
Abstract
Background and Objective
Increased glycine availability at the synaptic cleft may enhance N-methyl-D-aspartate receptor signalling and provide a promising therapeutic strategy for cognitive impairment associated with schizophrenia. These studies aimed to assess the pharmacokinetics of BI 425809, a potent glycine-transporter-1 inhibitor, when co-administered with a strong cytochrome P450 3A4 (CYP3A4) inhibitor (itraconazole) and inducer (rifampicin).
Methods
In vitro studies using recombinant CYPs, human liver microsomes, and human hepatocytes were conducted to determine the CYP isoforms responsible for BI 425809 metabolism. In addition, two open-label, fixed-treatment period, phase I studies in healthy male volunteers are described. Period 1: participants received oral BI 425809 25 mg (single dose) on day 1; period 2: participants received multiple doses, across 10 days, of oral itraconazole or rifampicin combined with a single dose of oral BI 425809 25 mg on day 4/7 of the itraconazole/rifampicin treatment, respectively. Pharmacokinetic and safety endpoints were assessed in the absence/presence of itraconazole/rifampicin and included area under the concentration-time curve (AUC) over the time interval 0–167 h (AUC0‒167; itraconazole), 0–168 h (AUC0‒168; rifampicin), or 0–infinity (AUC0-∞; rifampicin and itraconazole), maximum measured concentration (Cmax) of BI 425809, and adverse events.
Results
In vitro results suggested that CYP3A4 accounted for ≥ 90% of the metabolism of BI 425809. BI 425809 exposure (adjusted geometric mean ratio [%]) was higher in the presence of itraconazole (AUC0‒167: 265.3; AUC0−∞: 597.0; Cmax: 116.1) and lower in the presence of rifampicin (AUC0‒168: 10.3; AUC0−∞: 9.8; Cmax: 37.4) compared with BI 425809 alone. Investigational treatments were well tolerated.
Conclusions
Systemic exposure of BI 425809 was altered in the presence of strong CYP3A4 modulators, corroborating in vitro results that CYP3A4 mediates a major metabolic pathway for BI 425809.
Trial Registration Number
NCT02342717 (registered on 15 January 2015) and NCT03082183 (registered on 10 March 2017)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Systemic exposure of BI 425809, after a single oral dose in healthy males, is considerably increased when co-administered with itraconazole, a strong CYP3A4 inhibitor, and decreased when co-administered with rifampicin, a strong CYP3A4 inducer |
These drug-drug interaction findings are important when considering concomitant medication use in clinical settings |
BI 425809 alone or in combination with itraconazole/rifampicin was safe and well tolerated in the population included here |
1 Introduction
Cognitive impairment associated with schizophrenia is characterised by abnormalities in glutamatergic signalling related to N-methyl-D-aspartate (NMDA) receptor hypofunction in cortical and hippocampal brain areas [1] leading to cognitive impairment [2,3,4]. Glycine is an obligatory co-agonist for glutamate signalling at NMDA receptors; thus, inhibition of glycine transporter 1 (GlyT1) to increase glycine availability in the synaptic cleft may enhance NMDA receptor signalling, a promising therapeutic strategy for management of cognitive impairments [3, 5].
BI 425809 is a selective and potent inhibitor of GlyT1 that is being developed for the treatment of cognitive impairment associated with schizophrenia. It has been previously reported that BI 425809 is generally well tolerated at doses up to 75 mg in healthy volunteers [6,7,8] and that multiple oral administrations of BI 425809 can increase cerebrospinal fluid glycine levels dose-dependently by a mean of 50% at doses as low as 10 mg [6].
Drug-drug interaction (DDI) studies provide valuable information regarding potential treatment interactions that may occur as a result of concomitant medications [9]. Cytochrome P450 3A4 (CYP3A4) is a major drug-metabolizing enzyme in humans that metabolises > 50% of all administered pharmaceuticals [10]. CYP3A4 is the most highly expressed CYP isoform in both the intestines and liver; thus, it is particularly important for determining exposure to orally administered medications [11]. Furthermore, DDIs involving CYP3A4 may alter the efficacy and/or safety profile of BI 425809 due to changes in BI 425809 bioavailability and exposure. Therefore, it is important to understand whether induction or inhibition of CYP3A4 affects the pharmacokinetics and/or safety profile of BI 425809.
Preclinical studies aimed to identify the human CYP isoforms responsible for BI 425809 metabolism, in particular the contribution of CYP3A. Primary objectives of the clinical studies were to assess the effects of multiple doses of the strong CYP3A4 inhibitor, itraconazole, and inducer, rifampicin, on the pharmacokinetics of BI 425809 in healthy male volunteers.
2 Methods
2.1 Preclinical Studies
2.1.1 Metabolism of BI 425809 by Recombinant CYP Isoforms
To determine which human CYP isoforms were responsible for the metabolism of BI 425809, in vitro studies were conducted with human recombinant cytochromes P450 (rCYP) isoforms (including: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, and CYP3A5). Functional activity of each rCYP was provided by the vendor and based on the metabolism of probe substrates. Briefly, rCYPs were prepared at the desired concentration (pmol/ml; Supplementary Table 1) in potassium phosphate buffer (50 mM, pH 7.4). Final rCYP content used in each incubation ranged from 100 to 579 pmol/ml according to vendor-supplied information on content of rCYP/volume of suspension. BI 425809 was added to the rCYP suspension, and samples were equilibrated at 37 °C in a water bath for 5 min. Reactions were initiated by the addition of reduced nicotinamide adenine dinucleotide phosphate (NADPH; 2 mM). Metabolism of BI 425809 was followed by monitoring both the depletion of BI 425809 and formation of the metabolites M526, M530, and M544. After collecting samples at desired time points (up to 60 min), reactions were terminated with 100 μl of d6-BI 425809 0.1 μM in 80:20 acetonitrile:water and centrifuged through a glass fibre filter (0.25 mm) at 2500×g for 30 min at 4 °C. The resulting filtrate was analysed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a 4000 QTRAP mass spectrometer (Applied Biosystems) connected to a Shimadzu LC-10AD vp pump or an Agilent 1200 pump to determine the rates of depletion of BI 425809 and formation of its metabolites, M526, M530, and M544. Semi-quantitative data using the ratio of the peak area response of the analyte and internal standard were used. Studies were conducted in duplicate.
2.1.2 Inhibition of BI 425809 Metabolism by Human Liver Microsomes
Human liver microsomes were prepared as a pool from 150 donors (mixed sex). Functional activity of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A was provided by the vendor and based on metabolism of probe substrates. Incubation mixtures contained human liver microsomes, BI 425809 (0.2, 2, 10 μM), potassium phosphate buffer (50 mM; pH 7.4), and CYP isoform-selective inhibitors. Controls (no inhibitor) were prepared in the same way, but organic solvent (1% acetonitrile) was added instead of inhibitor. All samples were equilibrated at 37 °C in a water bath for 5 min and reactions were initiated by addition of reduced NADPH (2 mM). After drawing samples at 0, 5, 15, 25, 35, 45, and 60 min, the reactions were terminated by the addition of 100 µl of d6-BI 425809 0.1 μM in 80:20 acetonitrile:water solution) and centrifuged through a glass fibre filter (0.25 mm) at 2500×g for 30 min at 4 °C. The filtrates were analysed by LC-MS/MS using a 4000 QTRAP mass spectrometer (Applied Biosystems) connected to a Shimadzu LC-10AD vp pump or an Agilent 1200 pump to determine the formation rate of the BI 425809 metabolites M530 and M544; depletion of BI 425809 was also monitored. Studies were conducted in duplicate.
2.1.3 BI 425809 Metabolism by Human Hepatocytes
Micropatterned co-cultures of human hepatocytes (from a male donor, 31 years of age) and mouse fibroblasts (HepatoPac®, BioIVT Inc.) were used to assess the metabolism of BI 425809 by CYP3A [12]. The co-cultures were received in a 96-well plate in maintenance media containing 10% foetal bovine serum. Three days after receipt of the cells, wells were washed with 100 μl serum-free probing media (pre-warmed to 37 °C). Next, 32 μl probing medium was added to each well of the 96-well plate, which was then placed into an incubator for 15 min in 10% carbon dioxide at 37 °C. The incubation reaction was initiated by adding 32 μl of media containing BI 425809 (0.4, 4 or 210 μM) or alprazolam (0.2 µM; positive control) with or without erythromycin (final incubation concentration was 30 μM). At each time point, from 0 to 96 h, 192 µl quench solution (60% acetonitrile, 0.1% acetic acid and 0.1 μM 13C6-BI 425809 or 0.1 µM nevirapine in water) was added to stop the reaction. Samples were triturated three times and frozen at − 30 °C in a 96-well collection plate. Samples were centrifuged (1850×g) at 4 °C for 30 min through a glass fibre filter (0.25 mm). Filtrates were analysed by LC-MS/MS using a 4000 QTRAP mass spectrometer (Applied Biosystems) connected to a Waters Acquity ultraperformance liquid chromatography system. Duplicate experiments at each concentration of BI 425809 were completed. Each experiment consisted of two replicates, giving a total of four replicates across both experiments. Alprazolam (0.1 µM) was used as a positive control. Erythromycin (30 µM) inhibited the intrinsic clearance (CLint) of alprazolam by at least 89% over a 168 h incubation [12]. Semi-quantitative data using the ratio of the peak area response of the analyte and internal standard (nevirapine for alprazolam and 13C6-BI 425809) were used.
The elimination rate constants (kel) of BI 425809 and alprazolam were determined from the absolute value of the slope of the line when the natural logarithm of the percent remaining compound was plotted against time. The kel was then used in Eq. 1 to determine the Clint. Percent inhibition by erythromycin was determined using Eq. 2
where V is the volume of the incubation (0.064 ml).
where Clint,inh is the Clint value in the presence of erythromycin and Clint,uninh is the Clint value in the absence of erythromycin.
2.1.4 Data Analysis
The major CYP isoform(s) responsible for the metabolism of BI 425809 were determined to be those that showed the greatest turnover and metabolite formation rate in rCYP incubations. The estimated percent contribution of each CYP isoform to the in vivo metabolism of BI 425809 was determined by calculating the percent inhibition of metabolite formation in the presence of isoform-selective inhibitors in human liver microsomes and inhibition of BI 425809 depletion in human hepatocytes.
2.2 Clinical Studies
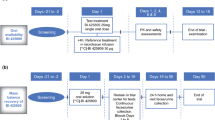
Two non-randomised, open-label, single-centre, fixed-sequence, phase I studies were also conducted (Clinicaltrials.gov identifiers: NCT02342717 [DDI with itraconazole] and NCT03082183 [DDI with rifampicin]), each consisting of two fixed treatment periods (Fig. 1).

Study design. BID twice daily, DDI drug-drug interaction, QD once daily, S screening
2.2.1 Clinical Study Design: DDI with Itraconazole
In period 1, participants received a single oral dose of BI 425809 25 mg on day 1. In period 2, participants received multiple oral doses of itraconazole for 10 days (day 1: 200 mg twice daily [BID]; days 2–10: 200 mg once daily [QD]) combined with a single dose of BI 425809 25 mg on day 4 of the itraconazole treatment. BI 425809 doses were separated by a washout period of ≥ 16 days.
2.2.2 Clinical Study Design: DDI with Rifampicin
In period 1, participants received a single oral dose of BI 425809 25 mg on day 1. In period 2, participants received multiple oral doses of rifampicin for 10 days (days 1–10: 600 mg QD) combined with a single dose of BI 425809 25 mg on day 7 of the rifampicin treatment. BI 425809 doses were separated by a washout period of ≥ 49 days.
2.2.3 Participants
For both studies, healthy male volunteers, 18–50 (DDI with itraconazole) or 18–55 (DDI with rifampicin) years of age, with a body mass index (BMI) of 18.5–29.9 kg/m2 were eligible for enrolment. A sample size of 16 volunteers per study was chosen to achieve sufficient precision for the 90% confidence interval of the bioavailability ratios, based on the observed intra-individual geometric coefficient of variation of BI 425809 in previous studies. Key exclusion criteria included any abnormal finding in the medical examination, laboratory values outside the reference range, or evidence of a concomitant disease, that were judged by the investigator to be clinically relevant. Other exclusion criteria included smoking (> 10 cigarettes per day); alcohol/drug abuse; intake of drugs with a long half-life (> 24 h) within 30 days, or < 10 half-lives, prior to the administration of study medications (DDI with itraconazole only); use of drugs that may have reasonably influenced the study results either 10 days (DDI with itraconazole) or 30 days (DDI with rifampicin) prior to the administration of study medication; or participation in another study where investigational drug was administered within 60 days prior to the administration of study medication. Participants were also excluded if they were unable to comply with the dietary regimen of the study site.
2.2.4 Ethical Considerations
The studies were conducted in compliance with the clinical study protocols and in accordance with the ethical principles of the Declaration of Helsinki [13], the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guideline for Good Clinical Practice (GCP) [14], applicable regulatory requirements, and standard operating procedures of the sponsor company (Boehringer Ingelheim International GmbH). All participants provided a signed and dated informed consent form prior to any study-related procedure. The study procedures, protocols, and documents were reviewed and approved by the Independent Ethics Committee of the study centre as well as the relevant local authorities.
2.2.5 Treatments
BI 425809 tablets were manufactured by Boehringer Ingelheim International GmbH, Germany. Itraconazole capsules and rifampicin film-coated tablets were manufactured by Janssen-Cilag GmbH, Neuss, Germany, and Riemser Pharma GmbH, Germany, respectively. Participants received BI 425809 as a 25 mg tablet (single dose) without (period 1) and in combination (period 2) with either itraconazole 100 mg capsules or rifampicin 600 mg film-coated tablets in a fixed sequence. All study medications were administered orally under the supervision of the investigating physician, or an authorised designated person, when participants were in the standing position.
For the DDI with itraconazole study, BI 425809 was administered after a standardised breakfast, while itraconazole was administered after a standardised breakfast or dinner. For the rifampicin study, BI 425809 was administered in the morning after an overnight fast, and rifampicin was given in the evening. All drugs were administered with 240 ml water. No meal was allowed within 4 h (itraconazole study) or 5 h (rifampicin study) of BI 425809 administration or 2 h before or 30 min after rifampicin intake.
2.2.6 Pharmacokinetic Endpoints
Pharmacokinetic endpoints and assessments were consistent between both studies unless otherwise stated. Primary endpoints included area under the concentration-time curve (AUC) over the time interval 0–167 h (DDI with itraconazole) or 0–168 h (DDI with rifampicin) (AUC0‒167/168) and maximum measured concentration (Cmax) of BI 425809 in plasma in the absence and presence of itraconazole or rifampicin. AUC of BI 425809 in plasma over the time interval from 0 extrapolated to infinity (AUC0−∞) in the absence and presence of itraconazole or rifampicin was also assessed as a secondary endpoint. Further pharmacokinetic parameters of interest for these clinical studies were time from dosing to Cmax (tmax), terminal half-life of the BI 425809 analyte (t1/2; time required for plasma concentration of analyte to decrease by 50%) in the absence and presence of itraconazole or rifampicin, and apparent clearance (CL/F) of analytes in plasma after extravascular administration. Additional further pharmacokinetic endpoints evaluated for BI 425809 that are not described here include the percentage of AUC obtained by extrapolation (% AUCtz–∞), terminal rate constant for analytes in the plasma (λz), AUC of the analytes in plasma over the time interval t1–t2 (AUCt1–t2), apparent volume of distribution during the terminal phase after extravascular administration (Vz/F), and mean residence time of the analytes in the body after oral administration (MRTpo). For further details of the pharmacokinetics of BI 452809 in healthy males, please refer to our previously published study [15].
2.2.7 Safety Endpoints
Safety and tolerability of the investigational drugs were assessed based on the frequency (n [%]) of participants with adverse events (AEs; including clinically relevant findings from physical examinations) and drug-related AEs, safety laboratory tests, vital signs (blood pressure, pulse rate), and 12-lead electrocardiogram. AEs were defined as any adverse medical occurrence, including an exacerbation of an existing condition, among participants of a clinical trial who received a pharmaceutical product, irrespective of being associated with or directly caused by treatment. AE intensity was graded as mild (easily tolerated signs or symptoms of AE), moderate (discomfort causing interference with usual activity), or severe (incapacitating or causing inability to work or perform usual activities). A serious AE was defined as any AE that resulted in death, was immediately life-threatening, required prolonged hospitalisation, or was deemed to pose immediate risk to the participant for any other medical reason. AEs were recorded for BI 425809 alone (period 1), itraconazole/rifampicin alone (treatment period after first dose of itraconazole/rifampicin [period 2 loading dose]), and BI 425809 in combination with itraconazole/rifampicin (treatment period after the second dose of BI 425809 [period 2]). Blood pressure and pulse rate were measured using ‘Dinamap Pro 100’ (GE Medical Systems, Germany), and electrocardiograms were recorded with ‘CardioSoft EKG System’ (GE Medical Systems, Germany).
2.2.8 Blood Sampling and Quantification
For quantification of BI 425809 plasma concentrations, 2.7 ml blood was taken from an antecubital or forearm vein into a tripotassium ethylenediaminetetraacetic acid (K3-EDTA) anticoagulant blood-drawing tube. The sampling schedule is detailed in Supplementary Tables 2 and 3.
Blood samples were centrifuged at 2000–4000×g for ~ 10 min at 4–8 °C. Two plasma aliquots were obtained from each blood sample and stored upright in polypropylene tubes at – 20 °C or below no later than 60 min after blood sampling (interim storage of blood samples was on ice). Frozen samples were transferred on dry ice to the analytical laboratory and stored at – 20 °C or below until analysis. BI 425809 concentrations in plasma were determined by a validated LC-MS/MS assay with a quantification range of 1.00–1000 nmol/l. Calibration standards and quality control samples used during sample analysis were prepared in human plasma containing tripotassium EDTA. Analyte stability has been demonstrated in human plasma samples containing tripotasssium EDTA for a period of 381 days at – 20 °C; therefore, all samples and calibration standards were analysed within this timeframe.
2.2.9 Pharmacokinetic Analysis
Pharmacokinetic parameters were derived using non-compartmental methods. Plasma exposure of BI 425809 with or without itraconazole or rifampicin was compared based on geometric mean (gMean) ratios for the pharmacokinetic endpoints using a (mixed effects) analysis of variance (ANOVA) model on log-transformed parameters with corresponding two-sided 90% confidence intervals (CIs), including participant as a random effect allowing for intra-participant comparisons. BI 425809 pharmacokinetic parameters were calculated using WinNonlin™ software professional Network version 5.2 and Network version Phoenix 6.3 (Pharsight Corporation, Mountain View, CA, USA) for the itraconazole and rifampicin studies, respectively. Statistical analyses were conducted using SAS version 9.4 (SAS Institute Inc., Cary, NC, USA). Descriptive statistics were calculated for all endpoints.
3 Results
3.1 Preclinical Metabolism Data
3.1.1 Metabolism of BI 425809 in Recombinant CYPs
Among all rCYPs assessed, significant depletion of BI 425809 (> 15%) and formation of metabolites were only observed with rCYP3A4 and rCYP3A5. Based on the depletion of BI 425809, CYP3A4 and CYP3A5 accounted for approximately 96% and 4%, respectively, of total CYP-mediated in vitro metabolism of BI 425809. Collectively, CYP3A was identified as the predominant pathway contributing to in vitro metabolism (Supplementary Table 4).
3.1.2 Metabolism of BI 425809 in Human Liver Microsomes
No detectable turnover of BI 425809 was observed in human liver microsomes. Hence, metabolism of BI 425809 was investigated by monitoring the formation of BI 425809 metabolites, M530 and M544. The formation of both metabolites was completely inhibited by the CYP3A4 inhibitor ketoconazole.
3.1.3 Metabolism of BI 425809 in Human Hepatocytes
Co-incubation with erythromycin (30 µM) significantly (92.1–100%) inhibited the depletion of BI 425809 at all concentrations evaluated. The effect of CYP3A inhibition by erythromycin on the intrinsic clearance (CLint) of BI 425809 in human hepatocytes is shown in Supplementary Table 5.
3.2 Clinical Study Results
3.2.1 Study Population and Disposition
A total of 16 healthy male volunteers were enrolled in each study and completed the planned observation period. Mean (standard deviation [SD]) ages were 40.1 (7.9; DDI with itraconazole) and 38.1 (10.8; DDI with rifampicin) years, and mean (SD) BMIs were 26.4 (1.9; DDI with itraconazole) and 25.0 (2.1; DDI with rifampicin) kg/m2. Further baseline demographic and clinical characteristics of the study population are summarised in Supplementary Table 6.
3.2.2 Pharmacokinetic Analyses: CYP3A4 Inhibition with Itraconazole
Plasma pharmacokinetic parameters of BI 425809 after single oral administration alone or in combination with itraconazole are presented in Table 1. Inferential analysis showed that co-administration of the strong CYP3A4 inhibitor itraconazole with a single dose of BI 425809 25 mg increased the overall systemic exposure of BI 425809. In the presence of itraconazole, gMean AUC0–167 and Cmax increased from 10,245 to 27,182 nmol·h/l and 316 to 367 nmol/l, respectively, and AUC0−∞ increased from 11,097 to 66,246 nmol·h/l. The adjusted gMean ratios (%) of AUC0–167, Cmax, and AUC0−∞ for BI 425809 co-administered with itraconazole relative to BI 425809 administered alone as a single dose were 265.3%, 116.1%, and 597.0%, respectively (Table 1). In the unadjusted analysis, BI 425809 plasma tmax (median [range]) values were similar in the presence and absence of itraconazole (2.5 [1.0, 4.5] vs 3.0 [1.5, 5.0] h, respectively). A decrease in CL/F from 73.3 to 12.4 ml/min and an increase in t1/2 from 45.5 to 222 h were observed when BI 425809 was co-administered with itraconazole compared with BI 425809 alone.
Graphical comparisons of individual and gMean AUC0–167 and Cmax values of BI 425809 after a single oral administration of BI 425809 25 mg alone and after concomitant administration with itraconazole are presented in Fig. 2b. The gMean plasma drug concentration-time profile of BI 425809 in plasma after single oral administration of BI 425809 25 mg with or without itraconazole is presented in Fig. 3a.

Comparison of individual and gMean AUC0–167/168 and Cmax of BI 425809 after a single oral administration of BI 425809 25 mg alone and after concomitant administration with a itraconazole or b rifampicin. AUC area under the concentration-time curve, AUC0‒167/168 area under the concentration-time curve of BI 425809 in the plasma over the time interval from 0 to 167/168 h, Cmax maximum measured concentration of BI 425809 in plasma, gMean geometric mean

gMean drug concentration-time profiles of BI 425809 in plasma after single oral administration of BI 425809 25 mg with or without itraconazole (a) or rifampicin (b) (semi-log scale). gMean geometric mean
3.2.3 Pharmacokinetic Analyses: CYP3A4 Induction with Rifampicin
Plasma pharmacokinetic parameters of BI 425809 after single oral administration alone or in combination with rifampicin are presented in Table 2. In the inferential analysis, induction of CYP3A4 expression by co-administration of rifampicin with BI 425809 25 mg led to a clinically relevant decrease in systemic exposure to BI 425809. In the presence of rifampicin, gMean AUC0–168 and Cmax decreased from 8222 to 844 nmol·h/l and 218 to 82 nmol/l, respectively, and gMean AUC0−∞ decreased from 8596 to 846 nmol·h/l. The adjusted gMean ratios (%) of AUC0–168, Cmax, and AUC0-∞ for BI 425809 co-administered with rifampicin relative to BI 425809 administered alone as a single dose were 10.3%, 37.4%, and 9.8%, respectively (Table 2). In the unadjusted analysis, BI 425809 plasma tmax (median [range]) was similar in the presence and absence of rifampicin (2.5 [1.5, 6.0] h vs 4.0 [2.0, 6.0] h, respectively). An increase in CL from 94.6 to 972000 ml/min and a decrease in t1/2 from 33.4 to 8.53 h were observed when BI 425809 was co-administered with rifampicin compared with BI 425809 alone.
Graphical comparisons of individual and gMean AUC0–168 and Cmax of BI 425809 after a single oral administration of BI 425809 25 mg alone and after concomitant administration with rifampicin are presented in Fig. 2b. The gMean plasma drug concentration-time profile of BI 425809 in plasma after single oral administration of BI 425809 25 mg with or without rifampicin is presented in Fig. 3b
3.2.4 Safety and Tolerability: CYP3A4 Inhibition with Itraconazole
Overall, 11 participants (68.8%) reported ≥ 1 AE (BI 425809 alone: n = 6 [37.5%]; itraconazole alone: n = 4 [25%]; BI 425809 + itraconazole: n = 6 [37.5%]; Table 3). In total, two (12.5%) participants reported ≥ 1 AE considered as drug-related by the investigator; of these, both reported drug-related AEs when BI 425809 was co-administered with itraconazole, and one (6.3%) also reported a drug-related AE following administration of itraconazole alone. All AEs were of mild intensity except for one case of vomiting (moderate) and one of nausea (severe), both reported by the same participant. No AEs of special interest (AESIs), serious AEs (SAEs), or clinically relevant changes in laboratory parameters were observed. Moreover, no AEs led to the discontinuation of study medication.
3.2.5 Safety and Tolerability: CYP3A4 Induction with Rifampicin
All 16 participants (100%) reported ≥ 1 AE (BI 425809 alone: 12.5%; rifampicin alone: 100%; BI 425809 + rifampicin: 18.8%) (Table 3). All participants reported AEs considered as drug-related by the investigator following administration of rifampicin alone (100%) and one participant reported a drug-related AE following treatment with BI 425808 + rifampicin (6.3%). All AEs were of mild to moderate intensity. No AESIs, SAEs, or clinically relevant changes in laboratory parameters were observed, and no AEs led to the discontinuation of study medication.
4 Discussion
In vitro CYP formation and depletion data from human liver microsomes and human hepatocytes were found to be in good agreement and strongly suggested that CYP3A is responsible for BI 425809 metabolism in the liver. In vitro contributions of recombinant CYP3A4 and CYP3A5 to BI 425809 metabolism, and the complete inhibition of the formation of BI 425809 metabolites by ketoconazole, further identified CYP3A4/5 as the isoforms responsible for most CYP-450-mediated BI 425809 metabolism in vivo. In support of this, BI 425809 metabolism was strongly inhibited (> 90%) by erythromycin in human hepatocytes.
Based on these preclinical findings, two phase I clinical studies were conducted to investigate the impact of CYP3A4 induction and inhibition on the pharmacokinetics of BI 425809. The strong CYP3A4 inhibitor itraconazole had minimal impact on BI 425809 absorption, with a slight increase in Cmax (∼ 16%), indicating no relevant first pass effect. However, there was a clear impact of itraconazole treatment on the metabolism of BI 425809; an increase in t1/2 was observed along with a decrease in CL and an overall increase in systemic exposure of BI 425809. Indeed, co-administration of BI 425809 with itraconazole resulted in an increase (gMean ratio: 597.0%) in exposure (AUC0−∞) compared with administration of BI 425809 without itraconazole.
The pharmacokinetic sampling duration was not sufficient to cover the complete plasma concentration-time profile of BI 425809 under itraconazole treatment, which is indicated by the high extrapolated area of 56%. Due to this uncertainty, the AUC0−∞ value and respective exposure ratio should be interpreted with caution but are considered closer to the real effect size than the increase in exposure (gMean ratio: 265.3%) that was observed based on AUC0‒167 values. A 265.3% increase in exposure based on AUC0‒167 would seem to indicate a lower contribution of CYP3A4, whereas the 597.0% increase in exposure based on AUC0−∞ matches the ~ 95% contribution compared with in vitro studies. Overall, the fold increase in AUC0−∞ in the presence of itraconazole along with the in vitro results suggests that CYP3A4 is a major metabolic enzyme of BI 425809 [7]. In the rifampicin DDI study, we observed a reduction in AUC0‒168 when BI 425809 was co-administered with rifampicin relative to BI 425809 alone, with a ratio of 0.10, further supporting a considerable contribution of CYP3A4 to BI 425809 metabolism. This is comparable to the effects of rifampicin on other sensitive CYP3A4 substrates such as midazolam and simvastatin [14, 16].
Overall, results from the present study suggest that BI 425809 plasma concentrations are significantly changed by CYP3A4 inhibitors and inducers, therefore indicating that caution should be exercised when BI 425809 is co-administered with either of the aforementioned drugs. However, it should be noted that firm dosing recommendations can only be made when the final dose and exposure-response correlations have been fully established with further study.
A potential limitation of these studies is that they were conducted in healthy volunteers and designed to report on the maximum pharmacokinetic DDI potential of BI 425809 by administering it alongside “worst-case scenario” drugs, which are less commonly used among the target population. Real-world pharmacokinetic parameters among patients with comorbidities and multiple comedications may differ. Furthermore, BI 425809 was only administered as a victim drug in a single dose; pharmacokinetic interactions between the victim and perpetrator drugs at steady state in real-world scenarios may also differ.
Nonetheless, BI 425809 was well tolerated overall in these studies. All observed drug-related AEs were considered to be of mild or moderate intensity and no new safety signals, AESIs, SAEs, or clinically relevant laboratory parameters were reported. All AEs observed in the study were consistent with the previously reported safety profile of BI 425809 and additional treatments [6, 7, 17, 18].
5 Conclusion
The DDI studies described here demonstrate that overall systemic exposure of BI 425809, after a single oral dose in healthy male volunteers, is considerably increased when co-administered with itraconazole, a strong CYP3A4 inhibitor, and decreased when co-administered with rifampicin, a strong CYP3A4 inducer. Moreover, BI 425809 alone or in combination with itraconazole/rifampicin was safe and well tolerated with no new safety signals identified. Overall, these findings provide invaluable data regarding potential drug interactions that may occur because of concomitant medication use in clinical settings.
References
Lin CH, Lane HY, Tsai GE. Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol Biochem Behav. 2012;100(4):665–77. https://doi.org/10.1016/j.pbb.2011.03.023.
Bowie CR, Reichenberg A, Patterson TL, Heaton RK, Harvey PD. Determinants of real-world functional performance in schizophrenia subjects: correlations with cognition, functional capacity, and symptoms. Am J Psychiatry. 2006;163:418–25. https://doi.org/10.1176/appi.ajp.163.3.418.
Hashimoto K. Glycine transport inhibitors for the treatment of schizophrenia. Open Med Chem J. 2010;4:10–9. https://doi.org/10.2174/1874104501004010010.
Lakhan SE, Caro M, Hadzimichalis N. NMDA receptor activity in neuropsychiatric disorders. Front Psychiatry. 2013;4:52. https://doi.org/10.3389/fpsyt.2013.00052.
Field JR, Walker AG, Conn PJ. Targeting glutamate synapses in schizophrenia. Trends Mol Med. 2011;17(12):689–98. https://doi.org/10.1016/j.molmed.2011.08.004.
Rosenbrock H, Desch M, Kleiner O, Dorner-Ciossek C, Schmid B, Keller S, Schlecker C, Moschetti V, Goetz S, Liesenfeld KH, Fillon G, Giovannini R, Ramael S, Wunderlich G, Wind S. Evaluation of pharmacokinetics and pharmacodynamics of BI 425809, a novel GlyT1 inhibitor: translational studies. Clin Transl Sci. 2018;11(6):616–23. https://doi.org/10.1111/cts.12578.
Moschetti V, Schlecker C, Wind S, Goetz S, Schmitt H, Schultz A, Liesenfeld KH, Wunderlich G, Desch M. Multiple rising doses of oral BI 425809, a GlyT1 inhibitor, in young and elderly healthy volunteers: a randomised, double-blind, phase I study investigating safety and pharmacokinetics. Clin Drug Investig. 2018;38(8):737–50. https://doi.org/10.1007/s40261-018-0660-2.
Tsuda Y, Ugai H, Wunderlich G, Shin J-G. Pharmacokinetics of single doses of BI 425809 in healthy Chinese and Japanese subjects: a randomized study. Clin Ther. 2019;41(5):961–71. https://doi.org/10.1016/j.clinthera.2019.03.014.
van Vollenhoven RF, Navarra SV, Levy RA, Thomas M, Heath A, Lustine T, Adamkovic A, Fettiplace J, Wang ML, Ji B, Roth D. Long-term safety and limited organ damage in patients with systemic lupus erythematosus treated with belimumab: a phase III study extension. Rheumatology (Oxford). 2020;59:281–91. https://doi.org/10.1093/rheumatology/kez279.
Sevrioukova IF, Poulos TL. Current approaches for investigating and predicting cytochrome P450 3A4-ligand interactions. Adv Exp Med Biol. 2015;851:83–105. https://doi.org/10.1007/978-3-319-16009-2_3.
Danielson P. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr Drug Metab. 2002;3(6):561–97. https://doi.org/10.2174/1389200023337054.
Chan TS, Scaringella YS, Raymond K, Taub ME. Evaluation of erythromycin as a tool to assess CYP3A contribution of low clearance compounds in a long-term hepatocyte culture. Drug Metab Dispos. 2020;48(8):690–7. https://doi.org/10.1124/dmd.120.090951.
World Medical Association (WMA). WMA Declaration of Helsinki—ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–4. https://doi.org/10.1001/jama.2013.281053.
Backman JT, Olkkola KT, Neuvonen PJ. Rifampin drastically reduces plasma concentrations and effects of oral midazolam. Clin Pharmacol Ther. 1996;59(1):7–13. https://doi.org/10.1016/s0009-9236(96)90018-1.
Moschetti V, Desch M, Goetz S, Liesenfeld K-H, Rosenbrock H, Kammerer K-P, Wunderlich G, Wind S. Safety, tolerability and pharmacokinetics of oral BI 425809, a glycine transporter 1 inhibitor, in healthy male volunteers: a partially randomised, single-blind, placebo-controlled, first-in-human study. Eur J Drug Metab Pharmacokinet. 2018;43(2):239–49.
Kyrklund C, Backman JT, Kivistö KT, Neuvonen M, Laitila J, Neuvonen PJ. Rifampin greatly reduces plasma simvastatin and simvastatin acid concentrations. Clin Pharmacol Ther. 2000;68(6):592–7. https://doi.org/10.1067/mcp.2000.111414.
Desch M, Schmitt H, Hohl K, Liesenfeld K, Chan T, Zhou J, Mueller F, Wunderlich G, Wind S. Pharmacokinetic interaction of BI 425809, a new glycine transporter 1 (glyt1) inhibitor, with cytochrome p450 (cyp) isoenzymes and p-glycoprotein (p-gp) probe drug. Cln Pharmacol Ther. 2017;101:S52–S52. https://doi.org/10.1002/cpt.570.
Desch MGM, Goetz S, et al. Effects of the potent cytochrome p450 3A4 inhibitor, itraconazole, on the pharmacokinetics of BI 425809, a new glycine transporter 1 (GlyT1) inhibitor. Clin Pharmacol Ther. 2017;101:S52. https://doi.org/10.1002/cpt.570.
Acknowledgements
Editorial support (in the form of initial preparation of the outline based on input from all authors, collation and incorporation of author feedback to develop subsequent drafts, assembling tables and figures, copy editing, and referencing) was provided by Sam Halliwell, PhD, and Eshvendar Reddy Kasala, PhD, of Fishawack Communications Ltd, part of Fishawack Health, which was funded by Boehringer Ingelheim International GmbH. The authors would also like to thank Andrea Whitcher-Johnstone of Boehringer Ingelheim Pharmaceuticals Inc. for her contributions to the conduct of the preclinical reaction phenotyping studies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The work presented here, including the conduct of the studies, data analysis, and interpretation, was funded by Boehringer Ingelheim International GmbH. The sponsor was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Conflicts of interest
MD, MG, SG, KHL, HR, RS, JL, SK, and SW are employees of Boehringer Ingelheim International GmbH, GW is an employee of Boehringer Ingelheim (Canada), and TSC is an employee of Boehringer Ingelheim Pharmaceuticals Inc. Authors did not receive any direct compensation relating to the development of this manuscript.
Ethics approval
The studies were conducted in compliance with the clinical study protocols and in accordance with the ethical principles of the Declaration of Helsinki [13], the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guideline for Good Clinical Practice (GCP) [14], applicable regulatory requirements, and standard operating procedures of the sponsor company (Boehringer Ingelheim International GmbH). The study procedures, protocols, and documents were reviewed and approved by the Independent Ethics Committee of the study centre as well as the relevant local authorities.
Consent to participate
All participants provided a signed and dated informed consent form prior to any study-related procedure.
Consent for publication
Not applicable
Availability of data and material
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to all relevant material, including participant-level clinical study data, and relevant material as needed by them to fulfill their role and obligations as authors under the ICMJE criteria.
Furthermore, clinical study documents (e.g. study report, study protocol, statistical analysis plan) and participant clinical study data are available to be shared after publication of the primary manuscript in a peer-reviewed journal and if regulatory activities are complete and other criteria met per the BI Policy on Transparency and Publication of Clinical Study Data: https://trials.boehringer-ingelheim.com/.
Prior to providing access, documents will be examined and, if necessary, redacted and the data will be de-identified to protect the personal data of study participants and personnel and to respect the boundaries of the informed consent of the study participants.
Clinical Study Reports and Related Clinical Documents can also be requested via the link https://trials.boehringer-ingelheim.com/.
All requests will be governed by a Document Sharing Agreement.
Bona fide, qualified scientific and medical researchers may request access to de-identified, analysable participant clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Data Sharing Agreement, data are shared in a secured data-access system for a limited period of 1 year, which may be extended upon request.
Researchers should use the https://trials.boehringer-ingelheim.com/ link to request access to study data.
Code availability
Not applicable.
Author contributions
All authors contributed to the development of the manuscript, have revised it critically for important intellectual content, and have approved the version to be submitted. MD, KHL, and SW made substantial contributions to the conception or design of the work and the analysis or interpretation of data. GW, SG, TSC, and HR made substantial contributions to the conception or design of the work. MG and RS made substantial contributions to the conception or design of the work and the acquisition of data. JL and SK made substantial contributions to the analysis or interpretation of data.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Desch, M., Wunderlich, G., Goettel, M. et al. Effects of Cytochrome P450 3A4 Induction and Inhibition on the Pharmacokinetics of BI 425809, a Novel Glycine Transporter 1 Inhibitor. Eur J Drug Metab Pharmacokinet 47, 91–103 (2022). https://doi.org/10.1007/s13318-021-00723-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-021-00723-y