
Abstract
Background and Objective
Clinical response to methotrexate in cancer is variable and depends on several factors including serum drug exposure. This study aimed to develop a population pharmacokinetic model describing methotrexate disposition in cancer patients using retrospective chart review data available from routine clinical practice.
Methods
A retrospective review of medical records was conducted for cancer patients in Qatar. Relevant data (methotrexate dosing/concentrations from multiple occasions, patient history, and laboratory values) were extracted and analyzed using NONMEM VII®. A population pharmacokinetic model was developed and used to estimate inter-individual and inter-occasion variability terms on methotrexate pharmacokinetic parameters, as well as patient factors affecting methotrexate pharmacokinetics.
Results
Methotrexate disposition was described by a two-compartment model with clearance (CL) of 15.7 L/h and central volume of distribution (V c) of 79.2 L. Patient weight and hematocrit levels were significant covariates on methotrexate V c and CL, respectively. Methotrexate CL changed by 50 % with changes in hematocrit levels from 23 to 50 %. Inter-occasion variability in methotrexate CL was estimated for patients administered the drug on multiple occasions (48 and 31 % for 2nd and 3rd visits, respectively).
Conclusion
Therapeutic drug monitoring data collected during routine clinical practice can provide a useful tool for understanding factors affecting methotrexate pharmacokinetics. Patient weight and hematocrit levels may play a clinically important role in determining methotrexate serum exposure and dosing requirements. Future prospective studies are needed to validate results of the developed model and evaluate its usefulness to predict methotrexate exposure and optimize dosing regimens.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Considerable interpatient and inter-occasion variability is observed in intravenous methotrexate pharmacokinetics. |
Intravenous methotrexate pharmacokinetics in patients with hematological malignancies are affected by patient weight and haematocrit levels. |
Routine methotrexate clinical therapeutic drug monitoring data can be used to provide understanding of factors affecting methotrexate pharmacokinetics and inform strategies for individualized patient therapy. |
1 Introduction
The concept of individualized medicine is a rapidly emerging global trend that strives to optimize both efficacy and safety of medications [1]. By designing regimens based on specialized patient-level data, better patient outcomes can be achieved. For instance, treatment of the Human Immunodeficiency Virus has been revolutionized by targeting therapy based on genotyping for resistant viral strains [2]. In addition, therapeutic drug monitoring and dosage adjustment based on patient and population pharmacokinetic data (i.e., through measurement of drug concentrations) have proved valuable for optimizing efficacy and safety of drugs with narrow therapeutic indices and in special populations [3].
The Middle East is a largely untouched region for pharmacokinetic-based dosing studies, although it is rich in diversity of ethnicity and other special populations. Within our clinical practice in Qatar, we have noticed great variability in patient response and have had considerable dosing problems when using the antimetabolite agent, methotrexate, for treatment of cancer patients. Methotrexate is a commonly used agent for many types of cancer in Qatar, especially for hematological tumors. However, it is highly toxic (renal, hepatic, and hematologic effects) and strict monitoring must occur through clinical symptoms, laboratory markers, and drug concentrations [4]. Specifically, drug concentrations guide administration of leucovorin for rescue and to avoid excessive destruction of host cells [4].
Therefore, optimization of methotrexate empiric dosing through determination of both patient and population pharmacokinetic parameters may lead to better patient outcomes and a healthier cancer population. Although methotrexate concentrations are usually monitored as part of routine clinical practice in the National Center for Cancer Care and Research (NCCCR) in Qatar, pharmacokinetic analyses and dose individualization approaches are not adequately implemented to optimize patient therapy. Prospective and retrospective use of routinely collected methotrexate therapeutic drug monitoring information can provide a powerful tool for better understanding of methotrexate pharmacokinetics and, hence, optimization of dosing regimens for cancer patients in the Middle East.
Previous models have been developed to describe methotrexate pharmacokinetics in cancer and non-cancer patients [5–8]. To our knowledge, none of these models have been developed in a diverse Asian/Middle Eastern population similar to the patient population in Qatar hospitals. Although previous studies have identified numerous factors associated with changes in methotrexate pharmacokinetics in cancer or non-cancer patients [e.g., renal function, coadministration of non-steroidal anti-inflammatory drugs (NSAIDs) or ciprofloxacin, and genetic polymorphisms in organic anion transporting polypeptide 1B (OATP1B) and multi-drug resistant protein 2 (MRP2) transporters] [9–12], similar studies are lacking for identification of such factors in cancer patients in the Middle Eastern population. Evaluation of factors affecting methotrexate pharmacokinetics in cancer patients in Qatar will help reduce the observed variability in methotrexate exposure and optimize treatment response. Therefore, population-based pharmacokinetic modeling approaches can provide a valuable tool for development of a framework for empiric dosing and dose adjustments in this special population.
The main goal of this project was to utilize routinely collected methotrexate monitoring information to understand and quantify methotrexate pharmacokinetics in cancer patients in Qatar as well as to identify any patient- or disease-related factors that affect methotrexate disposition in this patient population. The objectives were to develop a population pharmacokinetic model that describes and quantifies methotrexate disposition and pharmacokinetic parameters in cancer patients and to use the developed pharmacokinetic model to quantify the effects of patient and disease covariates on methotrexate distribution and elimination in cancer patients.
2 Methods
We employed a population pharmacokinetic modeling methodology using data collected from retrospective chart review of patient medical records. All study procedures were pre-approved by Qatar University and Hamad Medical Corporation institutional review boards.
2.1 Patients
Patient records were identified using pharmacy records for prescriptions of methotrexate between the dates of February 1, 2011 through February 1, 2014. Patient records were included in the study if there was at least one measured methotrexate serum concentration with corresponding sample collection time and dosage information (administered dose and administration time) during the study period. Only patients who received intravenous (IV) methotrexate were included in the study. No other inclusion or exclusion criteria were applied.
2.2 Procedures
A data extraction form was developed based on a comprehensive literature review of factors known to influence methotrexate pharmacokinetics and availability of data in NCCCR patient medical records. Methotrexate dosing information, methotrexate serum concentrations, and dosing administration and sample collection time records were collected for each patient within the pre-specified data collection period. Patient and disease factors to be included in the pharmacokinetic modeling analysis were collected including age, country of origin, gender, weight, height, body surface area, cancer type, concurrently administered medications, and documented comorbidities. In addition, laboratory test results were collected including bilirubin, hemoglobin, hematocrit, renal function tests (serum creatinine and blood urea nitrogen), and liver enzymes (Aspartate Transaminase “AST”, Alanine Transaminase “ALT”, and Alkaline Phosphatase “ALP”).
After identification of eligible patient records, electronic health records as well as paper charts were reviewed for collection of study-relevant data. At least two investigators reviewed each patient record to minimize data collection errors.
2.3 Population Pharmacokinetic Modeling Analysis
Population pharmacokinetic modeling was conducted using methotrexate dosing and serum concentration data. Most patients (n = 33/37) included in our study were administered methotrexate on multiple occasions (2–7 occasions) and data from all occasions were included in the analysis. Methotrexate doses ranged from 0.5 to 7 g/m2 infused over 4–6 h (n = 10) or 24 h (n = 27). A total of 530 samples were available with an average of 13 samples per patient. Samples were generally collected every 12 or 24 h after the methotrexate infusion until the first sample with methotrexate concentration below the limit of quantification. Samples with methotrexate concentrations below the limit of quantification (0.05 µM) were considered missing in this analysis. In addition, two more methotrexate concentrations were excluded from the analysis because they were considered physiologically implausible: a 4 h post-dose (2.9 g/m2) concentration of 246 µM for one subject and a 48 h post-dose concentration of 3 µM for another subject who had 36 and 60 h post-dose concentrations of 0.48 and 0.11 µM, respectively.
NONMEM VII® (Icon Development Solutions, Ellicott City, MD, USA) software was used to develop the population pharmacokinetic model using a nonlinear mixed effects modeling approach with the FOCE INTERACTION estimation routine. Model development process involved choice of structural model, inclusion of inter-individual variability (IIV), inter-occasion variability (IOV), and residual unexplained variability (RUV) terms, and finally testing of covariate effects on methotrexate pharmacokinetic parameters.
2.3.1 Model Development
One- and two-compartment models were compared to determine the most appropriate structural pharmacokinetic model to describe methotrexate disposition following IV administration. The random effects model was then developed through inclusion of IIV and IOV terms using an exponential model (Eq. 1), and RUV using a proportional error model (Eq. 2).
where P i is the true parameter value for individual i, \(\hat{P}\) is the typical value (population mean) of the parameter, η p i is the difference between the true value for individual i and the typical value for the population and is independently, identically distributed with a mean of 0 and a variance of ω 2.
where C ij is the jth measured concentration in individual i, \(\hat{C}_{ij}\) is the jth model-predicted concentration in individual i, ɛ ij is the proportional residual intra-individual error for the jth measurement in individual i, with a mean of 0 and a variance of σ 2.
Several approaches were evaluated for inclusion of IOV terms in the random effects model. The tested approaches included: a single IOV term for all visits after visit 1, a separate IOV term for each visit after visit 1, an IOV term for only one of the visits after visit 1 (i.e., visit 2 only, visit 3 only, etc.), as well as similar or different IOV terms for visits 2 and 3 only (visits with largest number of subjects). Performance of these alternative models was compared against that of the model without any IOV terms using the objective function value (OFV).
The developed mixed effects model (base model) was then used to evaluate and quantify the effects of different patient and disease covariates on methotrexate pharmacokinetic parameters and IIV/IOV terms. Covariate testing and inclusion in the final model was performed using forward selection–backward elimination process. The likelihood ratio test was used for hypothesis testing to discriminate among alternative nested models for covariate testing, with the differences between OFV used to guide model building.
The stepwise inclusion of covariates involved testing the effect of each covariate on all appropriate model parameters (CL and/or V c) in separate model runs. The covariates that were found to be significant (p < 0.01, drop in OFV of ≥6.64) were included in a model for further evaluation. The backward elimination process was then started by comparing the starting model to a model without the particular covariate–parameter relationship. If insignificant covariates (p > 0.001, increase in OFV of ≤10.83) were found, the covariate relationship that resulted in the least significant increase in OFV was discarded, and the reduced model was used as the starting model for the next iteration of the backward elimination process. This process was repeated until no insignificant covariates were left in the model.
Continuous covariates were entered into the model using a power function as shown in Eq. 3:
where TVVci is the typical value of methotrexate central volume of distribution for an individual i, θ 1 represents V c for an individual with median body weight, WTKGmedian, and θ 2 is a scaling exponent relating the fixed effect (body weight, WTKG i ) to V c.
Dichotomous covariates (gender, cancer type, origin) were entered into the model using a multiplicative shift factor as shown in in Eq. 4:
where TVCL i is the typical value of methotrexate clearance for individual i, θ 1 represents methotrexate clearance for a male subject, and θ 2 is a shift factor for the change in clearance in female subjects.
2.3.2 Model Evaluation
Throughout the model development process, the model adequacy was judged based on standard evaluation criteria including goodness-of-fit plots (observed concentrations versus individual and population predictions), residual plots (conditional weighted residuals versus time and population predictions), and precision of parameter estimates (relative standard error %) [13].
Additionally, a visual predictive check was used to evaluate the predictive performance of the final pharmacokinetic model. 500 simulated replicates of the dataset were generated using NONMEM®. Subsequently, the simulated predictions were compared to the observed data by superimposing observed concentrations on the median and 95 % prediction interval of simulated methotrexate concentrations.
3 Results
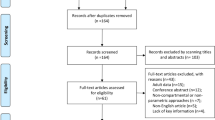
A total of 66 patients were initially identified for inclusion in the study based on administration of one or more methotrexate doses during the predetermined study period. After screening, 37 patient records met the inclusion criteria. Patients were excluded due to intrathecal or intramuscular methotrexate administration (n = 9), lack of dosing information or time (n = 16), and lack of methotrexate concentration records (n = 4). Demographics of patients included in the final analysis are given in Table 1.
3.1 Population Pharmacokinetic Modeling Results
Methotrexate disposition following IV administration was best described by a two-compartment model with first-order distribution and elimination. Methotrexate CL and V c estimates from the final model were 16 L/h and 79 L, respectively. The model estimated an IIV of 35 % (shrinkage: 1.23 %) and 63 % (shrinkage: 9.99 %) for methotrexate CL and V p (volume of distribution for peripheral compartment), respectively.
Most patients (33/37) included in the analysis administered methotrexate IV dosing on more than once occasion with doses ranging from 0.5 to 7 g/m2. Data from multiple patient visits were used to estimate IOV in methotrexate CL and/or V c. The final model included IOV of 48 % (shrinkage: 13.8 %) and 31 % (shrinkage: 25.7 %) on methotrexate CL for the second and third dosing occasions, respectively. Additional IOV terms for subsequent patient visits or for other methotrexate pharmacokinetic parameters did not improve the model or result in statistically significant changes in OFV. Similarly, inclusion of an equal IOV parameter for all dosing occasions (or for occasions 2 and 3) after the first visit resulted in worse model performance compared to the current model.
The final model was used to evaluate the effects of patient covariates on methotrexate CL and V c. Patient weight and hematocrit level were statistically significant covariates on methotrexate V c and CL, respectively (p < 0.001 for each). Inclusion of hematocrit level as a predictor of methotrexate CL resulted in 3 % decrease in IIV as well as IOV terms for CL. The relationship between hematocrit level and estimated methotrexate CL showed a slight increase in CL with the increase in hematocrit level (Fig. 1). Addition of IIV on methotrexate V c did not improve model predictions or result in significant drop in OFV. All model parameters were estimated with good precision [Relative standard error <30 % except for peripheral volume of distribution (V p) and distribution clearance (CLd)]. Final model parameter estimates are summarized in Table 2.

Relationship between individual hematocrit levels and methotrexate clearance estimates from population pharmacokinetic base model
3.1.1 Model Evaluation
The goodness-of-fit for the final model was evaluated graphically. The goodness-of-fit plots of the observed methotrexate concentrations versus individual- and population-predicted concentrations were randomly distributed across the line of identity and indicated that the model adequately described the observations over the entire methotrexate concentration range (Fig. 2). Similarly, the conditional weighted residuals for the final model did not show any major trends when plotted versus either time or population-predicted concentrations, suggesting that the model was appropriately unbiased (Fig. 3).

Goodness-of-fit plots of observed versus model-predicted log-transformed methotrexate concentrations (a Population predictions, b Individual predictions)

Conditional weighted residuals versus time (a) and population predictions (b)
Based on 500 simulations, the VPC for methotrexate serum concentrations versus time showed that the model adequately described the central tendency and variability of the observed data without marked systematic deviations as shown in Fig. 4.

Prediction-corrected visual predictive check on log scale
4 Discussion
To our knowledge, this is the first population pharmacokinetic study of methotrexate using retrospective chart review data in cancer patients in the Middle East. Nonlinear mixed effects modeling approach was used to quantify methotrexate pharmacokinetics in this patient population and determine patient and disease factors affecting methotrexate disposition.
Methotrexate CL estimated in our study (16 L/h) was slightly higher compared to that estimated in previous population pharmacokinetic studies in non-Middle Eastern populations (4–14 L/h) [5–8, 14, 16]. Several factors (e.g., age, weight, CLCR, genetic polymorphisms) may account for the observed differences. However, such a difference may not be attributed to differences in renal function between patients included in our study and those included in previous studies. Calculated creatinine clearance in patients included in our study ranged from 9 to 98 mL/min (median: 30 mL/min) which covers a wide range of renal function levels. Additionally, the age and body weight ranges for subjects included in our study are comparable to those reported for subjects in previous studies reporting lower estimates for methotrexate systemic clearance. Hence, future larger prospective studies may be needed to evaluate factors (e.g., genetic polymorphisms) contributing to the higher estimated methotrexate clearance in this Middle Eastern population.
Despite the inclusion of patients with a wide range of estimated glomerular filtration rate, serum creatinine or creatinine clearance (CLCR) was not identified as significant covariates on methotrexate CL in our model. Given the high precision (RSE: 17 %) in CL estimate and the very low eta-shrinkage for IIV on CL (1.23 %), the observation of no significant effect of CLCR on methotrexate CL may be a true finding. Although, previous population pharmacokinetic studies of methotrexate in cancer patients have reported significant effects of CLCR or serum creatinine on methotrexate CL [5, 7, 8, 14–17], the effects were mostly minimal. For example, Dupuis et al. reported only 10 % difference in methotrexate CL for patients with creatinine clearance above or below 89 mL/min [15]. Similarly, Fukuhara et al. and Johansson et al. reported only 16 and 17 % decrease in methotrexate CL (for the drop in CLCR from 100 to 20 mL/min or increase in serum creatinine from 0.3 to 3 mg/dL), respectively [5, 16].
Hematocrit level was determined to be a significant covariate on methotrexate CL in our model. To our knowledge, this is the first report of the effect of hematocrit on methotrexate CL. Dupuis et al. have previously reported an effect of hemoglobin level on methotrexate volume of distribution with only 3 % change between patients with hemoglobin levels above and below 13.45 g/dL [15]. Hemoglobin was initially detected as a significant covariate on methotrexate CL in the forward selection process in our model. However, due to its high correlation with hematocrit levels, it was not retained in the model following inclusion of hematocrit effect on methotrexate CL.
According to the estimated hematocrit–CL covariate relationship, a 50 % difference in methotrexate CL is predicted for patients with lowest and highest hematocrit levels in our study (23 and 50 %, respectively). Such a difference in methotrexate CL may have clinically important effects on methotrexate exposure and response to treatment. Differences in hematocrit levels between patients or changes within the same patient during the course of treatment may require dose adjustments to account for differences in methotrexate CL.
The effect of hematocrit on methotrexate CL may be a result of methotrexate distribution into red blood cells; resulting in decreased serum concentrations and a higher estimated CL. Methotrexate is known to be transported into cells through the reduced folate carrier as well as folate receptors α and β and methotrexate metabolite concentrations in red blood cells have been linked to treatment outcomes in rheumatoid arthritis patients [18]. Similar results have been reported before for the effects of hematocrit levels on tacrolimus pharmacokinetics (also known to distribute to blood cells). In 2004, Minematsu et al. showed that hematocrit levels affect tacrolimus pharmacokinetics and recommended that such effects should be taken into consideration upon dosing tacrolimus to transplant patients [19].
Patient body weight was found to be a significant covariate in our model on methotrexate V c. Similar effects of body size on methotrexate volume of distribution have been reported before [8, 15]. The estimated covariate relationship for patient weight and V c results in 2.7-fold difference in V c between patients with lowest and highest body weights enrolled in our study (50 and 109 kg, respectively).
In addition to estimation of IIV and RUV in methotrexate pharmacokinetics, our population pharmacokinetic model included IOV terms on methotrexate CL. Our final model estimated 47 and 31 % IOV in methotrexate CL for the second and third patient visits, respectively. Previous studies have also reported IOV on methotrexate CL with estimates ranging from 12 to 17 % [7, 14, 16]. Although most patients included in our study administered methotrexate on multiple occasions (n = 33/37), only a few administered the drug on more than 3 occasions (4 occasions: 15/37, ≥5 occasions: 4/37). Consequently, inclusion of additional IOV terms for subsequent visits beyond the third visit (as similar or different from IOV for visit 3) did not result in significant model improvement or markedly better prediction of observed concentrations.
Individualized medicine is trending as a popular concept worldwide. While standard empirical dosing is likely sufficient for the majority of medications, our results show that methotrexate pharmacokinetics is dependent on patient factors and that therapeutic drug monitoring should remain as an important strategy to provide further information regarding pharmacokinetics in special populations. Future studies in the Middle Eastern population should include relevant clinical outcomes and should be designed to further investigate patient and disease factors that may influence clinical efficacy of methotrexate through impact on drug exposure. This concept provides a great opportunity for clinical pharmacists and other healthcare professionals to work collaboratively with an aim of improving dosing regimens to optimize outcomes achieved.
Some limitations of our study should be noted. Firstly, the retrospective nature of the study enabled only collection of data available in electronic and paper medical records. For example, the impact of genetic polymorphisms in drug transporters previously reported to impact methotrexate disposition could not be studied due to lack of such information. In addition, assessment of the impact of ethnicity on methotrexate pharmacokinetics was not performed due to lack of such information in patient medical records at the time of data collection. However, the country of origin was tested as a covariate in the population pharmacokinetic model and did not result in statistically significant effects on methotrexate pharmacokinetics. This can be partially explained by the fact that most of subjects included in the study (24/37) were from the same geographical area. Secondly, our study did not include assessment of methotrexate exposure–response relationship in cancer patients in Qatar. Although such analysis was initially planned, it was determined to be unfeasible due to lack of reliable clinical outcome data in patient medical records. Thirdly, our study relied on routine clinical monitoring practices at the NCCCR in Qatar; which does not include measurement of methotrexate metabolite concentrations. Inclusion of methotrexate metabolite information in the population pharmacokinetic model could provide more insight into methotrexate disposition. However, it is important to note that the main objective of our study was to demonstrate usefulness of routine clinical monitoring parameters rather than requiring additional measurements or sampling times in addition to what is already being done in hospital protocols. Lastly, the sample size included in our final analysis was relatively small due to several exclusions for lack of complete data. While the sample size was small, the subjects included in our analysis provide good representation of the general population in this area over the specific time period. The current study serves as an exploratory and hypothesis generating analysis that calls for future studies to further assess potential differences in methotrexate pharmacokinetics between the Middle Eastern population and other previously studied patient populations.
5 Conclusion
In conclusion, our study demonstrates the usefulness of routinely collected TDM data together with pharmacokinetic analyses to provide better understanding of methotrexate pharmacokinetics in cancer patients and identify clinically important factors affecting methotrexate exposure. Optimization of methotrexate dosing and clinical outcomes is an emerging role for clinical pharmacists that should be further explored in future prospective studies.
References
Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363(4):301–4. doi:10.1056/NEJMp1006304.
Durant J, Clevenbergh P, Halfon P, Delgiudice P, Porsin S, Simonet P, et al. Drug-resistance genotyping in HIV-1 therapy: the VIRADAPT randomised controlled trial. Lancet. 1999;353(9171):2195–9.
Schumacher GE, Barr JT. Therapeutic drug monitoring: do the improved outcomes justify the costs? Clin Pharmacokinet. 2001;40(6):405–9. doi:10.2165/00003088-200140060-00002.
Holmboe L, Andersen AM, Morkrid L, Slordal L, Hall KS. High dose methotrexate chemotherapy: pharmacokinetics, folate and toxicity in osteosarcoma patients. Br J Clin Pharmacol. 2012;73(1):106–14. doi:10.1111/j.1365-2125.2011.04054.x.
Fukuhara K, Ikawa K, Morikawa N, Kumagai K. Population pharmacokinetics of high-dose methotrexate in Japanese adult patients with malignancies: a concurrent analysis of the serum and urine concentration data. J Clin Pharm Ther. 2008;33(6):677–84. doi:10.1111/j.1365-2710.2008.00966.x.
Nagulu M, Kiran VU, Nalini Y, Reddy YN, Krishna DR. Population pharmacokinetics of methotrexate in Indian cancer patients. Asian Pac J Cancer Prev. 2010;11(2):403–7.
Ruhs H, Becker A, Drescher A, Panetta JC, Pui CH, Relling MV, et al. Population PK/PD model of homocysteine concentrations after high-dose methotrexate treatment in patients with acute lymphoblastic leukemia. PLoS One. 2012;7(9):e46015. doi:10.1371/journal.pone.0046015.
Min Y, Qiang F, Peng L, Zhu Z. High dose methotrexate population pharmacokinetics and Bayesian estimation in patients with lymphoid malignancy. Biopharm Drug Dispos. 2009;30(8):437–47. doi:10.1002/bdd.678.
Aouinti I, Gaies E, Trabelsi S, Salouage I, Jebabli N, Charfi R, et al. Delayed elimination of methotrexate in a patient receiving ciprofloxacin. Therapie. 2013;68(3):175–7. doi:10.2515/therapie/2013031.
Frenia ML, Long KS. Methotrexate and nonsteroidal antiinflammatory drug interactions. Ann Pharmacother. 1992;26(2):234–7.
van de Steeg E, van Esch A, Wagenaar E, Kenworthy KE, Schinkel AH. Influence of human OATP1B1, OATP1B3, and OATP1A2 on the pharmacokinetics of methotrexate and paclitaxel in humanized transgenic mice. Clin Cancer Res. 2013;19(4):821–32. doi:10.1158/1078-0432.CCR-12-2080.
Simon N, Marsot A, Villard E, Choquet S, Khe HX, Zahr N, et al. Impact of ABCC2 polymorphisms on high-dose methotrexate pharmacokinetics in patients with lymphoid malignancy. Pharmacogenomics J. 2013;13(6):507–13. doi:10.1038/tpj.2012.37.
Beal S, Sheiner L, Boeckmann A, et al. NONMEM User’s Guide. Ellicott City: Icon Development Solutions; 1989–2009.
Joerger M, Huitema AD, van den Bongard HJ, Baas P, Schornagel JH, Schellens JH, et al. Determinants of the elimination of methotrexate and 7-hydroxy-methotrexate following high-dose infusional therapy to cancer patients. Br J Clin Pharmacol. 2006;62(1):71–80. doi:10.1111/j.1365-2125.2005.02513.x.
Dupuis C, Mercier C, Yang C, Monjanel-Mouterde S, Ciccolini J, Fanciullino R, et al. High-dose methotrexate in adults with osteosarcoma: a population pharmacokinetics study and validation of a new limited sampling strategy. Anticancer Drugs. 2008;19(3):267–73.
Johansson AM, Hill N, Perisoglou M, Whelan J, Karlsson MO, Standing JF. A population pharmacokinetic/pharmacodynamic model of methotrexate and mucositis scores in osteosarcoma. Ther Drug Monit. 2011;33(6):711–8. doi:10.1097/FTD.0b013e31823615e1.
Mao J, Zhang L, Shen H, Tang Y, Song H, Zhao F, et al. Creatinine clearance rate and serum creatinine concentration are related to delayed methotrexate elimination in children with lymphoblastic malignancies. Neoplasma. 2013;. doi:10.4149/neo_2014_011.
Pan S, Stamp LK, Duffull SB, Barclay ML, Dalrymple JM, Drake J, et al. Assessment of the relationship between methotrexate polyglutamates in red blood cells and clinical response in patients commencing methotrexate for rheumatoid arthritis. Clin Pharmacokinet. 2014;53(12):1161–70. doi:10.1007/s40262-014-0179-5.
Minematsu T, Sugiyama E, Kusama M, Hori S, Yamada Y, Ohtani H, et al. Effect of hematocrit on pharmacokinetics of tacrolimus in adult living donor liver transplant recipients. Transplant Proc. 2004;36(5):1506–11. doi:10.1016/j.transproceed.2004.04.097.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by a UREP award [UREP15-019-3-008] from the Qatar National Research Fund (a member of The Qatar Foundation).
Conflict of interest
Dr. Ahmed Nader, Noran Zahran, Aya Alshammaa, Heba Altaweel, Nancy Kassem and Dr. Kyle John Wilby have no conflicts of interest to disclose.
Ethical approval
All procedures in this study were in accordance with the 1964 Helsinki declaration (and its amendments) and were approved by Qatar University and Hamad Medical Corporation institutional review boards.
Informed consent
No informed consent was required for this study given the study design (retrospective chart review) and the reporting of de-identified patient data.
Rights and permissions
About this article

Cite this article
Nader, A., Zahran, N., Alshammaa, A. et al. Population Pharmacokinetics of Intravenous Methotrexate in Patients with Hematological Malignancies: Utilization of Routine Clinical Monitoring Parameters. Eur J Drug Metab Pharmacokinet 42, 221–228 (2017). https://doi.org/10.1007/s13318-016-0338-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-016-0338-1