitated by 300 μL of methanol containing internal standard (IS, raloxifene), followed by vortex for 5 min and then centrifuged at 12,000 rpm for 10 min [28]. An aliquot of 200 μL of supernatants was separated and 7 μL was injected onto analytical column. Analysis was carried out using UPLC-MS/MS consisting of Waters UPLC and API 4000 mass spectrometer (Applied Biosystems, MDS Sciex Toronto, Canada). Chromatographic separations of analytes were achieved using a gradient UPLC method at a flow rate of 0.6 mL/min using C18 (100 × 2.1 mm, 1.8 μm) column. The mobile phase was consisted of acetonitrile and 0.1% formic acid in Milli Q water. The detection of ions was performed in a multiple reaction monitoring (MRM) mode, monitoring the transition of m/z 924.7 precursor ion [M + H] + to the m/z 743.5 product ion for AmB and m/z 474.2 precursor ion [M + H] + to the m/z 112.2 product ion for IS. The lower limit of quantification for AmB in plasma and tissue samples was 3.9 and 7.8 ng/mL, respectively. The standard curves were linear up to the concentration of 1000 ng/mL. The accuracy and precision was found to be between 85 and 115%.
Serum creatinine assay for nephrotoxicity assessment
Picric acid-based method was used for estimating serum creatinine [30]. Picric acid in an alkaline medium reacts with creatinine to form an orange colored complex. Intensity of the color formed is directly proportional to the amount of creatinine present in the sample.
Serum samples were processed as below:
Serum samples (10 μL) were added to wells of a 96-well plate. Then 120 μL of double distilled water was added to each sample and the samples were mixed by pipetting. Thereafter, 20 μL of freshly prepared alkaline picrate solution was added to it and the samples were mixed properly. The plate was incubated at room temperature (25 °C) for 45 min with slow shaking. Thereafter, the absorbance was taken at 505 nm using a plate reader and the concentration of creatinine was calculated using a previously plotted standard curve.
Statistical analysis
All data were summarized as mean ± standard deviation (SD) or standard error (SE) of at least three or four independent experiments. When comparing more than two mean values of groups, one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test using Prism software (GraphPad Software Inc., Version 3.0, CA, USA) was performed. P value less than 0.05 was considered as statistically significant.
Results and discussion
Low bioavailability and nephrotoxicity are the major concerns associated with AmB-based therapy for VL treatment. To address these inadequacies, we combined Pip, a natural bioenhancer, with AmB by entrapping both of them in guar gum based NPs. We hypothesized that the guar gum-based formulation would actively target the macrophages of the reticuloendothelial system (RES) and release Pip and AmB in a controlled manner which would increase the bioavailability, alleviate the toxicity, and enhance the therapeutic efficacy of AmB due to site specific delivery. Moreover, coating of eudragit L30D would protect the nanoformulation from extreme acidic environment of stomach, enabling these nanoparticles to be delivered via oral route. The lead formulation named Eu-HDGG-AmB-Pip-NPs (Nanoformulation 6) exhibited controlled release of both the loaded agents, protected the drug in acidic conditions, increased the bioavailability of AmB, demonstrated non-nephrotoxic nature, and exhibited potential antileishmanial effect in vitro and in vivo.
Synthesis and characterization of AmB loaded targeted nanoparticles
Guar gum was selected as a carrier for drug delivery because of its biodegradable and biocompatible nature and its ability to target macrophages through mannose-like receptors [16]. We synthesized amphiphilic hexadecylated guar gum by reacting 3-(hexadecyloxy)-1-chloropropan-2-ol with guar gum, where 3-(hexadecyloxy)-1-chloropropan-2-ol is converted into its epoxide in situ by reacting with hydroxyl functions of guar gum in the presence of an alkali, and obtained O-(3-(hexadecyloxy-2-hydroxypropyl) substituted guar gum (HDGG) (Scheme 1). The percent substitution of hexadecyl groups on guar gum was found to be 18% (35% attempted) by 1H NMR spectroscopy [measurement of the relative peak area of -CαH(Galactose)- and –C16H33-HD]. AmB loaded NPs [HDGG-AmB-NPs (2)] were prepared by self-assembly of the substituted guar gum in double distilled water at 25 ± 2 °C with drug:polymer ratio of 1:10. HDGG-AmB-NPs (2) were obtained in ~ 92% yield. Entrapment efficiency of AmB in substituted guar gum was found to be 48%. We speculated that the drug, AmB interacted with the C-16 chains by physical interactions in self-assembled substituted guar gum NPs.

Schematic presentation of the preparation of 6-O-(3-hexadecyloxy-2-hydroxypropyl)-guar gum (HDGG) (1)
AmB and Pip dual-drug loaded nanoparticles [HDGG-AmB-Pip-NPs] (3) and blank nanoparticles without drug [HDGG-NPs] (4) were also prepared following similar procedures to compare their therapeutic efficacies with HDGG-AmB-NPs (2). The yields were found to be ~ 54% and ~ 77% for 3 and 4, respectively. Interestingly, the incorporation of AmB along with Pip reduces the Pip payload but increases the AmB payload. It is known that AmB is more hydrophobic than Pip. Since, the core of HD grafted GG-NP is hydrophobic, the chance of accumulation of AmB in the core of NP would be more than Pip. Similar observation also obtained by Katiyar et al. [31] where they have reported about the co-delivery of rapamycin and piperine as a NPs. They have also reported that entrapment efficiency of rapamycin was increased after incorporation of Pip inside the NP. However, only rapamycin entrapment efficiency was lower than both drug entrapped NPs.
Coating of eudragit LD30 on AmB-loaded nanoparticles
Enteric polymer coated nanoparticles were prepared for oral delivery to provide protection against the gastric environment of GI track [25]. Enteric polymer coated nanoparticles [Eu-HDGG-AmB-NPs (5)] were prepared by addition of eudragit LD30 during preparation of AmB loaded HDGG nanoparticles and the yield of 5 was found to be ~ 87%. Similarly, eudragit-coated AmB and Pip loaded HDGG nanoparticles [Eu-HDGG-AmB-Pip-NPs (6)] and eudragit-coated blank HDGG nanoparticles [Eu-HDGG-NPs (7)] were prepared. The yields of 6 and 7 were found to be ~ 80% and ~ 82%, respectively. Encapsulation efficiencies of all the nanoparticles 2, 3, 4, 5, 6, and 7 are listed in Table 1.
Size measurements
Size of the synthesized NPs is one of the deciding factors for its efficient delivery to the target site. The sizes of all the synthesized NPs were determined by dynamic light scattering (DLS) and the results revealed that the average diameters of all the NPs (2, 3, 4, 5, 6, and 7) were < 200 nm (Fig. 1) as listed in Table 1. The low PDI values of all the NPs indicated the monodisperse nature of the synthesized formulations. We deliberately kept the size of the NPs < 200 nm assuming that smaller sized particles will have better cellular uptake and thereby, would have better therapeutic efficacy.

Size distributions of prepared NPs by dynamic light scattering (DLS) (a) AmB loaded HDGG-NPs (2), (b) AmB and Pip loaded HDGG-NPs (3), (c) HDGG-NPs (4), (d) Eu-HDGG-AmB-NPs (5), (e) Eu-HDGG-AmB-Pip-NPs (6), and (f) Eu-HDGG-NPs (7)
In vitro release profile of the NPs
Drug release behavior is one the important parameters to be studied for the development of an efficient drug delivery system. We studied the in vitro drug release profiles of the drug loaded NPs at pH 7.4, the physiological pH. Moreover, the enteric coated NPs (5 and 6) were also characterized for their drug release behavior at acidic pH (1.5), to mimic the pH of GI track which ranges from 1.5 to 6.5, as they were prepared to be administrated via oral route [32]. We studied the release kinetics of AmB from HDGG-NPs (2 and 3) for 10 days at 37 °C (Fig. 2a and c). In both the cases, AmB was released in a slow and controlled manner indicating the availability of the drug over a period of time [2, 24]. We observed that drug release is faster from non-coated NPs than the eudragit-coated NPs at a particular time. After 48 h, the drug release was ~ 25% and ~ 16% at pH 1.5, from the non-coated (2) and eudragit-coated AmB (5) NPs, respectively, which indicated that eudragit coating inhibited the release of drug at acidic pH, thereby, demonstrating to possess the ability to protect the drug from degradation in acidic medium. Likewise, the release of both AmB and Pip from 3 and 6 were ~ 17% and 3%, respectively, after 48 h at pH 1.5 (Fig. 2b and d), which also indicated that eutragit has potentially protected the NPs from acidic pH of GI track, rendering them to reach safely to the site of delivery.

In vitro release profile of AmB loaded nanoparticles (a) AmB-loaded HDGG-NPs (2) at pH 7.4. b Eudragit-coated AmB loaded HDGG-NPs (5) at pH 1.5. c AmB and Pip loaded HDGG-NPs (3) at pH 7.4. d Eudragit-coated AmB and Pip loaded HDGG-NPs (6) at pH 1.5. Each experiment was performed thrice (n = 3). e Calibration curve of AmB as a function of concentration. f Calibration curve of Pip as a function of concentration
In vitro antileishmanial efficacy of AmB NPs on L. donovani
In vitro antileishmanial effect of the different nanoformulations was studied on promastigote and amastigotes of L. donovani. The IC50 values of different NPs are listed in Table 2. All the formulations exhibited antileishmanial efficacy against both promastigote and amastigote stages of parasite. HDGG-AmB-Pip-NPs (3) exhibited maximum efficacy in vitro as indicated by minimum IC50 values in both promastigotes and amastigotes. The eightfold improvement in antileishmanial efficacy of nanoformulation 3 as compared to 2 is expected to be a consequence of improved bioavailability of AmB in the presence of the bioenhancer, Pip (Table 2). However, the IC50 values of Eu-HDGG-AmB-Pip-NPs (6) were found almost equivalent to bulk AmB, especially in case of amastigotes, might be due to slow release of the therapeutic agents from the eudragit-coated NPs (Fig. 2d) affecting its in vitro therapeutic effect.
In vivo efficacy of the NPs in hamster-L. donovani model
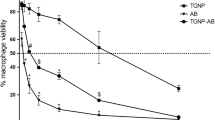
To translate the in vitro results, we examined the in vivo antileishmanial effect of the synthesized nanoformulations on goleden hamster-L.donovani model. Dose optimization of drug loaded NPs was carried out as mentioned in Table 3. Bulk AmB exhibited up to 93% inhibition of the parasite load at 5 mg/Kg by intraperitoneal route, whereas nanoformulation 3 (HDGG-AmB-Pip-NPs) exhibited up to 95% parasite inhibition at same dose and same route of administration (Table 2, Fig. 3a). Interestingly, eudragit-coated NPs (Eu-HDGG-AmB-Pip-NPs) showed very high efficacy via oral route with up to 96% parasite inhibition at a dose of 10 mg/kg (Table 3, Fig. 3b). Parasite burden was significantly reduced in naoformultaion (5 and 6) as compared to untreated controls (***P < 0.001) and bulk AmB treated animals by oral route (###P < 0.001). Very low therapeutic activity of bulk AmB is due its low bioavailability [7]. The inhibition of parasite load by the drug loaded NPs was consistent for 28 days post treatment. Increased parasitic inhibition by Eu-HDGG-AmB-Pip-NPs (6) by oral administration clearly indicates the potentiation of antileishmanial efficacy of nanoformulation having AmB and Pip.

Leishmania Parasite burden on 28th day post treatment AmB loaded naoparticles. a Treatment with nanopformulation 2, 3,and 4 by intraperitoneal route. b Treatment with nanoformulation 5, 6, and 7 by oral route. Values are mean parasite burden (number of amastigotes/500 macrophage cell nuclei) ± SD of six animals. *Statistical significance as compared to untreated control with respect to their P value (***P value < 0.001, **P value < 0.01, *P value < 0.05, ns no significance). #Statistical significance as compared to treated group with bulk AmB with respect to their P value (###P value < 0.001, ##P value < 0.01, #P value < 0.05, ns no significance)
Pharmacokinetic studies of AmB formulations
As we aimed to increase the bioavailability of AmB by combing it with Pip in a nanotized formulation, we studied the pharmacokinetics of AmB to examine its bioavaiability. Hamster plasma concentration-time profiles of AmB following a single oral administration of bulk AmB and Eu-HDGG-AmB-Pip-NPs (6) were studied (Fig. 4). The pharmacokinetics parameters are summarized in Table 4. We observed that the oral administration of bulk AmB at a 2.5 mg/kg dose in hamsters showed very low levels of AmB in plasma (Fig. 4a). The area under the curve (AUC0-t) was 78.29 ± 48.16 ng.h/mL. The maximum concentration of AmB in plasma was 7.5 ng/mL at 4 h. However, hamsters that received nanoformulation Eu-HDGG-AmB-Pip-NPs (6) showed a much higher concentration of AmB, i.e., 252 ng/mL at 4 h and significant levels of AmB remain upto 24 h in the range between 188 and 136 ng/mL (Fig. 4b). Plasma pharmacokinetics clearly demonstrated the significant improvement in the exposure of AmB upon oral administration of its nanoformulation as compared to bulk AmB administration (Table 4). Improved plasma concentration of AmB in Eu-HDGG-AmB-Pip-NPs (6) indicated the improved bioavailability of AmB. We speculated that bioavailability of AmB in Eu-HDGG-AmB-Pip-NPs was increased due to slow release of AmB from the nanotized formulation as well as owing to the presence of the bioenhancer Pip. It is well known that piperin acts as bioenhancer and the following are the probable mechanism of action of piperin as a bioenhancer in case of gastrointestinal absorption.
-
i.
Solubility enhancer: Bile acid usually forms micelle with lipids and lipid soluble drugs for required absorption. Pip boosts the bile acids secretion and inhibits bile acid metabolism. Therefore, micelle formation increases and thus enhances the solubility and absorption of hydrophobic drugs [33].
-
ii.
Increased blood supply: Annamalai et al. [34] proposed that Pip increases gastrointestinal blood flow and thereby absorption of drugs increases from the digestive tract.
-
iii.
Pip increases fluidity of brush border membrane and increases microvilli length [35]. Therefore, Pip enhances the drug uptake inside the cells and increases bioavailability of particular drug.
-
iv.
Inhibition of solubilizer attachment: Attachment of glucuronic acid with water soluble compound helps to excrete the compound either into the urine or by small intestine. It is reported that Pip inhibits glucuronic acid attachment with water soluble substances, thus facilitates enhanced uptake into the cell [36]. We presume that Pip, co-delivered with AmB, increases the bioavailability of AmB by a forementioned probable mechanisms.

Pharmacokinetic studies of a bulk AmB and b Eu-HDGG-AmB-Pip-NPs (6) at 2.5 mg/kg administered orally. Values are mean ± SE of 4 animals
For successful antileishmanial therapy, it was important to determine the AmB tissue distribution and penetration, especially in the liver and spleen, where parasite resides within the macrophages. Besides liver and spleen, tissue penetration was also investigated in kidney. We did not perform the tissue distribution study for bulk AmB due to very poor levels in plasma. However, the tissue distribution study of Eu-HDGG-AmB-Pip-NPs (6) was performed which showed highest AmB exposure (area under the curve, AUC0-t) in liver (18,416 h.ng/g) followed by the spleen (10,409.0 h.ng/g) and the kidney (5979.5 h.ng/g) (Table 4). The nanoformulation exhibited peak tissue concentrations at 8 h after dosing, which were 976.2 ng/g in liver, 652.9 ng/g in spleen, and 316.5 ng/g in kidney. Since AmB is a nephrotoxic drug, it was crucial to determine the plasma to kidney ratio and its comparison with the target organs (liver and spleen) of pathology. The liver presented the highest AmB tissue to plasma ratio (4.3), followed by the spleen (2.5). The kidney presented the lowest value (1.4). Besides, some AmB was also detected in brain at a few time points in few animals, but the concentration was very low. These data indicated that the NPs delivered relatively more drug to the target organs (liver and spleen) as compared to kidney. NPs formulated by guar gum target mannose-like receptors on macrophages [15], hence can easily reach immune cells where parasites reside. Further, piperin acts as a bioenhancer [33] which increases the absorption of drug from GI track. Detection of relatively more concentration of AmB in macrophage rich organs like liver and spleen clearly indicated the site specific delivery of AmB by Eu-HDGG-AmB-Pip-NPs, which were targeted to macrophages’ mannose receptors.
Serum creatinine assay for nephrotoxicity assessment
Creatinine is the breakdown product of creatinine phosphate which is used by skeletal muscles. There is a constant rate of production and urinary excretion of creatinine. Increased levels may indicate renal dysfunction. Abnormal creatinine level in the serum is widely studied as a marker for kidney damage [37]. Among healthy animals serum creatinine level is maintained at a certain concentration due to constant rate of production and urinary excretion of creatinine. Though considerable level of AmB was detected in kidney, no significant changes were observed in the serum creatinine level (Fig. 5). All the values were in range for creatinine levels found in healthy animals suggesting the safety of the nanoformulation for kidney. The serum creatinine level in the normal range indicated the non-nephrotoxic nature of the synthesized nanoformulation.

Serum creatinine concentration after oral administration of Eu-HDGG-AmB-Pip-NPs (6) and equivalent bulk AmB for 5 days once daily. Control is vehicle control. Normal values for serum creatinine in golden hamster are between 0.4 to 1 mg/dL. No significant difference in serum creatinine levels were observed suggesting absence of any toxicity. Standard error represented as error bars. n = 4 animals per group
Overall, the formulated nanoparticles exhibited improved AmB bioavailability, non-nephrotoxic nature, delivered the drug to the target organs and demonstrated highly efficient antileishmanial activity.
Conclusions
VL demands alternative treatment strategies for its efficient eradication. In the present study, we prepared eudragit-coated guar gum NPs loaded with AmB and Pip to deliver these therapeutic agents to the macrophages, where the parasite grows and proliferates. The formulated NPs protected the drugs in acidic pH and demonstrated controlled drug release. These NPs exhibited potential suppression of the L. donovani parasite in vitro as well as in vivo, improved AmB bioavailability, delivered the drug to the target organs, and exhibited non-nephrotoxic nature. Conclusively, the developed NPs possess great potential to be utilized as favorable and cost effective therapy against VL and call on to further investigations.
Change history
06 March 2020
In the original version of this article, Vikas Srivastava’s last name was spelled incorrectly.
References
Kumar R, Hafner LM, Engwerda CR. Immune regulation during chronic visceral leishmaniasis. PLoS Negl Trop Dis. 2014;8(7):e2914.
Kole L, Das L, Das PK. Synergistic effect of interferon-g and mannosylated liposome incorporated doxorubicin in the therapy of experimental visceral leishmaniasis. J Infect Dis. 1999;180:811–20.
Elizabeth M, Contreras M. Chemotherapy used in the treatment of visceral leishmaniasis. CPQ Microbiol. 2019;3(1):01–14.
Soto J, Soto P. Miltefosine: oral treatment of leishmaniasis. Expert Rev Anti-Infect Ther. 2006;4:177–85.
Sundar S, Chakravarty J. Single-dose liposomal amphotericin B for visceral leishmaniasis in India. Expert Opin Investig Drugs. 2015;24:43–59.
Paila YD, Bhaskar Saha B, Chattopadhyay A. Amphotericin B inhibits entry of Leishmania donovani into primary macrophages. Biochem Biophys Res Commun. 2010;399:429–33.
Italia JL, Yahya MM, Singh D, Ravi Kumar MN. Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone. Pharm Res. 2009;26(6):1324–31.
Laniado-Laborín R, Cabrales-Vargas MN. Amphotericin B: side effects and toxicity. Rev Iberoam Micol. 2009;26(4):223–7.
Zaioncz S, Khalil NM, Mainardes RM. Exploring the role of nanoparticles in amphotericin B delivery. Curr Pharm Des. 2017;23(3):509–21.
Malani PN, Depestel DD, Riddell J, Bickley S, Klein LR, Kauffman CA. Experience with community-based amphotericin B infusion therapy. Pharmacotherapy. 2005;25:690–7.
Polonio T, Efferth T. Leishmaniasis: drug resistance and natural products. Int J Mol Med. 2008;22:277–86.
Mishra BB, Kale RR, Singh RK, Tiwari VK. Alkaloids: future prospective to combat leishmaniasis. Fitoterapia. 2009;80:81–90.
Wadhwa S, Singhal S, Rawat S. Bioavailability enhancement by piperine: a review. Asian J Biomed Pharm Sci. 2014;04(36):1–8.
Randhawa KG, Kullar SJ, Rajkumar. Bioenhancer from mother nature and their applicability to modern medicines. International journal of applied and basic medicinal research. Int J App Basic Med Res. 2001;1(1):5–10.
Moretton MA, Chiappetta DA, Andrade F, Das Neves J, Ferreira D, Sarmento B, et al. Hydrolyzed galactomannan-modified nanoparticles and flower-like polymeric micelles for the active targeting of rifampicin to macrophages. J Biomed Nanotechnol. 2013;9(6):1076–87.
Bansal R, Singh A, Gandhi R, Pant AB, Kumar P, Gupta KC. Galactomannan-PEI based non-viral vectors for targeted delivery of plasmid to macrophages and hepatocytes. Eur J Pharm Biopharm. 2014;87(3):461–71.
Coviello T, Alhaique F, Dorigo A, Matricardi P, Grassi M. Two galactomannans and scleroglucan as matrices for drug delivery: preparation and release studies. Eur J Pharm Biopharm. 2007;66(2):200–9.
Nurnadiah R, Kamarun D, Li AR, Ahmad MR. Synthesis and characterization of crosslinked galactomannan nanoparticles for drug delivery application. Adv Mater Res. 2013;812:12–9.
Duncan CJ, Pugh N, Pasco DS, Ross SA. Isolation of a galactomannan that enhances macrophage activation from the edible fungus Morchella esculenta. J Agric Food Chem. 2002;50(20):5683–5.
Y-m H, Chun S-H, S-t H, Y-c K, H-d C, Lee K-w. Immune enhancing effect of a Maillard-type lysozyme-galactomannan conjugate via signaling pathways. Int J Biol Macromol. 2013;60:399–404.
Noleto GR, Mercê ALR, Iacomini M, Gorin PA, Soccol VT, Oliveira MBM. Effects of a lichen galactomannan and its vanadyl (IV) complex on peritoneal macrophages and leishmanicidal activity. Mol Cell Biochem. 2002;233(1–2):73–83.
Hosny KM. Alendronate sodium as enteric coated solid lipid nanoparticles; preparation, optimization, and in vivo evaluation to enhance its oral bioavailability. PlosOne. 2016;11(5):e0154926.
Ray L, Kumar P, Gupta KC. The activity against Ehrlich’s ascites tumors of doxorubicin contained in self assembled, cell receptor targeted nanoparticles with simultaneous oral delivery of the green tea polyphenol epigallocatechin-3-gallate. Biomaterials. 2013;34:3064–76.
Ray L, Pal MK, Ray RS. Synergism of co-delivered nanosized antioxidants displayed enhanced anticancer efficacy in human colon cancer cell lines. Bioact Mater. 2017;2:1–14.
Ashutosh, Gupta S, Ramesh, Sundar S, Goyal N. Use of Leishmania donovani field isolates expressing the luciferase reporter gene in in vitro drug screening. Antimicrob Agents Chemother. 2005;49:3776–83.
Seifert K, Croft SL. In vitro and in vivo interactions between sodium stibogluconate, miltefosine and other antileishmanial drug. Antimicrob Agents Chemother. 2006;50:73–9.
Bhatnagar S, Guru PY, Katiyar JC, Srivastava R, Mukherjee A, Akhtar MS. Exploration of antileishmanial activity in heterocycles; results of their in vivo & in vitro bioevaluations. Indian J Med Res. 1989;89:439–43.
Gershkovich P, Sivak O, Wasan EK, Magil AB, Owen D, Clement JG, et al. Biodistribution and tissue toxicity of amphotericin B in mice following multiple dose administration of a novel oral lipid-based formulation (iCo-009) J. Antimicrob Chemother. 2010;65:2610–3.
Deshpande NM, Gangrade MG, Kekarea MB, Vaidya VV. Determination of free and liposomal amphotericin B in human plasma by liquid chromatography–mass spectroscopy with solid phase extraction and protein precipitation techniques. J Chromatogr B. 2010;878:315–26.
Peters JH. The determination of creatinine and creatine in blood and urine with the photoelectric colorimeter. J. biol. Chem, 1942;146: 179-186.
Katiyar SS, Muntimadugu E, Rafeeqi AT, Domb AJ, Khan W. Co-delivery of rapamycin- and piperine-loaded polymeric nanoparticles for breast cancer treatment. Drug Deliv. 2016;23:2608–16.
Fallingborg J. Intraluminal pH of the human gastrointestinal tract. Dan Med Bull. 1999;46:183–96.
Lee KW, Everts H, Beynen AC. Essential oils in broiler nutrition. Int J Poult Sci. 2004;3:738–52.
Annamalai AR, Manavlan R. Trikatu- A bioavailability enhancer. Indian Drugs. 1989;27:595–604.
Khajuria A, Thusu N, Zutshi U. Piperine modulates permeability characteristics of intestine by inducing alterations in membrane dynamics: influence on brush border membrane fluidity, ultrastructure and enzyme kinetics. Phytomedicine. 2002;9:224–31.
Reen RK, Singh J. In vitro and in vivo inhibition of pulmonary cytochrome P450 activities by piperine, a major ingredient of piper species. Indian J Exp Biol. 1991;29:568–73.
Sobh MA, Moustafa FE, Ramzy RM, Deelder AM, Ghoneim MA. Schistosoma haematobium-induced glomerular disease: an experiment study in the golden hamster. Nephron. 1991;57:216–24.
Acknowledgments
KCG thanks, Indian Council of Medical Research (ICMR), New Delhi, for awarding Dr. A.S. Paintal Distinguished Scientist Chair at CSIR-IGIB, Delhi. LR thanks CSIR, New Delhi, for her senior research associate (SRA) fellowship CSIR (Pool Section) (IA-27607) at CSIR-CDRI. NG acknowledges Council of Scientific and Industrial Research Network Project, HOPE (BSC0114) for financial support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethical statement
Institutional Animal Ethics Committee (IAEC) of CSIR-Central Drug Research Institute, Lucknow, reviewed and approved the animal protocol [IAEC/2012/87/Renewed 05(5/13) and IAEC/2012/87/Renewed 06(15/14)] which was adhered to National guidelines CPCSEA (Committee For the Purpose of Control and Supervision of Experiments on Animals) of Government of India. Animals were housed in plastic cages at 23 ± 2 °C, humidity 60–63%, and fed standard rodent pellet and water ad libitum and fed with standard rodent food pellet (Lipton India, Bombay).
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original version of the article has been corrected: Vikas Srivastava’s last name was spelled incorrectly.
Rights and permissions
About this article

Cite this article
Ray, L., Karthik, R., Srivastava, V. et al. Efficient antileishmanial activity of amphotericin B and piperine entrapped in enteric coated guar gum nanoparticles. Drug Deliv. and Transl. Res. 11, 118–130 (2021). https://doi.org/10.1007/s13346-020-00712-9
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-020-00712-9