
Abstract
Lipid-based drug delivery systems, a well-tolerated class of formulations, have been evaluated extensively to enhance the bioavailability of poorly soluble drugs. However, it has been difficult to predict the in vivo performance of lipid dosage forms based on conventional in vitro techniques such as cell monolayer permeability studies because of the complexity of the gastrointestinal processing of lipid formulations. In the current study, we explored the feasibility of coupling Caco-2 and Madin-Darby canine kidney monolayer permeability studies with lipolysis, a promising in vitro technique to evaluate lipid systems. A self-emulsifying lipid delivery system was formulated using a blend of oil (castor oil), surfactant (Labrasol® or PL497), and co-surfactant (lecithin). Formulations demonstrating high drug solubility and rapid self-emulsification were selected to study the effect of lipolysis on in vitro cell permeability. Lipolysis of the formulations was carried out using pancreatin as the digestive enzyme. All the digested formulations compromised monolayer integrity as indicated by lowered trans-epithelial electrical resistance (TEER) and enhanced Lucifer yellow (LY) permeability. Further, the changes in TEER value and LY permeability were attributable to the digestion products of the formulation rather than the individual lipid excipients, drug, digestion enzyme, or the digestion buffer. The digested formulations were fractionated into pellet, oily phase, and aqueous phase, and the effect of each of these on cell viability was examined. Interestingly, the aqueous phase, which is considered important for in vivo drug absorption, was responsible for cytotoxicity. Because lipid digestion products lead to disruption of cell monolayer, it may not be appropriate to combine lipolysis with cell monolayer permeability studies. Additional in vivo studies are needed to determine any potential side effects of the lipolysis products on the intestinal permeability barrier, which could determine the suitability of lipid-based systems for oral drug delivery.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The oral route of administration continues to be the preferred route of drug delivery for most chronic diseases. Therapeutic effectiveness of an orally administered drug is critically dependent upon its bioavailability, which in turn is affected by drug solubility. However, poorly water-soluble drug candidates have become increasingly prevalent. It has been projected that approximately 60–70% of newly discovered drug candidates lack adequate aqueous solubility and/or permeability [1, 2]. The poor aqueous solubility of a drug molecule leads to insufficient solubilization in the gastrointestinal (GI) fluids, resulting in low and variable oral bioavailability [3]. In recent years, numerous lipid-based delivery systems have been developed to improve the oral bioavailability of poorly water-soluble drugs. These formulations include simple lipid solutions or dispersion of drugs [4] to more advanced delivery systems such as microemulsions [5, 6], solid lipid nanoparticles [7, 8], and self-emulsifying drug delivery systems (SEDDS) [9,10,11].
Of particular interest in our studies are the SEDDS, which have been used extensively to increase solubility and oral absorption of hydrophobic compounds [12, 13]. SEDDS are composed of one or more surfactants dispersed in an oil phase [10, 13,14,15]. These formulations, when added to an aqueous phase under gentle agitation (either prior to dosing or in situ in the gut), result in thermodynamically stable nano-emulsions [16]. These nano-emulsions disperse readily in the gut, and their small droplet size contributes to large interfacial surface area and better drug absorption [17]. Several SEDDS have been investigated for the delivery of lipophilic drugs such as halofantrine [18], vitamin E [19], and coenzyme Q10 [20], and are used in commercially available formulations such as Neoral® [21].
The primary mechanism through which lipids and lipophilic excipients increase bioavailability following oral administration is a unique combination of dispersion, digestion, and solubilization of the co-administered drug in formulation-derived lipids in the GI milieu [22]. Stimulation of bile salt secretion in the presence of lipids and lipid-digestion products promotes emulsification and increased drug solubility in the GI fluid [23]. The lipid excipients can further improve the bioavailability of co-administered drugs by prolonging the GI residence time [24, 25], increasing the intestinal permeability [26], facilitating the lymphatic passage [27], and inhibiting the efflux transport [28] or pre-systemic metabolism [23].
An important challenge associated with the development of effective lipid-based formulations is the lack of a satisfactory in vitro model for oral bioavailability prediction [29]. This is primarily due to the complex relationship between formulation dispersion and digestion in the gut, and the subsequent absorption of the solubilized drug and lipid components. Numerous studies have attempted to characterize the physicochemical properties of lipid digestion products in humans following lipid-rich test meals [30,31,32]. Traditionally, lipid-based formulations are often designed to optimize the solubility of the drug in lipid vehicle, enhancing its in vitro dispersion ability [18] and increasing the digestibility of the lipid excipients [33]. However, the digestion and phase behavior of typical lipid excipients and the fate of co-administered drugs as the digestion progresses remain key variables that can affect drug absorption. Interactions between lipid excipients and co-administered drug when the formulation is exposed to and digested in the GI environment will largely depend on the nature of lipids involved as well as the physicochemical characteristics of the individual drug molecule. Therefore, additional mechanistic studies are required to predict the biological performance of lipid-based formulations.
The goal of the current study was to explore the feasibility of coupling cell monolayer permeability studies with lipolysis, an in vitro technique that has been shown to be of value in predicting the in vivo absorption for some drugs. Elacridar, a P-glycoprotein (P-gp) inhibitor [34], was selected as the model drug. The low bioavailability of elacridar, which is due to its low aqueous solubility and poor permeability, limits its clinical use as P-gp inhibitor. A SEDDS formulation of elacridar has been shown to improve its oral bioavailability [35], hence making it a suitable candidate for our studies. In the current studies, SEDDS formulations were prepared by dispersing elacridar in a blend of suitable oils, surfactant, and co-surfactants. In order to test the feasibility and limitations of coupling lipolysis with in vitro permeability models, we evaluated the permeability of the drug from digested lipid formulations across Caco-2 and Madin-Darby canine kidney (MDCK) cell monolayers. These two cell lines are well-established in vitro models to screen the drugs for oral absorption and evaluate the mechanism of drug transport [36,37,38,39].
Materials and methods
Materials
Castor oil, sodium deoxytaurocholate, pancreatin, calcium chloride, magnesium chloride, and phosphatidyl choline were purchased from Sigma (St. Louis, MO, USA). Plurol® Oleique CC497 (PL497) and Labrasol® were received as a gift from Gattefosse (Paramus, NJ, USA). Solec® 70 SB (premium refined soy lecithin) was received from Solae LLC (St. Louis, MO, USA). Elacridar was purchased from Hangzhou Sage Chemical Co., Ltd. (Hangzhou, China).
Preparation of lipid formulations
Lipid formulations comprising castor oil as the oily vehicle and PL497, Labrasol®, or lecithin as surfactant/co-surfactant were prepared based on partial factorial design and at concentrations that were within the inactive ingredient limits (IIG) set by the FDA. Table 1 lists the chemical composition, function, and IIG limits for each of the excipients used. The range of concentrations tested for the different excipients is provided in Table 2. For lecithin-containing formulations, castor oil and lecithin were warmed at 37 °C and mixed first by gentle vortexing. PL497 or Labrasol® was then added and mixed by vortexing. For other formulations, castor oil, PL497, and Labrasol® were added together and mixed by vortexing. For drug-loaded formulations, a calculated amount of premixed blank formulation was added to elacridar and mixed by sonication using a probe sonicator set at 10 W power output for 30 s. Drug-loaded formulations were freshly prepared on the day of the study. Blank formulations were used within 2 weeks of preparation.
Solubility of elacridar in the individual excipients and lipid formulations
Solubility of elacridar in each lipid excipient as well as in different formulations was determined using the shake flask method [40]. Briefly, 4 mg of elacridar was added to 500 μL of the lipid or the formulation being tested and mixed by magnetic stirring at 500 rpm at room temperature. At the end of 24 h, the mixture was centrifuged at 16,000×g for 15 min to remove undissolved elacridar. Then, 50 μL of the supernatant was withdrawn, diluted in methanol, and the concentration of elacridar was quantified by high-performance liquid chromatography (HPLC) or liquid chromatography tandem-mass spectrometry (LC-MS/MS). Due to the high solubility of elacridar in Labrasol®, 10 mg of elacridar was added to 500 μL of Labrasol® and processed as described above.
HPLC method for quantification of elacridar
HPLC analysis of elacridar was performed using a Shimadzu HPLC system equipped with SCL-10A VP controller, SIL-10AD VP autosampler, LC-10AT VP pump, CTO-10AS VP column oven, SPD-10AV VP UV-Vis absorbance detector, and RF-10A XL fluorescence detector. Chromatographic separation was achieved using a Zorbax Eclipse XDB-C18 column (4.6 mm × 50 mm, particle size 1.8 μm; Agilent Technologies, CA, USA). The isocratic mobile phase consisted of a mixture of acetonitrile and 20 mM ammonium formate with 0.1% (v/v) formic acid (42:58, v/v). The injection volume was 10 μL and the elution was carried out at a flow rate of 0.25 mL/min. The ultraviolet detection of elacridar was performed at 260 nm wavelength and fluorescence detection carried out using an excitation wavelength of 413 nm and emission wavelength of 491 nm.
LC-MS/MS method for quantification of elacridar
LC-MS/MS was used to measure the elacridar concentrations that were below the detection limit of the HPLC method (10 ng/mL). A simple liquid-liquid extraction procedure was used for extraction of elacridar from formulation. To an aliquot of 100 μL sample, internal standard (IS) solution (10 μL of 2 μg/mL tyrphostin AG 1478) along with 150 μL of pH 10 buffer was added and mixed thoroughly. After the addition of 1.2 mL of ethyl acetate, the mixture was vortexed for 5 min, followed by centrifugation at 10,000 rpm for 10 min (Centrifuge 5810 R; Eppendorf AG, Hamburg, Germany). The organic layer (1 mL) was transferred to a glass tube and evaporated to dryness at room temperature using a gentle stream of nitrogen (N-EVAP™; Organomation, Berlin, MA, USA). The residue was reconstituted in 100 μL of mobile phase. The LC system consisted of an Aquity UPLC® System (Waters, Milford, MA, USA) coupled with a degasser, quaternary pump, and a temperature-controlled autosampler maintained at 10 °C. Five microliters of the processed sample was injected onto a Zorbax Eclipse XDB-C18 column. The isocratic mobile phase consisted of a mixture of acetonitrile and 20 mM ammonium formate with 0.1% (v/v) formic acid (42:58, v/v) at a flow rate of 0.25 mL/min. Quantification was performed by MS/MS detection in positive ion mode using a Quattro Ultima triple quadrupole mass spectrometer (Waters, Milford, MA, USA) equipped with an electrospray ionization source operating at a capillary voltage of 3000 V. The sheath gas pressure and auxiliary gas pressure was maintained at 50 and 20 arbitrary units, respectively. The capillary temperature was maintained at 300 °C throughout the run. The collision energy for elacridar and IS was 40 and 33 V, respectively. Detection of the ions was performed in multiple-reaction monitoring mode. The mass spectrometer was programmed to monitor the transition pair of elacridar at the m/z 564.6 precursor ion to the m/z 251.99 product ion and m/z 316.01 precursor ion to the m/z 299.97 product ion for the IS. Data acquisition and analysis were achieved by MassLynx software, version 4.1.
Emulsification of SEDDS formulations in water and particle size analysis
Self-emulsification time for the different formulations was assessed by determining the time taken for the dispersion of the formulation in a large volume of water. In a beaker containing 25 mL of deionized water at 37 °C, ~0.5 g of the formulations was added under mechanical shaking at 125 rpm. The time taken for the emulsion droplets to form (the dispersion turning cloudy) was recorded as self-emulsification time.
The particle size of the different formulations following emulsification in water was measured by Delsa™ Nano C Particle Analyzer (Beckman, Brea, CA, USA) operating at a 165° constant scattering angle at 25 °C.
Digestion of lipid formulations
In vitro lipolysis of selected formulations was carried out using a method described by Porter et al. [22]. The lipolysis medium consisted of a digestion buffer (150 mM NaCl, 2 mM Tris, pH 6.5), bile salts (sodium taurodeoxycholate, 20 mM), and phosphatidylcholine (5 mM). For simulation of fasted state condition, 5 mM phosphatidylcholine was used. Test formulations (250 mg) containing 1 mg of elacridar was added into 9 mL digestion medium, and digestion was initiated by adding 1 mL of pancreatin extract (containing 1000 tributyrin units of pancreatic lipase activity). The digestion experiment was carried out for 20 min at 37 °C. The fatty acids produced during lipid digestion were titrated with 0.2 M NaOH solution. The total volume of NaOH added to adjust the pH of digestion medium to 7.4 and the rate of addition were recorded. The NaOH addition rate was used as the primary estimate of the rate of lipid digestion. Following the completion of lipolysis, digested formulations were tested for permeability in Caco-2 and MDCK monolayer models.
Cell lines
Caco-2 cells were purchased from American Type Culture Collection (Rockville, MD, USA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% (v/v) non-essential amino acids, 15% (v/v) fetal bovine serum (FBS), 1× sodium pyruvate, and 1% (v/v) penicillin–streptomycin antibiotic solution at 37 °C under 5% carbon dioxide. MDCK cells were received as a kind gift from Dr. William Elmquist, University of Minnesota. MDCK cells were propagated in DMEM containing 10 mM HEPES, 1% (v/v) non-essential amino acids, 10% (v/v) FBS, 1× sodium pyruvate, and 1% (v/v) penicillin–streptomycin antibiotic solution at 37 °C and 5% carbon dioxide.
Caco-2 and MDCK monolayer formation and integrity determination
Caco-2 cells (84,000 cells/well) were suspended in 200 μL of growth medium and placed on the top chamber of a 24-well HTS Transwell plate with a 0.2-μm polyester membrane (Corning Life Sciences, Lowell, MA, USA). The bottom chamber was filled with 800 μL of growth medium. Cells were incubated with fresh medium every 3 days and cultured at 37 °C and 5% carbon dioxide for 21 days to allow for the formation of a confluent monolayer. Similarly, MDCK cell monolayer was formed by adding 65,000 cells in the top chamber of the 24-well HTS Transwell plate and supplemented with MDCK cell growth media. Cell media was changed on day 3 and permeability assay was performed on day 5. The Caco-2 cells grow slowly compared to MDCK cells, which increases the time required for Caco-2 cells to form a confluent monolayer with tight junctions [37, 41].
Monolayer integrity was tested by measuring the trans-epithelial electrical resistance (TEER) values of the wells across the monolayer after equilibrating the plate to room temperature for 10 min. In general, MDCK cells exhibited lower TEER values than Caco-2 cells, which is attributed to the relatively loose junctions between adjacent MDCK cells [37]. However, it is a commonly used alternative permeability model because cell confluency is reached within 3–5 days and the TEER values are more comparable to the intestinal TEER values [42]. Typical Caco-2 monolayers have a TEER value in the range of 300 to 800 Ω.cm2 [43,44,45,46] and MDCK monolayers have a TEER value about 200 Ω.cm2 [47]. Improperly formed or compromised monolayers demonstrate a lower TEER value compared to the well-formed monolayers.
In addition to TEER measurement, monolayer integrity was also determined by measuring the permeability of Lucifer yellow (LY) (Sigma-Aldrich, St. Louis, MO, USA) across the monolayer. LY solution (100 μM) was added on the top chamber of the Transwell and maintained under gentle agitation at 37 °C. After 90 min, the media was withdrawn from the bottom chambers to measure LY concentration. The typical LY permeability across intact MDCK and Caco-2 monolayers is approximately 1.5 to 3 ⨯ 10−6 cm/s and 1 to 7 × 10−7 cm/s, respectively [37].
Permeability of elacridar-containing formulations
Caco-2 and MDCK monolayers were washed with the assay buffer comprising Hank’s Balanced Salt Solution containing 10 mM HEPES. The wells were filled with 200 μL of assay buffer on the apical side and 800 μL of assay buffer containing 4% bovine serum albumin in the basal side. The plates were maintained in a 37 °C incubator. Permeability samples comprised digested or undigested test formulations, solution, and suspension having a final elacridar concentration between 0 and 5 μM. Elacridar solution was prepared by dissolving elacridar in a vehicle comprising dimethyl sulfoxide, propylene glycol, and saline in 2:2:1 (v/v/v) ratio, and a suspension formulation was prepared by suspending elacridar in an aqueous mixture of 0.5% hydroxypropylmethylcellulose and 1% Tween 80 [48]. For each set of samples, one well received 100 μM LY in addition to the test formulations. The test formulations were incubated for 2 h and the concentrations of drug in the apical and basolateral side was analyzed using HPLC or LC-MS/MS. The apparent permeability (P app) was calculated using following formula (1):
where dQ/dt is the mass transport rate obtained from the slope of the amount transported versus time plot, A is the surface area of the cell monolayer, and C 0 is the initial donor concentration. Integrity of the monolayer was determined by recording the TEER value of the monolayer in the treatment wells, before and after the study.
Effect of different digested formulations on viability of MDCK cells
The cytotoxicity of digested and undigested formulations was assessed by MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay. MDCK cells were seeded onto a 96-well plate (65,000 cells/well) in 200 μL of complete growth medium and cultured for 5 days. Cells were then treated with 1 μL of the test formulation diluted with 200 μL medium and incubated for different time periods (1, 2, 5, and 24 h). After incubation for designated time periods, the medium was replaced with 200 μL of MTS-containing medium. The MTS medium consisted of complete growth medium, MTS solution (2 mg/mL in Dulbecco’s phosphate-buffered saline (DPBS); Promega, Madison, WI, USA), and phenazine methosulfate (0.92 mg/mL in DPBS) in the ratio of 100:20:1. The cells were then incubated at 37 °C under 5% carbon dioxide for 2 h, allowing conversion of MTS tetrazolium into aqueous soluble colored formazan product by NAD(P)H-dependent dehydrogenase enzymes found in metabolically active cells. The optical density (OD) of the formazan product was measured at 490 nm using a microplate reader (Biotek Inc., Winooski, VT, USA). Cells without test formulation treatment were used as controls. The cell viability was calculated using the following Eq. (2):
where ODsample is the optical density of treated cells and ODcontrol is the optical density of control cells.
Digestion inhibitor and component separation
In vitro digestion of CPL4 formulation was carried out using the procedure described earlier. At 5, 10, 15, 20, 25, and 30 min, the excess pancreatin was inhibited by addition of the lipase inhibitor 4-bromobenzeneboronic acid (BBBA) [49]. The resulting mixture was used to study the effect of digestion inhibitor on cell viability. MDCK cells were cultured in a 96-well plate (65,000 cells/well) in 200 μL of complete growth medium for 5 days. Cells were then treated with 1 μL aliquot (diluted in 200 μL of assay buffer) of above digested CPL4 formulations (5 min BBBA, 10 min BBBA, 15 min BBBA, 20 min BBBA, 25 min BBBA, and 30 min BBBA). In addition to BBBA-mediated inhibition, enzyme activity was also blocked by incubating cells at 4 °C and/or by increasing the medium pH to 9. Digested formulations without BBBA, with BBBA, and at pH 9 were used as positive controls. Additional controls included cells incubated with BBBA, BBBA at pH 9, at 4 °C, 4 °C at pH 9, and pancreatin. After 24 h, the presence of metabolically active MDCK cells after treatment with digested formulations was determined using the MTS assay.
To investigate the effect of each phase of the lipolytic products on MDCK cell viability, the different phases of the digested samples were separated following the procedure described by Sek et al. [50]. The digested CPL4 formulation was ultra-centrifuged at 35,000 rpm for 30 min (Beckman Optima™ LE-80K, Palo Alto, CA, USA) to separate the digested lipid into an upper oily layer, an aqueous phase in the middle, and an insoluble pellet at the bottom. Each of the components was diluted in assay buffer and added to the cells in 96-well plates. After 24 h, cell viability was determined by MTS assay.
Statistical analysis
All results were expressed as mean ± SD. Statistical analysis was performed using two-tailed Student’s t test. A probability level of p < 0.05 was considered significant.
Results and discussion
Lipid-based drug delivery systems provide an exciting formulation strategy for poorly water-soluble, lipophilic drugs. The primary role of lipids is to dissolve the drug in the formulation and keep it in a solubilized form in the GI tract. Therefore, any incidence of precipitation before reaching the duodenum is likely to reduce bioavailability of incorporated drug molecule by reintroducing the need for drug solubilization in the GI fluids [51]. Even in the case of physically stable formulations, the drug precipitation may occur upon digestion of the formulation owing to lower solvation of the drug in digested components than in the undigested parent formulation [52]. This is particularly important for drug molecules with poor aqueous solubility and low bioavailability [53]. Thus, subjecting the lipid formulation to an in vitro digestion test is important for predicting the GI absorption and bioavailability of the drug from lipid-based delivery systems.
In a typical lipolysis experiment, in vivo digestion conditions are simulated by the addition of a lipase enzyme (typically pancreatin) in a temperature-controlled reactor containing the lipid formulation dispersed in bile salt–lecithin mixed micelles [54, 55]. Lipid digestion results in the release of fatty acids, which in turn causes a drop in pH of the reaction mixture. The fatty acids are titrated using a base of known molarity to allow indirect quantification of the rate and extent of digestion.
The digested product is composed of three phases, which can be separated by high-speed ultracentrifugation. These include an undigested or partially digested oil phase on top (the “oil phase”); an aqueous micellar phase in the middle comprising of bile salt, cholesterol, phospholipid, and digested lipids (the “aqueous phase”); and a solid precipitate at the bottom (the “pellet phase”) [51]. Traditionally, the ability to maximize aqueous phase drug concentration (and minimize drug precipitation) following digestion is considered an indicator of the formulation’s ability to increase drug absorption in vivo [51]. However, the correlation between in vitro solubilization and in vivo absorption has been found to be highly dependent on the property of the drug being investigated [49, 56, 57]. In addition, accurate measurement of the free drug concentration (i.e., the fraction accessible for absorption) in a dynamic digestion setting is difficult as separation of free drug from solubilized drug is not trivial. Thus, the in vitro digestion testing may not be sufficient as a stand-alone characterization technique for screening of lipid-based formulations. We hypothesized that combining lipolysis with in vitro cell permeability studies will allow direct determination of the effect of lipid digestion on drug permeability and eliminate the need to measure the rate of change of drug concentrations in various phases. Based on this rationale, we evaluated the feasibility of combining lipolysis with in vitro Caco-2 and MDCK monolayer permeability models.
Formulation and characterization of lipid formulations
A series of lipid formulations were prepared using an appropriate combination of oil (castor oil), surfactants (Labrasol® and PL497), and co-surfactant (lecithin) (Table 2). The initial study design consisted of 27 unique formulations manufactured using different concentrations of each excipient as outlined in Table 2. Development of a suitable formulation having maximum solubilizing potential for the drug molecule is critical to achieve high drug loading. In order to identify an appropriate lipid blend, the saturation solubility of the model drug elacridar was determined in each of the excipients. Saturation solubility of elacridar in castor oil, Labrasol®, PL497, and castor oil containing 2% lecithin was 0.43, 12.50, 0.22, and 0.54 mg/mL, respectively. Solubility of elacridar in different formulations is presented in Table 3. Elacridar had significantly higher solubility in all the formulations tested than in water (aqueous solubility of elacridar is 1 μg/mL [58]).
Self-emulsification time is an indicator of the efficiency of emulsion formation. Lipid-based formulations should disperse rapidly and completely when they encounter aqueous dilution under mild agitation. Therefore, self-emulsification time was determined for each of the formulations, and the formulations that emulsified within a minute were selected for further studies. Table 3 summarizes the physical characteristics evaluated for each of the formulations. The self-emulsification time study demonstrated that the selected formulations could emulsify within 45 s, which indicates rapid emulsification. In addition, we studied the particle size of the emulsion formed. Particle size plays an important role in enhancing the available surface area (for a given volume) for absorption and the bioavailability of the drug. All the formulations resulted in micron-sized emulsions when introduced in water. The results are presented in Table 3.
Lipolysis
Since the small intestine is the primary site of lipid digestion and the target for drug absorption, the in vitro lipolysis studies were performed under simulated intestinal conditions. We first studied the effect of lipid composition on the rate of lipid digestion. It has been reported that the composition of lipid formulation (i.e., long chain vs. medium chain glycerides, type and amount of surfactants, and presence of cosolvent) can affect the rate and extent of lipid digestion and thereby influence the absorption of incorporated drug molecule [59]. The rate of lipid digestion was estimated from the time and volume of NaOH used for the lipolysis as described in previous reports [22]. The plots of 0.2 M NaOH volume versus time (Fig. 1a–c) were similar for the different groups of lipids as well as for different compositions of the same lipid components. This suggests that there are no discernible differences in the digestion rates for the various formulations tested.

The consumption of 0.2 M NaOH as a function of time for different a CPL, b CLeP, and c CLeL lipid formulations. The lipolysis was carried out for 20 min at 37 °C using pancreatin extract. The pH was maintained constant at 7.4
Caco-2 permeability assay
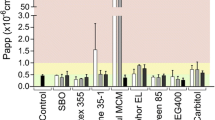
The intestinal permeability of both digested and undigested lipid formulations was determined using Caco-2 monolayer as an in vitro model. The permeability of the different formulations is shown in Fig. 2a. Among the different digested formulations, CPL4, CPL5, CLeP1, CLeP2, CLeL2, CLeL3, and CLeL4 showed significantly (p ˂ 0.05) higher permeability compared to their respective undigested formulation.

Effect of different digested formulations on a apparent permeability (P app) of elacridar across Caco-2 cell monolayer. Data represents mean ± SD, n = 4, ∗p < 0.05 compared to respective undigested group; b the integrity of Caco-2 cell monolayer determined by trans-epithelial electrical resistance (TEER) values. Data represents mean ± SD, n = 4. “D” denotes digested formulation and “U” denotes undigested formulation
To determine the effect of different formulations on the integrity of Caco-2 cell monolayer, we monitored TEER values of the monolayer before and after the permeability studies. Figure 2b demonstrates that the digested formulations resulted in compromised monolayer integrity as shown by decreased TEER values at the end of the study. Since a compromised monolayer offers little resistance to drug transport, an accurate evaluation of the permeability of digested formulation was, thus, not possible.
MDCK permeability assay
MDCK cells form monolayers more quickly than Caco-2 cells. However, similar to Caco-2 cells, they form tight junctions and serve as an alternate model for studying intestinal permeability [37]. Hence, the MDCK monolayer was also used to estimate the permeability of elacridar from digested and undigested lipid formulations. Similar to that seen in the Caco-2 study, digested CPL2, CPL5, and CLeL2 had significantly (p < 0.05) higher permeability compared to the respective undigested formulations (Fig. 3a). We next examined the effect of different formulations on the integrity of MDCK cell monolayer by measuring the TEER values and LY permeability. Digested formulations demonstrated lower TEER values and higher LY permeability compared to the respective undigested formulations (Fig. 3b, c). MDCK monolayers treated with various digested lipid formulations showed LY permeability higher than 4 × 10−6 cm/s, which clearly indicates the presence of compromised monolayer [37]. However, in the case of elacridar solution, no significant difference in LY permeability was observed under digested and undigested conditions. This data suggest that the digestion products of the lipid formulation rather than the drug or the digestion buffer were responsible for changes in LY permeability and TEER values.

Effect of different digested formulations on a apparent permeability of elacridar across MDCK cell monolayer and the integrity of MDCK cell monolayer determined by b TEER values (“Blank” represents TEER value of membrane without cells), and c Lucifer yellow (LY) permeability. Data represents mean ± SD, n = 4. “D” denotes digested formulation and “U” denotes undigested formulation. p < 0.05 compared to respective undigested formulation
Effect of different digested formulations on cell viability
Since digestion products appeared to affect the cell monolayer integrity, we determined the effect of digested and undigested formulations on cell viability using the MTS assay. The MDCK cell monolayer was used for this experiment because of its short culture time relative to Caco-2 cells. Time-dependent effect of digested and undigested formulations on cell viability is shown in Fig. 4. Incubation with digested formulations resulted in rapid loss of cell viability, with about 60% loss within 1 h. These results suggest that digested formulations induce some kind of catastrophic cell damage that results in rapid cell death in these cells.

Time-dependent toxicities of digested and undigested formulations on MDCK cells characterized by MTS assay. Data represents mean ± SD, n = 4
Effect of digestion inhibitors on cell viability
To investigate the possible cause of cytotoxicity of the digested formulations, MDCK cells were treated with formulations undergoing lipid digestion and the digestion was inhibited at different time intervals by the addition of BBBA, by elevating pH to 9, or lowering temperature to 4 °C. The results from this study (Fig. 5) suggested that lipolysis results in rather rapid generation of toxic byproducts, as shown by more than 50% reduction in cell viability even after neutralizing the excess pancreatin by the addition of lipolysis inhibitor BBBA, elevating pH, or lowering temperature. The control cells treated with BBBA, pH 9, low temperature, or a combination of these parameters showed no reduction in cell viability. Finally, treatment with pancreatin alone had no effect on the cell viability, suggesting that the digestion products are responsible for cell death and not the digestion enzyme.

Influence of various digestion inhibitors on MDCK cell viability. Data represents mean ± SD, n = 4. “D” denotes digested formulation
Effect of digested formulation components on cell viability
The digested formulations were divided into a pellet phase (containing insoluble fatty acids), an oily phase (containing fatty acids and mono-, di-, and triglycerides), and an aqueous phase (containing soluble fatty acids, surfactants, and some monoglycerides). MTS assay revealed that the aqueous soluble part of the digested lipid formulations was responsible for toxicity (Fig. 6). Surfactants are known to induce acute cell damage by compromising the cell membrane integrity [60, 61]. Such an effect of surfactants would explain the rather rapid effect of digested formulations on cell monolayer viability. Similarly, monoglycerides can exhibit significant cytotoxicity at high concentrations. A study by Jia and Wasan showed that monoolein induced ~80% loss in cell viability at concentrations greater than 500 μM [62].

Effect of digested formulation components on MDCK cell viability. Data represents mean ± SD, n = 4
Conclusions
Lipolysis is considered an important in vitro method to evaluate the in vivo performance of lipid-based drug delivery systems [63]. However, our studies indicate that the lipid digestion products lead to disruption of cell monolayer, and thus, it may not be appropriate to combine lipolysis with cell monolayer permeability studies. Further, it would be of importance to determine whether the cytotoxicity of lipolysis products observed in our in vitro studies translates into negative effects on intestinal epithelial barrier in vivo. There are a number of protective mechanisms in vivo such as the presence of a thick mucus layer as well as rapid dilution and clearance of these byproducts that could mitigate some of the side effects. Additional in vivo toxicology studies are needed to evaluate any potential toxicity of lipid formulations.
References
Lindenberg M, Kopp S, Dressman JB. Classification of orally administered drugs on the World Health Organization model list of essential medicines according to the biopharmaceutics classification system. Eur J Pharm Biopharm. 2004;58:265–78.
Gupta S, Kesarla R, Omri A. Formulation strategies to improve the bioavailability of poorly absorbed drugs with special emphasis on self-emulsifying systems. ISRN Pharmaceutics. 2013;2013:848043.
Stegemann S, Leveiller F, Franchi D, de Jong H, Linden H. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31:249–61.
Elsayed A, Remawi MA, Qinna N, Farouk A, Badwan A. Formulation and characterization of an oily-based system for oral delivery of insulin. Eur J Pharm Biopharm. 2009;73:269–79.
Kawakami K, Yoshikawa T, Hayashi T, Nishihara Y, Masuda K. Microemulsion formulation for enhanced absorption of poorly soluble drugs. II. In vivo study. J Control Release. 2002;81:75–82.
Patel V, Kukadiya H, Mashru R, Surti N, Mandal S. Development of microemulsion for solubility enhancement of clopidogrel. Iran J Pharm Res. 2010;9:327–34.
Luo Y, Chen D, Ren L, Zhao X, Qin J. Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Release. 2006;114:53–9.
Potta SG, Minemi S, Nukala RK, Peinado C, Lamprou DA, Urquhart A, et al. Development of solid lipid nanoparticles for enhanced solubility of poorly soluble drugs. J Biomed Nanotechnol. 2010;6:634–40.
Borkar N, Holm R, Yang M, Mullertz A, Mu H. In vivo evaluation of lipid-based formulations for oral delivery of apomorphine and its diester prodrugs. Int J Pharm. 2016;513:211–7.
Charman SA, Charman WN, Rogge MC, Wilson TD, Dutko FJ, Pouton CW. Self-emulsifying drug delivery systems: formulation and biopharmaceutic evaluation of an investigational lipophilic compound. Pharm Res. 1992;9:87–93.
Pouton CW. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur J Pharm Sci. 2000;11(Suppl 2):S93–8.
Gershanik T, Benita S. Self-dispersing lipid formulations for improving oral absorption of lipophilic drugs. Eur J Pharm Biopharm. 2000;50:179–88.
Gursoy RN, Benita S. Self-emulsifying drug delivery systems (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–82.
Constantinides PP. Lipid microemulsions for improving drug dissolution and oral absorption: physical and biopharmaceutical aspects. Pharm Res. 1995;12:1561–72.
Wakerly MG, Pouton CW, Meakin BJ, Morton FS. Self-emulsification of vegetable oil-nonionic surfactant mixtures. In: Phenomena in mixed surfactant systems. American Chemical Society. 1986. p. 242–55.
Weerapol Y, Limmatvapirat S, Nunthanid J, Sriamornsak P. Self-nanoemulsifying drug delivery system of nifedipine: impact of hydrophilic-lipophilic balance and molecular structure of mixed surfactants. AAPS PharmSciTech. 2014;15:456–64.
Carriere F. Impact of gastrointestinal lipolysis on oral lipid-based formulations and bioavailability of lipophilic drugs. Biochimie. 2016;125:297–305.
Khoo S-M, Humberstone AJ, Porter CJH, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167:155–64.
Julianto T, Yuen KH, Noor AM. Improved bioavailability of vitamin E with a self emulsifying formulation. Int J Pharm. 2000;200:53–7.
Kommuru TR, Gurley B, Khan MA, Reddy IK. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int J Pharm. 2001;212:233–46.
Mueller EA, Kovarik JM, van Bree JB, Tetzloff W, Grevel J, Kutz K. Improved dose linearity of cyclosporine pharmacokinetics from a microemulsion formulation. Pharm Res. 1994;11:301–4.
Porter CJ, Trevaskis NL, Charman WN. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat Rev Drug Discov. 2007;6:231–48.
Kalepu S, Manthina M, Padavala V. Oral lipid-based drug delivery systems—an overview. Acta Pharm Sin B. 2013;3:361–72.
Feinle C, Rades T, Otto B, Fried M. Fat digestion modulates gastrointestinal sensations induced by gastric distention and duodenal lipid in humans. Gastroenterology. 2001;120:1100–7.
Fried M, Mayer EA, Jansen JB, Lamers CB, Taylor IL, Bloom SR, et al. Temporal relationships of cholecystokinin release, pancreatobiliary secretion, and gastric emptying of a mixed meal. Gastroenterology. 1988;95:1344–50.
Chakraborty S, Shukla D, Mishra B, Singh S. Lipid—an emerging platform for oral delivery of drugs with poor bioavailability. Eur J Pharm Biopharm. 2009;73:1–15.
Khoo SM, Shackleford DM, Porter CJ, Edwards GA, Charman WN. Intestinal lymphatic transport of halofantrine occurs after oral administration of a unit-dose lipid-based formulation to fasted dogs. Pharm Res. 2003;20:1460–5.
Constantinides PP, Wasan KM. Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: in vitro/in vivo case studies. J Pharm Sci. 2007;96:235–48.
Sek L, Porter CJ, Charman WN. Characterisation and quantification of medium chain and long chain triglycerides and their in vitro digestion products, by HPTLC coupled with in situ densitometric analysis. J Pharm Biomed Anal. 2001;25:651–61.
Armand M, Borel P, Pasquier B, Dubois C, Senft M, Andre M, et al. Physicochemical characteristics of emulsions during fat digestion in human stomach and duodenum. Am J Phys. 1996;271:G172–83.
Hernell O, Staggers JE, Carey MC. Physical-chemical behavior of dietary and biliary lipids during intestinal digestion and absorption. 2. Phase analysis and aggregation states of luminal lipids during duodenal fat digestion in healthy adult human beings. Biochemistry. 1990;29:2041–56.
Hofmann AF, Roda A. Physicochemical properties of bile acids and their relationship to biological properties: an overview of the problem. J Lipid Res. 1984;25:1477–89.
MacGregor KJ, Embleton JK, Lacy JE, Perry EA, Solomon LJ, Seager H, et al. Influence of lipolysis on drug absorption from the gastro-intestinal tract. Adv Drug Deliv Rev. 1997;25:33–46.
Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res. 1993;53:4595–602.
Sane R, Mittapalli RK, Elmquist WF. Development and evaluation of a novel microemulsion formulation of elacridar to improve its bioavailability. J Pharm Sci. 2013;102:1343–54.
Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (Caco-2) cells. Biochem Biophys Res Commun. 1991;175:880–5.
Irvine JD, Takahashi L, Lockhart K, Cheong J, Tolan JW, Selick HE, et al. MDCK (Madin-Darby canine kidney) cells: a tool for membrane permeability screening. J Pharm Sci. 1999;88:28–33.
Miret S, Abrahamse L, de Groene EM. Comparison of in vitro models for the prediction of compound absorption across the human intestinal mucosa. J Biomol Screen. 2004;9:598–606.
Volpe DA. Drug-permeability and transporter assays in Caco-2 and MDCK cell lines. Future Med Chem. 2011;3:2063–77.
Baka E, Comer JEA, Takács-Novák K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J Pharm Biomed Anal. 2008;46:335–41.
Jin X, Luong T-L, Reese N, Gaona H, Collazo-Velez V, Vuong C, et al. Comparison of MDCK-MDR1 and Caco-2 cell based permeability assays for anti-malarial drug screening and drug investigations. J Pharmacol Toxicol Methods. 2014;70:188–94.
Di L, Kerns EH, Fan K, McConnell OJ, Carter GT. High throughput artificial membrane permeability assay for blood-brain barrier. Eur J Med Chem. 2003;38:223–32.
Feighery LM, Cochrane SW, Quinn T, Baird AW, O'Toole D, Owens SE, et al. Myosin light chain kinase inhibition: correction of increased intestinal epithelial permeability in vitro. Pharm Res. 2008;25:1377–86.
Ma B, Wang J, Sun J, Li M, Xu H, Sun G, et al. Permeability of rhynchophylline across human intestinal cell in vitro. Int J Clin Exp Pathol. 2014;7:1957–66.
Li C, Liu T, Cui X, Uss AS, Cheng KC. Development of in vitro pharmacokinetic screens using Caco-2, human hepatocyte, and Caco-2/human hepatocyte hybrid systems for the prediction of oral bioavailability in humans. J Biomol Screen. 2007;12:1084–91.
Skolnik S, Lin X, Wang J, Chen XH, He T, Zhang B. Towards prediction of in vivo intestinal absorption using a 96-well Caco-2 assay. J Pharm Sci. 2010;99:3246–65.
Cho MJ, Thompson DP, Cramer CT, Vidmar TJ, Scieszka JF. The Madin Darby canine kidney (MDCK) epithelial cell monolayer as a model cellular transport barrier. Pharm Res. 1989;6:71–7.
Sane R, Agarwal S, Elmquist WF. Brain distribution and bioavailability of elacridar after different routes of administration in the mouse. Drug Metab Dispos. 2012;40:1612–9.
Thomas N, Richter K, Pedersen TB, Holm R, Mullertz A, Rades T. In vitro lipolysis data does not adequately predict the in vivo performance of lipid-based drug delivery systems containing fenofibrate. AAPS J. 2014;16:539–49.
Sek L, Porter CJ, Kaukonen AM, Charman WN. Evaluation of the in-vitro digestion profiles of long and medium chain glycerides and the phase behaviour of their lipolytic products. J Pharm Pharmacol. 2002;54:29–41.
Feeney OM, Crum MF, McEvoy CL, Trevaskis NL, Williams HD, Pouton CW, et al. 50 years of oral lipid-based formulations: provenance, progress and future perspectives. Adv Drug Deliv Rev. 2016;101:167–94.
Anby MU, Williams HD, McIntosh M, Benameur H, Edwards GA, Pouton CW, et al. Lipid digestion as a trigger for supersaturation: evaluation of the impact of supersaturation stabilization on the in vitro and in vivo performance of self-emulsifying drug delivery systems. Mol Pharm. 2012;9:2063–79.
Carlert S, Palsson A, Hanisch G, von Corswant C, Nilsson C, Lindfors L, et al. Predicting intestinal precipitation—a case example for a basic BCS class II drug. Pharm Res. 2010;27:2119–30.
Zangenberg NH, Mullertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model. I. Controlling the rate of lipolysis by continuous addition of calcium. Eur J Pharm Sci. 2001;14:115–22.
Zangenberg NH, Mullertz A, Kristensen HG, Hovgaard L. A dynamic in vitro lipolysis model. II: Evaluation of the model. Eur J Pharm Sci. 2001;14:237–44.
Griffin BT, Kuentz M, Vertzoni M, Kostewicz ES, Fei Y, Faisal W, et al. Comparison of in vitro tests at various levels of complexity for the prediction of in vivo performance of lipid-based formulations: case studies with fenofibrate. Eur J Pharm Biopharm. 2014;86:427–37.
McEvoy CL, Trevaskis NL, Edwards GA, Perlman ME, Ambler CM, Mack MC, et al. In vitro–in vivo evaluation of lipid based formulations of the CETP inhibitors CP-529414 (torcetrapib) and CP-532623. Eur J Pharm Biopharm. 2014;88:973–85.
Kallem R, Kulkarni CP, Patel D, Thakur M, Sinz M, Singh SP, et al. A simplified protocol employing elacridar in rodents: a screening model in drug discovery to assess P-gp mediated efflux at the blood brain barrier. Drug Metab Lett. 2012;6:134–44.
Williams HD, Sassene P, Kleberg K, Bakala-N'Goma J-C, Calderone M, Jannin V, et al. Toward the establishment of standardized in vitro tests for lipid-based formulations, part 1: method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J Pharm Sci. 2012;101:3360–80.
Aungst BJ. Absorption enhancers: applications and advances. AAPS J. 2012;14:10–8.
Koga K, Kusawake Y, Ito Y, Sugioka N, Shibata N, Takada K. Enhancing mechanism of Labrasol on intestinal membrane permeability of the hydrophilic drug gentamicin sulfate. Eur J Pharm Biopharm. 2006;64:82–91.
Jia JX, Wasan KM. Effects of monoglycerides on rhodamine 123 accumulation, estradiol 17 beta-D-glucuronide bidirectional transport and MRP2 protein expression within Caco-2 cells. J Pharm Pharm Sci. 2008;11:45–62.
Kollipara S, Gandhi RK. Pharmacokinetic aspects and in vitro–in vivo correlation potential for lipid-based formulations. Acta Pharm Sin B. 2014;4:333–49.
Acknowledgments
The authors would like to greatly acknowledge the financial support provided by College of Pharmacy Seed Grant, University of Minnesota.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Rights and permissions
About this article

Cite this article
Sadhukha, T., Layek, B. & Prabha, S. Incorporation of lipolysis in monolayer permeability studies of lipid-based oral drug delivery systems. Drug Deliv. and Transl. Res. 8, 375–386 (2018). https://doi.org/10.1007/s13346-017-0383-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13346-017-0383-6