
Abstract
Human infection with H7 influenza subtypes usually resulted in mild disease with a rare mortalities, however, human infection with the avian low pathogenic H7N9 influenza virus resulted in about 38.6 % human fatality. Due to the new cross-species barrier of this virus subtype, there is an urgent need to better understand the susceptibility to commercially available antivirals and their relation to the structural changes of the viral neuraminidase. Neuraminidases derived from 2013 H7N9, H5N1 and H1N1 were subjected to a structural analysis of their catalytic and framework binding sites. The modeling structure of selected neuraminidases from H7N9 and influenza A subtypes were solved and the docking studies with oseltamivir, zanamivir, laninamivir and peramivir were conducted. The active site residues that are responsible for both binding and cleavage of the terminally linked sialic acid receptors were found conserved. Docking studies with oseltamivir, zanamivir, laninamivir and peramivir revealed that the laninamivir and peramivir showed superior energy binding activities in comparison to the commonly used oseltamivir and zanamivir. The results presented in the current study provide data that are useful for the future treatment of different influenza A subtypes including the recently emerged H7N9.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Influenza A viruses belong to the Family Orthomyxoviridae and include viruses that affect humans, birds and different animal species. They are subtyped according to antigenic differences between the two surface glycoproteins haemagglutinin [H1–H18] and neuraminidase [N1–N11] [17, 20, 21]. Crossing of the influenza A viruses from the aquatic birds to other avian or mammalian hosts do occur and the viruses mutate rapidly causing mild or in certain subtypes severe respiratory disease [8].
Avian influenza viruses (AIVs) continue to constitute challenging threats to public health. H5, H7, and H9 are the common avian influenza viruses that are circulating in domestic poultry, and accidently jump to humans causing mild to serious fatal diseases [1].The severity of the disease appears to vary with the infecting AIV subtype. H5N1 resulted in more than 57.5 % case fatality, a total of 676 laboratory-confirmed infections, 389 of which were fatal (http://www.who.int/influenza/human_animal_interface/EN_GIP_20141223CumulativeNumberH5N1cases.pdf?ua=1) [WHO, 4 Dec 2014]. AIV subtype H7 did not result in severe disease in humans [12], however, the recently record influenza A virus subtype H7N9 resulted in dozens of human cases with a case fatality of about 38.6 % [175/453] (http://www.who.int/influenza/human_animal_interface/influenza_h7n9/riskassessment_h7n9_2Oct14.pdf?ua=1), providing insight into unexpected virulence of H7 subtype to human beyond the predominant hypothesis of the mild nature of H7 infection to humans.
The viral neuraminidase (NA) is a receptor destroying enzyme that cleaves the terminal linkage of the sialic acid receptor resulting in the release of the progeny viral particles from the infected cells. NA may also facilitate the early processing of influenza virus infection in lung epithelial cells [16]. With the exception of N10, the nine NA subtypes are classified into two groups based on the structure and the phylogenetic analysis. Group 1 NA included N1, N4, N5 and N8, while group 2 included N2–N3, N6–N7 and N9 [18].The three dimensional structures revealed the variable conformations of areas adjacent to the enzymatic active site between group 1 and group 2 members [18]. NA is an attractive target for the anti-influenza drugs due to its role in virus release from infected cells [4]. Oseltamivir and zanamivir are commercially available NA inhibitors which are active against both group 1 and group 2 NA as well as influenza B NA [3]. Meanwhile, laninamivir is another long-acting NA inhibitor including oseltamivir-resistant viruses in adults [24, 25]. Recently, peramivir has been approved in Japan for use in over 1 month of age [11].
In the present study, we intended to study the sensitivity of the H7N9 and other influenza A subtypes to different neuraminidase inhibitors and to screen whether there are structural variations in the binding site that may affect the binding forces.
Materials and methods
Multiple sequence analysis
Multiple sequence alignment program, Mega 4.1 [13] was used to align different influenza NA sequences. Selected AIV NA sequences used for the alignments were obtained from the GenBank database. NA deduced amino acid sequences of recent H7N9 human strains were screened and compared them with 501 H1N1 and 164 human H5N1 and other H7N9 strains available in the flu database. The comparison was conducted to screen the amino acid variability in the catalytic and framework catalytic active sites.
Protein structure modeling
Target NA amino acid sequences for protein structure modeling were obtained from the NCBI-flu database. Influenza A subtypes H7N9 [A/Hangzhou/1/2013], mutant H5N1-N294S [A/Egypt/14724-NAMRU3/2006], sensitive H5N1 [A/Egypt/12374-NAMRU3/2006] and H1N1-H274Y mutant [A/Arkansas/01/2009] were included in the protein modeling. Modeling of each protein sequence was performed after minimizing and equilibration by steric clashes caused by the addition of hydrogen atoms, alleviation of water and ions prior to performing molecular dynamics. Sequence alignments of the target and template proteins were performed. This was followed by three dimensional [3D] structure of the target protein with the molsoft modeling software. In the modeling process, Molsoft moved the main chain and the side-chain atoms of the target protein alternatively in maintaining the conformational space between the model and the template 3D structure, and conducted conformational search close to the native structure in the packing state of the main and side chains. Neuraminidase proteins were modeled as the protein including the low molecular weight compounds.
Molecular docking
All docking studies were performed using ‘Internal Coordinate Mechanics [Molsoft ICM 3.4–8C]. We conducted in silico screening and molecular docking for the targeted NA against two sets of compounds, the first set included four different known NA inhibitors namely Ostelmavir [Tamiflu], Zanamavir [Relenza], Peramivir and Laninamivir (Suppl. 1). The second set included four different NA subtypes namely A subtypes H7N9 [A/Hangzhou/1/2013], mutant H5N1-N294S [A/Egypt/14724-NAMRU3/2006], sensitive H5N1 [A/Egypt/12374-NAMRU3/2006] and H1N1-H274Y mutant [A/Arkansas/01/2009] against N-acetylneuraminic acid [Nue5Ac], the most abundant natural ligand for binding to NA. All ligands were compiled by us using ChemDraw, 3D structures were constructed using Chem 3D ultra 12.0 software Molecular Modeling and Analysis; Cambridge Soft Corporation, USA [2010], then they were energetically minimized by using MOPAC [semi-empirical quantum mechanics], Job Type with 100 iterations and minimum RMS gradient of 0.01, and saved as MDL MolFile [*.mol]. Ligands were then used for docking process against tested neuraminidases. ICM stochastic global optimization algorithm attempts to find the global minimum of the energy function that included five grid potentials describing interaction of the flexible ligand with the receptor and internal conformational energy of the ligand. A stack of alternative low energy conformations was saved during the latter process. All inhibitors were compared according to the best binding free energy [minimum] obtained among all the run.
Results
Multiple sequence analysis
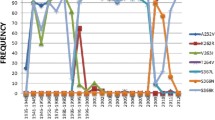
The NA of the recent H7N9 showed 5 amino acid deletions and two unique amino acids at 85 Gly/Ser/Thr to Asn and Thr to Ala at 400 (Suppl. 2). Amino acid residues Glu 276 and Tyr 406 as well as in the Arg tirade [R118, R292, and R371] were found conserved in the H7N9 (Suppl. 2, Table 1). Arg 292 to Lys was detected in six H7N9 strains (Table 1).Val 116 and Ile117 were found in new H7N9 human strains (Suppl. 2).
Protein structure modeling
The N2-Neu5Ac complex of the reference structure: PDB ID code 2BAT, was used as a standard classical model. All the residues in the cataylic and the framework sites of the canonical influenza NAs were found to be conserved in N9 (Table 1). The charged residues’ pocket for the sialic acid binding in the influenza A NA were divided into catalytic and framework sites (Table 1).
Molecular docking
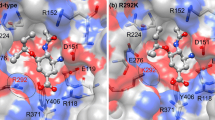
The functional activity of the novel N9 as a canonical sialidase was not altered with good fitting to sialic acid. The positive charged triple arginines at amino acid residues 118, 292 and 371 form important high-energy bridges with the sialic acid [negatively charged C1 carboxylate]. Neu5Ac was found to bind with 13 H-bonds: Arg 118 [3 bonds], Glu119 [2 bonds], Asp 151 [2 bonds], Trp 178, Glu 227, Arg 292, Arg 371 [2 bonds] and Tyr 406 with the NA of the original H7N9 classical strain. Meanwhile, Neu5Ac was found to bind to the NA of mutant strain with 19 H-bonds: Arg 118, Asp 151 [2 bonds], Arg 1152 [2 bonds], Lys 292 [7 bonds], Glu 276, Glu 277 [2 bonds], Asn 294, Arg 371 and Tyr 406 [2 bonds] [data not shown]. The total free energy of binding was higher in the non mutant stain in comparison to mutant one. Meanwhile arginine at 152 residue binds with the sialic acid N-acetyl group with a hydrogen bond. On the other hand, glutamic acid residues at 119, 227, 276, and 277 form a negatively charged “platform” region located below the binding residues to sialic acid. Arg to Lys 292 mutant showed lower receptor affinity and altered pattern of amino acid binding affinity to Neu5Ac receptor and the binding free energy was higher in the non mutant strain in comparison to the mutant strain (Fig. 1).
Virtual studying of the binding activity of N9 with ostlemaivir showed the lowest docking score energy binding among the tested drugs while peramivir and laninamivir showed the highest docking score energy binding (Table 2, Fig. 2). Virtual studying of the binding activity of N9 with zanamivir revealed that the C4 guanidine group of zanamivir interact with four of the catalytic sites: Arg 292 [6 bonds], Arg 371 [4 bonds], Asp 151 and Tyr 406 as well as one of the framework sites; Asn 294 in addition to a new binding site: Asn 347 (Fig. 2). The number of amino acids that shared in the drug binding activity was the lowest compared to sensitive and resistant group 1 strains and the docking score energy binding was lower than that of the H5N1 and H1N1 susceptible strains (Table 2).

Docking of H7N9 neuraminidase protein to Neu5Ac receptor. a Neuraminidase of the H7N9 strain. b Neuraminidase of the H7N9 strain R to K292 mutant strain. The NAs of the classical and mutant H7N9 strains were depicted in white, whereas the Molsoft plot of the ligand was depicted as mutli-colour stick model inside the binding pocket of the neuraminidase proteins. The binding free energy was −61.49 in mutant strain but −66.80 in non mutant strain

Molecular docking of different NA substrates and inhibitors. a Oseltamivir versus H7N9 original strain, b Oseltamivir versus H7N9 R to K 292 mutant strain, c Zanamivir versus H7N9 original strain, d Zanamivir versus H7N9 R to K 292 mutant strain, e Peramivir versus H7N9 original strain, f Peramivir versus H7N9 R to K 292 mutant strain, g Laninamivir versus H7N9 original strain, h Laninamivir versus H7N9 R to K 292 mutant strain. N7 amino acid numbering was used to allocate amino acid in the binding pocket of the neuraminidase
Discussion
Investigation of the NA active site architecture of N9 for both binding and hydrolyzing capacities to the sialidase substrate was conducted. Amino acid residues at 276 and 406 were found conserved in the H7N9. Glu 276 residue was assumed to interact with Tyr 406 thus playing a critical role in the catalytic mechanism [22]. The conserved key catalytic residues supported the finding that N9 possesses a canonical sialidase and revealed an efficient sialic acid-binding capability of N9. The N9 was found to interact with the silaic acid carboxylate group with Arg 118, Glu 119, Arg 152, Arg 292 and Arg 371 [data not shown]. Arginine residue at 371 was considered a crucial residue among the Arg tirade [R118, R292, and R371] that interacts with the sialic acid [2, 26]. However, Arg 118 residue was actually considered the most important Arg residue due to its interaction with Glu 425 [26]. This interaction is conserved in all NA subtypes including the novel H7N9 strains. Mutations responsible for conferring clinical resistance were found in different NA subtypes: Arg 292 to Lys and Glu 119 to Val were recorded in H3N2 while His 274 to Tyr and Asp 294 to Ser were recorded in H5N1 and H1N1 virus subtypes [6, 10, 14, 16]. Arg 292 to Lys was first recorded in the novel H7N9 strain by Gao et al. [5] then six full length sequences were found to harbour the same mutation. In the current study, it resulted in lower receptor affinity and altered pattern of amino acid binding affinity to Neu5Ac receptor. The same mutation was found to confer both in vitro and in vivo resistance to NA inhibitors [7, 16]. However, the effect of such substitution on the structure around it might be minimal [26]. On the other hand, the substitution at Arg to Lys at 292 was found to be compensated as oseltamivir bind with hydrogen bond from Tyr 344 of N1 of H5N1 viruses [18]. The conservation of the catalytic active sites could lead to the conserved interactions essential for the recognition of the sialidase substrate. It is apt to mention that, the proposed key nucleophilic residue; Y406 was found to be conserved among H7N9 strains.
Virtual studying of the binding activity of N9 with ostlemaivir showed the lowest docking score energy binding among the tested drugs while peramivir and laninamivir showed the highest docking score energy binding. This finding was supported with a recent study proved that the peramivir treatment possessed the rapid fever alleviation [19]. However, no considerable difference was found in the fever reduction time with oseltamivir, zanamivir, and laninamivir. Interestingly, none of the tested strains in that study showed His 275 to Tyr mutation [19]. Although still effective, both oseltamivir and zanamivir showed lower activities in reducing the duration of illness in children with influenza [23]. Interestingly, Arg 292 to Lys mutation resulted in lowered-drugsensitivity to both laninamivir and peramivir but not oseltamivir or zanamivir. Although not present in the new H7N9 human strains, Arg 152 to Lys mutation was the only reported instance of clinical resistance to zanamivir in influenza B [16, 6]. Furthermore, Val 116 to Ala and Ile 117 to Val mutations were not found in the new H7N9 human strains. Such substations were found to decrease the sensitivity to zanamivir and/or oseltamivir to H5N1 isolates [9, 15].
In conclusion, the structural analysis of the novel N9 did not alter its functional activity as a canonical sialidase. Due to significantly stable key active site residues and favourable surface electrostatic potential, N9 was able to bind to sialic acid. It is assumed that the N9 influenza virus fits very well with the human receptor. The good legend-receptor fitting helps in rapid virus release from the infected cells and shortening the virus replication cycles with subsequent positive impact on the virus virulence. The currently available influenza NA inhibitors are likely to be effective against the newly emerged H7N9 strains with oseltamivir showed the lowest activity and peramivir and laninamivir showed the highest activities.
References
Cardona CJ, Xing Z, Sandrock CE, Davis CE. Avian influenza in birds and mammals. Comp Immunol Microbiol Infect Dis. 2009;32(4):255–73.
Chong AK, Pegg MS, Taylor NR, von Itzstein M. Evidence for a sialosyl cation transition-state complex in the reaction of sialidase from influenza virus. Eur J Biochem. 1992;207(1):335–43.
Colman PM. New antivirals and drug resistance. Annu Rev Biochem. 2009;78:95–118.
Du QS, Wang SQ, Chou KC. Study of drug resistance of chicken influenza A virus (H5N1) from homology-modeled 3D structures of neuraminidases. Biochem Biophys Res Commun. 2007;354:634–40.
Gao R, Cao B, Hu Y, Feng Z, Wang D, Hu W, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;. doi:10.1056/NEJMoa1304459.
Gubareva LV. Molecular mechanisms of influenza virus resistance to neuraminidase inhibitors. Virus Res. 2004;103:199–203.
Gubareva LV, Webster RG, Hayden FG. Comparison of the activities of zanamivir, oseltamivir, and RWJ-270201 against clinical isolates of influenza virus and neuraminidase inhibitor-resistant variants. Antimicrob Agents Chemother. 2001;45:3403–8.
Horimoto T, Kawaoka Y. Pandemic threat posed by avian influenza A viruses. Clin Microbiol Rev. 2001;14(1):129–49.
Hurt AC, Selleck P, Komadina N, Shaw R, Brown L, Barr IG. Susceptibility of highly pathogenic A (H5N1) avian influenza viruses to the neuraminidase inhibitors and adamantanes. Antiviral Res. 2007;73:228–31.
Kiso M, Mitamura K, Sakai-Tagawa Y, Shiraishi K, Kawakami C, Kimura K, et al. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759–65.
Kohno SH, Kida H, Mizuguchi M, Shimada J. Efficacy and safety of intravenous peramivir for treatment of seasonal influenza virus infection. Antimicrob Agents Chemother. 2010;54(11):4568–74.
Koopmans M, Wilbrink B, Conyn M, Natrop G, van der Nat H, Vennema H, et al. Transmission of H7N7 avian influenza A virus to human beings during a large outbreak in commercial poultry farms in the Netherlands. Lancet. 2004;363(9409):587–93.
Kumar S, Tamura K, Jakobsen IB, Nei M. Molecular evolutionary genetics analysis software. Bioinformatics. 2001;17:1244–5.
Le QM, Kiso M, Someya K, Sakai YT, Nguyen TH, Nguyen KH, et al. Avian flu: isolation of drug-resistant H5N1 virus. Nature. 2005;437(7062):1108.
Le MT, Wertheim HF, Nguyen HD, Taylor W, Hoang PV, Vuong CD, et al. Influenza A H5N1 clade 2.3.4 virus with a different antiviral susceptibility profile replaced clade 1 virus in humans in northern Vietnam. PLoS ONE. 2008;3(10):e3339.
McKimm-Breschkin JL. Resistance of influenza viruses to neuraminidase inhibitors-a review. Antiviral Res. 2000;47:1–17.
Murphy BR, Webster RG. Orthomyxoviruses. Fields Virology. Philadelphia: Lippincott-Raven; 1996.
Russell J, Haire F, Stevens J, Collins J, Lin P, Blackburn M, et al. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature. 2006;443:45–9.
Shobugawa Y, Saito R, Sato I, Kawashima T, Dapat C, Dapat IC, et al. Clinical effectiveness of neuraminidase inhibitors–oseltamivir, zanamivir, laninamivir, and peramivir–for treatment of influenza A(H3N2) and A(H1N1)pdm09 infection: an observational study in the 2010-2011 influenza season in Japan. J Infect Chemother. 2012;18(6):858–64.
Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, et al. A distinct lineage of influenza A virus from bats. Proc Natl Acad Sci USA. 2012;109(11):4269–74.
Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013;9(10):e1003657.
von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Discov. 2007;6:967–74.
Wang K, Shun-Shin M, Gill P, Perera R, Harnden A. Neuraminidase inhibitors for preventing and treating influenza in children (published trials only). Cochrane Database Syst Rev. 2012;18(4):CD002744.
Watanabe A, Chang SC, Kim MJ, Chu DWS, Ohashi Y. Long-acting neuraminidase inhibitor laninamivir octanoate versus oseltamivir for treatment of influenza: a double-blind, randomized, noninferiority clinical trial. Clin Infect Dis. 2010;51(10):1167–75.
Yamashita M, Tomozawa T, Kakuta M, Tokumitsu A, Nasu H, Kubo S. CS-8958, a prodrug of the new neuraminidase inhibitor R-125489, shows long-acting anti-influenza virus activity. Antimicrob Agents Chemother. 2009;53(1):186–92.
Yen HL, Hoffmann E, Taylor G, Scholtissek C, Monto AS, Webster RG, et al. Importance of neuraminidase active-site residues to the neuraminidase inhibitor resistance of influenza viruses. J Virol. 2006;80(17):8787–95.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

{kind=link}
Cite this article
Eweas, A.F., Abdel-Moneim, A.S. In-silico structural analysis of the influenza A subtype H7N9 neuraminidase and molecular docking with different neuraminidase inhibitors. VirusDis. 26, 27–32 (2015). https://doi.org/10.1007/s13337-014-0245-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13337-014-0245-5