
Abstract
Background and Objectives
Triptolide is an active component derived from Tripterygium wilfordii and it possesses numerous pharmacological activities. However, it remains unclear how triptolide influences the activity of human liver cytochrome P450 (CYP) enzymes and P-glycoprotein (P-gp).
Methods
In this study, the inhibitory effects of triptolide on the eight human liver CYP isoforms (i.e., 1A2, 3A4, 2A6, 2E1, 2D6, 2C9, 2C19, and 2C8) were investigated in vitro using human liver microsomes (HLMs), and the effects of triptolide on the activity of P-gp were investigated using a rhodamine-123 uptake assay.
Results
The results showed that triptolide inhibited the activity of CYP1A2 and CYP3A4, with 50 % inhibitory concentration (IC50) values of 14.18 and 8.36 μM, respectively, but that other CYP isoforms were not affected. Enzyme kinetic studies showed that triptolide was not only a non-competitive inhibitor of CYP1A2, but also a competitive inhibitor of CYP3A4, with inhibition constant (K i) values of 7.32 and 5.67 μM, respectively. In addition, triptolide is a time-dependent inhibitor for CYP1A2, and the concentration at 50 % maximum inactivation (K I) and maximum inactivation (K inact) values were 286.5 μM and 0.024 min−1, respectively. The rhodamine-123 uptake assay showed that triptolide could not affect the activity of P-gp.
Conclusions
The in vitro studies of triptolide with CYP isoforms and P-gp indicate that triptolide has the potential to cause pharmacokinetic drug interactions with other co-administered drugs metabolized by CYP1A2 and CYP3A4. Further clinical studies are needed to evaluate the significance of this interaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
First, the effects of triptolide on the activity of CYP enzymes were investigated. |
Then, the effects of triptolide on the activity of P-gp were investigated. |
Triptolide did inhibit the activity of CYP1A2 and CYP3A4. |
1 Introduction
Triptolide (Fig. 1), a diterpenoid triepoxide, is a major pharmacological component isolated from Tripterygium wilfordii Hook F [1, 2]. Triptolide has been primarily used to treat inflammatory and autoimmune diseases, such as rheumatoid arthritis and systemic lupus erythematosus [3, 4]. Recently, some studies have indicated that triptolide can influence several anti-tumor target genes and inhibit tumors by altering multiple signaling pathways [5–7].

Chemical structure of triptolide
Cytochrome P450 (CYP) enzymes, a superfamily of heme-containing isoenzymes located primarily in hepatocytes, are important phase I enzymes in the biotransformation of xenobiotics, which include drugs, environmental pollutants, carcinogens and endogenous substrates [8, 9]. CYP1A, CYP2C, CYP2D, CYP3A and CYP2E are major CYP enzymes in drug metabolism [10]. Most CYP enzymes can be inhibited or induced by a variety of drugs and chemicals that can give rise to toxicity or treatment failure, and many drug–drug interactions have been generated from the concurrent use of herbal prescription and over-the-counter drugs [11]. St. John’s wort, ginkgo, ginseng, and licorice have all been reported to interact with anticoagulants, antiretroviral drugs, anticancer drugs, immunosuppressants, or antidepressants [12–15]. However, undesired drug–drug interactions can also be caused by intestine membrane transporters [16]. Some drug interactions, which are previously believed to be mediated by CYP enzymes, are now considered to be at least partially due to the inhibition of membrane transport proteins [17]. For example, in the case of talinolol, which undergoes little metabolism, the increased bioavailability in the presence of verapamil is most likely due to the inhibition of P-glycoprotein (P-gp) by verapamil [18]. Therefore, the effects of triptolide on the activity of CYP enzymes and P-gp should be investigated.
Previous studies have indicated that triptolide can be metabolized by CYP3A4 and CYP2C19 in rats and humans [19, 20]. However, to the best of our knowledge, few studies have investigated the effects of triptolide on CYP enzymes, particularly the inhibitory effects, which will increase the risk of therapeutic applications of triptolide and its medical preparations. Several research articles have indicated that the transport of triptolide in the small intestines was mediated by P-gp, and triptolide was a substrate of P-gp [21, 22]. However, little data are available concerning the effects of triptolide on the activity of P-gp, particularly whether triptolide can inhibit or induce the activity of P-gp.
The aim of this study was to investigate the effects of triptolide on eight major CYP isoforms in human liver microsomes (HLMs) and P-gp using a rhodamine-123 uptake assay. In vitro, phenacetin (CYP1A2), testosterone (CYP3A4), coumarin (CYP2A6), chlorzoxazone (CYP2E1), dextromethorphan (CYP2D6), diclofenac (CYP2C9), S-mephenytoin (CYP2C19) and paclitaxel (CYP2C8) were used as probe substrates to determine the effects of triptolide on eight CYP enzymes. Enzyme kinetic studies were also conducted to determine the inhibition mode of triptolide on CYP enzymes. Additionally, the effects of triptolide on the activity of P-gp were investigated using a rhodamine-123 uptake assay.
2 Materials and Methods
2.1 Chemicals
Triptolide (≥98 %) and testosterone (≥98 %) were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). d-glucose-6-phosphate, glucose-6-phosphate dehydrogenase, corticosterone, NADP+, phenacetin, acetaminophen, 4-hydroxymephenytoin, 7-hydroxycoumarin, 4′-hydroxydiclofenac, sulfaphenazole, quinidine, tranylcypromine, chlorzoxazone, 6-hydroxychlorzoxazone, paclitaxel, 6β-hydroxytestosterone, clomethiazole, furafylline, and rhodamine-123 were obtained from Sigma Chemical Co. (St. Louis, MO, USA). Montelukast was obtained from Beijing Aleznova Pharmaceutical (Beijing, China). Coumarin, diclofenac, dextromethorphan, and ketoconazole were purchased from ICN Biomedicals (Aurora, OH, USA). Pooled HLMs were purchased from BD Biosciences Discovery Labware (Woburn, MA, USA). The Alkaline Phosphatase Assay Kit was provided by Nanjing Jiancheng Bioengineering Institute (Nanjing, China). All other reagents and solvents were of analytical reagent grade.
Dulbecco’s modified Eagle’s medium (DMEM) and non-essential amino acid (NEAA) solution were purchased from Thermo Scientific Corp. (Logan, UT, USA). Fetal bovine serum (FBS) was obtained from GIBCO BRL (Grand Island, NY, US). Penicillin G (10,000 U/ml) and Streptomycin (10 mg/ml) were purchased from Amresco (Solon, OH, USA). Hanks’ balanced salt solution (HBSS) was purchased from GIBCO (Grand Island, NY, USA).
2.2 Assay with HLM
As shown in Table 1, to investigate the inhibitory effects of triptolide on different CYP isoforms in HLM, the following probe reactions were used, according to previously described method [11, 23]: phenacetin O-deethylation for CYP1A2, testosterone 6β-hydroxylation for CYP3A4, coumarin 7-hydroxylation for CYP2A6, chlorzoxazone 6-hydroxylation for CYP2E1, dextromethorphan O-demethylation for CYP2D6, diclofenac 4′-hydroxylation for CYP2C9, S-Mephenytoin 4-hydroxylation for CYP2C19, and paclitaxel 6α-hydroxylation for CYP2C8. All incubations were performed in triplicate, and the mean values were utilized. The typical incubation systems contained 100 mM potassium phosphate buffer (pH 7.4), NADPH-generating system (1 mM NADP+, 10 mM glucose-6-phosphate, 1 U/mL of glucose-6-phosphate dehydrogenase, and 4 mM MgCl2), the appropriate concentration of HLM, a corresponding probe substrate and triptolide (or positive inhibitor for different probe reactions) in a final volume of 200 μL.
The concentration of triptolide was 100 μM, and the positive inhibitor concentrations were as follows: 10 μM furafylline for CYP1A2, 1 μM ketoconazole for CYP3A4, 10 μM tranylcypromine for CYP2A6, 50 μM clomethiazole for CYP2E1, 10 μM quinidine for CYP2D6, 10 μM sulfaphenazole for CYP2C9, 50 μM tranylcypromine for CYP2C19, 5 μM montelukast for CYP2C8. Probe substrates, positive inhibitors (except for dextromethorphan and quinidine which were dissolved in water) and triptolide were dissolved in methanol, with a final concentration of 1 % (v/v), and 1 % neat methanol was added to the incubations without inhibitor. The final microsomal protein concentration and incubation times for the different probe reactions are shown in Table 1 [23]. There was a 3-min preincubation period (at 37 °C) before the reaction was initiated by adding a NADPH-generating system. The reaction was terminated by adding a 100 μL acetonitrile (10 % trichloroacetic acid for CYP2A6) internal standard mix, and the solution was placed on ice. The mixture was centrifuged at 12,000 rpm for 10 min, and an aliquot (50 μL) of supernatant was transferred for HPLC analysis. The instrument used in this study was Agilent 1260 series instrument with DAD and FLD detector, and the quantitative assay for the corresponding metabolites was performed as previously reported [23], and the method is shown in Table 2. The concentration range of quantification for corresponding metabolites was as follows: phenacetin (0.027–27 μg/mL), testosterone (0.254–25.4 μg/mL), coumarin (0.015–15 μg/mL) chlorzoxazone (0.052–26 μg/mL), dextromethorphan (0.031–31 μg/mL), diclofenac (0.075–37.5 μg/mL), S-Mephenytoin (0.046–23 μg/mL), and paclitaxel (0.068–34 μg/mL). The accuracy and precision were all acceptable for determination (≤15 %).
2.3 Enzyme Inhibition and Kinetic Studies of Triptolide
A 100 μM triptolide was used to initially screen for its direct inhibitory effects toward different human CYP isoforms. For the CYP isoforms whose activities were strongly inhibited, secondary studies were performed to obtain the half inhibition concentration (IC50). Inhibition constant (K i) values were obtained by incubating various concentrations of different probe substrates (20–100 μM phenacetin, or 20–100 μM testosterone) in the presence of 0–30 μM triptolide.
2.4 Time-Dependent Inhibition Study of Triptolide
To determine whether triptolide could inhibit the activity of CYP3A4 or CYP1A2 in a time-dependent manner, triptolide (10 μM) was pre-incubated with HLMs (1 mg/mL) in the presence of an NADPH-generating system for 30 min at 37 °C. After incubation, an aliquot (20 μL) was transferred to another incubation tube (final volume 200 μL) containing an NADPH-generating system and probe substrates whose final concentrations were approximate to concentration at half the maximum velocity (K m). Then, further incubations were performed to measure the residual activity. After being incubated for 10 and 30 min, the reactions were terminated by adding a 100 μL acetonitrile internal standard mix and then placed on ice; the corresponding metabolites were determined by HPLC.
To determine the concentration at 50 % maximum inactivation (K I) and maximum inactivation (k inact) values for the inactivation of CYP1A2, the incubations were conducted using higher probe substrate concentrations (approximately fourfold K m values) and various concentrations of triptolide (0–50 μM) after different preincubation times (0–30 min), with a two-step incubation scheme, as described above. The K I and K inact values were calculated as described by Qi et al. [11].
2.5 Cell Culture
The Caco-2 cell line was provided by the American Type Culture Collection (Manassas, VA, USA). The Caco-2 cells were cultured in DMEM high glucose medium containing 15 % FBS, 1 % NEAA and 100 U/mL penicillin and streptomycin. The cells were grown at 37 °C with 5 % CO2.
2.6 Measurement of Cellular Accumulation of Rhodamine 123 in Caco-2 Cell
The accumulation of rhodamine 123, a fluorescent substrate of P-gp, was measured, and the effects of triptolide were determined, as previously described [24]. Briefly, Caco-2 cells (plated at 1 × 105 cells/well in 24-well plates) were incubated with 20 μM rhodamine 123 in the absence or presence of triptolide (50 μM) for 1 h in a CO2 incubator at 37 °C. After the incubation, the medium was removed through aspiration, and the cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed with 0.1 % triton-X100 in PBS. Fluorescence intensity was measured with a microplate fluorometer (Fluoroskan Ascent, Thermo Fisher Scientific, Waltham, MA, USA). The excitation and emission wavelengths were 485 and 538 nm, respectively. Protein concentrations were measured with the Lowry method using a Bio-Rad DC protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA), with bovine serum albumin as the standard. Accumulation ratios were calculated using the accumulation of rhodamine 123 in cells incubated without triptolide as a control.
2.7 Statistical Analysis
The enzyme kinetic parameters for the probe reaction were estimated from the best fit line using least-squares linear regression of the inverse substrate concentration versus the inverse velocity (Lineweaver–Burk plots), and the mean values were used to calculate maximum velocity (V max) and K m. Inhibition data from the experiments that were conducted using multiple compound concentrations were represented by Dixon plots, and values were calculated using non-linear regression according to equation 1:
where I is the concentration of the compound, K i is the inhibition constant, S is the concentration of the substrate, and K m is the substrate concentration at half the maximum velocity (V max) of the reaction. The mechanism of the inhibition was inspected using the Lineweaver–Burk plots and the enzyme inhibition models. The data comparison was performed using Student’s t test and performed using IBM SPSS statistics 20 (SPSS Inc. Chicago, IL, USA). IC50 values were determined using Microsoft Excel software (Microsoft Inc, USA).
3 Results
3.1 Inhibition of CYP Activities
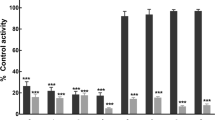
To investigate whether the triptolide affects the catalytic activity of CYP enzymes, the probe reaction assays were conducted with varied concentrations of triptolide. Specific inhibitors of CYP1A2, 3A4, 2A6, 2E1, 2D6, 2C9, 2C19 and 2C8 were used as positive controls. As shown in Fig. 2, triptolide did not inhibit the activities of CYP2A6, 2E1, 2D6, 2C9, 2C19 and 2C8 at a concentration of 100 μM. In contrast, the activities of CYP1A2 and 3A4 were inhibited to 22.4 and 12.7 % of their control activities, respectively.

Inhibition of triptolide on cytochrome P450 (CYP) enzymes in pooled human liver microsomes. All data represent mean (SD). of the triplicate incubations. *P < 0.05, significantly different from the negative control. Negative control: incubation systems without triptolide; Triptolide: incubation systems with triptolide; Positive control: incubation systems with their corresponding positive inhibitors
The enzyme inhibition study showed that inhibition of CYP1A2 (Fig. 3) and CYP3A4 (Fig. 4) by triptolide was concentration dependent, with IC50 values of 14.18 and 8.36 μM, respectively.

Lineweaver–Burk plots (a) and the secondary plot for K i (b) of inhibition of triptolide on CYP1A2 catalyzed reactions (phenacetin O-deethylation) in pooled HLM. Data are obtained from a 30 min incubation with phenacetin (20–100 μM) in the absence or presence of triptolide (0–20 μM). The data represent the mean ± SD of the incubations (performed in triplicate). CYP cytochrome P450, HLM human liver microsomes, K i inhibition constant

Lineweaver–Burk plots (a) and the secondary plot for K i (b) of inhibition of triptolide on CYP3A4 catalyzed reactions (testosterone 6β-hydroxylation) in pooled HLM. Data are obtained from a 30 min incubation with phenacetin (20–100 μM) in the absence or presence of triptolide (0–30 μM). All data represent the mean ± SD of the incubations (performed in triplicate). CYP cytochrome P450, HLM human liver microsomes, K i inhibition constant
Lineweaver–Burk plots of inhibitory kinetic data suggested that the inhibition of CYP1A2 by triptolide was best fit in a non-competitive manner (Fig. 3a), whereas the inhibition of CYP3A4 by triptolide was best fit in a competitive manner (Fig. 4a). The K i values of triptolide on CYP1A2 (Fig. 3b) and CYP3A4 (Fig. 4b) were obtained from the secondary Lineweaver–Burk plot for K i, with values of 7.32 and 5.67 μM, respectively.
3.2 Time-Dependent Inhibition Study
As shown in Fig. 5, after preincubation of triptolide with HLM for 30 min, the activity of CYP1A2 decreased with the incubation time; however, the activity of CYP3A4 was not affected. To characterize the time-dependent inhibition of CYP1A2 by triptolide, inactivation parameters of K I and K inact values were calculated using non-linear regression analysis in HLM. As calculated from the inactivation plot of Fig. 6, the K I and K inact values for CYP1A2 were 286.5 μM and 0.024 min−1, respectively. The K inact values imply that approximately 2.4 % of CYP1A2 is inactivated each minute when a saturating concentration of triptolide is incubated with HLM.

Time-dependent inhibition investigations of CYP1A2 and CYP3A4 catalyzed reactions by triptolide (10 μM). All data represent the mean ± SD of the incubations (performed in triplicate). CYP cytochrome P450

Time and concentration inactivation of microsomal CYP1A2 activity by triptolide in the presence of NADPH. The initial rate constant of inactivation of CYP1A2 by each concentration (K obs) was determined through linear regression analysis of the natural logarithm of the percentage of remaining activity versus preincubation time (a). The K I and K inact values were determined through non-linear analysis of the K obs versus the triptolide concentration (b). CYP cytochrome P450, K I concentration at 50 % maximum inactivation, K inact maximum inactivation
3.3 Effects of Triptolide on the Accumulation of Rhodamine-123 in Caco-2 cells
Figure 7 shows the accumulation of rhodamine-123 in Caco-2 cell in the presence of 50 μM of the triptolide. Verapamil, which is a known inhibitor of P-gp, increased the cellular accumulation of rhodamine-123. However, triptolide could not increase the cellular accumulation of rhodamine-123, which indicates that triptolide could not affect the P-gp-mediated efflux of rhodamine-123, and it had little influence on the activity of P-gp.

Effects of the triptolide (50 μM) on the accumulation of rhodamine-123 in Caco-2 cells. Each column represents the mean (SD). of six experiments. *P < 0.05, significantly different from control
4 Discussion
In clinical practice, two or more drugs are often concurrently administered to a patient to treat diseases; therefore, a patient may be at a risk of adverse drug–drug interactions (DDIs) [25]. It is common knowledge that DDIs are often caused by one drug inhibiting the metabolism of another drug, which leads to an increased plasma concentration. For example, ketoconazole, an antifungal agent, is a potent inhibitor of CYP3A4; when ketoconazole is co-administered with drugs that are substrates of CYP3A4, the metabolism of the drug will be inhibited, the plasma concentration of the drug will increase, and adverse DDIs might occur [26, 27]. For drugs with narrow therapeutic index, adverse DDIs might occur; therefore, it is important to investigate the drug–drug interaction potentials of some common drugs.
CYP enzymes, particularly CYP1A2, 3A4, 2A6, 2E1, 2D6, 2C9, 2C19 and 2C8, are responsible for most drug metabolism in humans [8]. The inhibition of these enzymes by co-administered drugs has led to the removal of several drugs from the market; therefore, in drug development, it is necessary to study the inhibition of CYP enzymes to evaluate the DDI because the inhibition of CYP enzymes is one of the most common causes of adverse DDIs [28]. To the best of our knowledge, this study is the first to investigate the effects of triptolide on the metabolism of probe substrates of several CYP isoforms, including CYP1A2, 3A4, 2A6, 2E1, 2D6, 2C9, 2C19 and 2C8.
CYP1A2 is one of the major CYP enzymes in the human liver, which accounting for approximately 13 % of the total content of this enzyme group [29]. CYP1A2 plays an important role in the metabolism of several clinically used drugs and foodborne procarcinogens [30, 31]. Our study showed that triptolide noncompetitively inhibited human liver microsomal CYP1A2 activity, with K i and IC50 values of 7.32 and 14.18 μM, respectively. The results also indicated that triptolide is a time-dependent inhibitor for CYP1A2, and the K I and K inact values were 286.5 μM and 0.024 min−1, respectively. These results indicated that triptolide could inhibit the activity of CYP1A2, and therefore, triptolide should be used carefully with drugs metabolized by CYP1A2 such as clozapine, theophylline, and amitriptyline to avoid possible drug interactions [32]. As triptolide was a weak CYP1A2 inhibitor, and the potential of drug–drug interaction with CYP1A2 would be low.
The CYP3A subfamily is one of the dominant CYP enzymes in the liver and extra-hepatic tissues, such as the intestines, and it plays an important role in the oxidation of xenobiotics and contributes to the biotransformation of approximately 60 % of currently used therapeutic drugs [33]. Human CYP3A4 is one of the most abundant drug-metabolizing CYP isoforms in human liver microsomes, accounting for approximately 40 % of the total CYP enzymes [34]. In fact, characterization of the CYP3A4 isoform responsible for the metabolism of drugs and herbal constituents is important for identifying potential drug–drug or herbdrug interactions in humans. The present study showed that triptolide had inhibitory effects in vitro on CYP3A4 isoform, with K i and IC50 values of 5.67 and 8.36 μM, respectively. Thus, to avoid adverse drug interactions, it is recommended that triptolide should be carefully used with drugs metabolized by CYP3A4. However, triptolide was still a weak CYP3A4 inhibitor, and the potential of drug–drug interaction with CYP3A4 would also be low.
As we know, in vitro data are essential for understanding a potential enzyme inhibition and DDI in vivo. However, an observed in vitro inhibition of a CYP enzyme does not mean that the drug will cause clinically relevant interactions. Many other factors might influence drug interactions mediated by CYP inhibition, including the contribution of the hepatic clearance to the total clearance of the affected drug, the fraction of the hepatic clearance which is subject to metabolic inhibition, and the ratio of the inhibition constant (K i) over the in vivo concentration of the inhibitor [35, 36]. Therefore, further in vivo system studies are needed to identify the interactions of triptolide with CYP isoform in humans.
The results of this study indicate that triptolide may influence the in vitro metabolism of drugs that are substrates of CYP1A2 and CYP3A4. Some research articles have reported that the plasma C max values of triptolide in rats treated with an intravenous dose (0.6 mg/kg) and an oral dose (2.4 mg/kg) were less than 600 ng/mL [37]. Therefore, the rat plasma concentrations of triptolide were much lower than the K i and IC50 values of triptolide determined in this study, and severe drug–drug interaction might not occur if triptolide were co-administered with the substrates of the CYP1A2 and CYP3A4. However, due to the pharmacokinetic differences of human and rats, further in vivo system studies are needed to identify the interactions of triptolide with CYP isoform in humans.
The importance of drug transporters in influencing the pharmacokinetics of orally administered drugs has attracted much attention since the discovery of P-gp in 1976 [38]. Drug transporters played an important role in the absorption, distribution and secretion of drugs in both animals and humans [16, 39]. The pharmacokinetic characteristics of drugs that are substrates of these transporters are expected to be influenced by co-administered drugs that act as inhibitors or enhancers of the transporter function. In this study, we found that triptolide could not affect the activity of P-gp. However, Zhang et al. reported that triptolide was a substrate of P-gp, and therefore, drug–drug interaction might also occur when triptolide was co-administered with P-gp inhibitor or inducer because triptolide has a narrow therapeutic index. Li et al. [40] have also reported that triptolide was a substrate of breast cancer resistance protein (BCRP), and triptolide could also decrease the transcript and protein levels of BCRP in the testis, which indicated that drug–drug interaction might occur when triptolide was co-administered with BCRP inhibitor or inducer or substrate of BCRP.
However, this study had some limitations. First, UDP-glucuronosyltransferases also play an important role in the biotransformation of drugs, while the inhibitory effects of triptolide on the activity of UDP-glucuronosyltransferases were not investigated. Second, other membrane transporters may also be involved in the transport of triptolide, while the effects of triptolide on the activity of other membrane transporters were not investigated in this study. In future research, the effects of triptolide on the activity of UDP-glucuronosyltransferases and other membrane transporters should be investigated in vitro and in vivo.
5 Conclusion
In conclusion, the effects of triptolide on the activity of CYP enzymes and P-gp were systematically investigated. The results showed that triptolide could inhibit the activity of CYP3A4 and CYP1A2, while the activity of other CYP enzymes and P-gp was not affected. The drug–drug interaction of triptolide should be monitored if triptolide is co-administered with CYP3A4 or CYP1A2 substrates.
References
Wei D, Huang Z. Anti-inflammatory effects of triptolide in LPS-induced acute lung injury in mice. Inflammation. 2014;37:1307–16.
Li XJ, Jiang ZZ, Zhang LY. Triptolide: progress on research in pharmacodynamics and toxicology. J Ethnopharmacol. 2014;155:67–79.
Grzegorzewska AE, Frankiewicz D, Breborowicz D, Matlawska I, Bylka W. Disseminated cutaneous Kaposi sarcoma in a patient receiving triptolide/tripdiolide for rheumatoid arthritis. Med Sci Monit. 2012;18:CS67–71.
Liu C, Zhang Y, Kong X, Zhu L, Pang J, Xu Y, et al. Triptolide prevents bone destruction in the collagen-induced arthritis model of rheumatoid arthritis by targeting RANKL/RANK/OPG signal pathway. Evid Based Complement Altern Med. 2013;2013:626038.
Park B. Triptolide, a diterpene, inhibits osteoclastogenesis, induced by RANKL signaling and human cancer cells. Biochimie. 2014;105:129–36.
Zhou J, Xi C, Wang W, Fu X, Jinqiang L, Qiu Y, et al. Triptolide-induced oxidative stress involved with Nrf2 contribute to cardiomyocyte apoptosis through mitochondrial dependent pathways. Toxicol Lett. 2014;230:454–66.
Chen Z, Sangwan V, Banerjee S, Chugh R, Dudeja V, Vickers SM, et al. Triptolide sensitizes pancreatic cancer cells to TRAIL-induced activation of the death receptor pathway. Cancer Lett. 2014;348:156–66.
Wrighton SA, Stevens JC. The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol. 1992;22:1–21.
Yan Z, Caldwell GW. Metabolism profiling, and cytochrome P450 inhibition and induction in drug discovery. Curr Top Med Chem. 2001;1:403–25.
Li AP. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today. 2001;6:357–66.
Qi XY, Liang SC, Ge GB, Liu Y, Dong PP, Zhang JW, et al. Inhibitory effects of sanguinarine on human liver cytochrome P450 enzymes. Food Chem Toxicol. 2013;56:392–7.
Meng Q, Liu K. Pharmacokinetic interactions between herbal medicines and prescribed drugs: focus on drug metabolic enzymes and transporters. Curr Drug Metab. 2014;15:791–807.
Pirotta M, Willis K, Carter M, Forsdike K, Newton D, Gunn J. ‘Less like a drug than a drug’: the use of St John’s wort among people who self-identify as having depression and/or anxiety symptoms. Complement Ther Med. 2014;22:870–6.
Wang ZY, Chen M, Zhu LL, Yu LS, Zeng S, Xiang MX, et al. Pharmacokinetic drug interactions with clopidogrel: updated review and risk management in combination therapy. Ther Clin Risk Manag. 2015;11:449–67.
Unger M. Pharmacokinetic drug interactions involving Ginkgo biloba. Drug Metab Rev. 2013;45:353–85.
Binkhathlan Z, Lavasanifar A. P-glycoprotein inhibition as a therapeutic approach for overcoming multidrug resistance in cancer: current status and future perspectives. Curr Cancer Drug Targets. 2013;13:326–46.
Takahashi R, Ma S, Yue Q, Kim-Kang H, Yi Y, Lyssikatos JP, et al. Dose-dependent exposure and metabolism of GNE-892, a beta-secretase inhibitor, in monkeys: contributions by P450, AO, and P-gp. Eur J Drug Metab Pharmacokinet. 2015;40:171–85.
Nguyen MA, Staubach P, Wolffram S, Langguth P. Effect of single-dose and short-term administration of quercetin on the pharmacokinetics of talinolol in humans—implications for the evaluation of transporter-mediated flavonoid-drug interactions. Eur J Pharm Sci Off J Eur Fed Pharm Sci. 2014;61:54–60.
Li W, Liu Y, He YQ, Zhang JW, Gao Y, Ge GB, et al. Characterization of triptolide hydroxylation by cytochrome P450 in human and rat liver microsomes. Xenobiotica. 2008;38:1551–65.
Liang Y, Zhou Y, Zhang J, Liu Y, Guan T, Wang Y, et al. In vitro to in vivo evidence of the inhibitor characteristics of Schisandra lignans toward P-glycoprotein. Phytomedicine. 2013;20:1030–8.
Gong X, Chen Y, Wu Y. Absorption and metabolism characteristics of triptolide as determined by a sensitive and reliable LC-MS/MS method. Molecules (Basel, Switzerland). 2015;20:8928–40.
Zhang Y, Li J, Lei X, Zhang T, Liu G, Yang M, et al. Influence of verapamil on pharmacokinetics of triptolide in rats. Eur J Drug Metab Pharmacokinet. 2015. doi:10.1007/s13318-015-0275-4.
Zhang JW, Liu Y, Cheng J, Li W, Ma H, Liu HT, et al. Inhibition of human liver cytochrome P450 by star fruit juice. J Pharm Pharm Sci. 2007;10:496–503.
Nabekura T, Yamaki T, Kitagawa S. Effects of chemopreventive citrus phytochemicals on human P-glycoprotein and multidrug resistance protein 1. Eur J Pharmacol. 2008;600:45–9.
Zhou S, Chan E, Li SC, Huang M, Chen X, Li X, et al. Predicting pharmacokinetic herb-drug interactions. Drug Metabol Drug Interact. 2004;20:143–58.
Hu X, Huang W, Yang Y. Cytochrome P450 isoenzymes in rat and human liver microsomes associate with the metabolism of total coumarins in Fructus Cnidii. Eur J Drug Metab Pharmacokinet. 2015;40:373–7.
Liu T, Qian G, Wang W, Zhang Y. Molecular docking to understand the metabolic behavior of GNF-351 by CYP3A4 and its potential drug-drug interaction with ketoconazole. Eur J Drug Metab Pharmacokinet. 2015;40:235–8.
Lee SY, Lee JY, Kang W, Kwon KI, Park SK, Oh SJ, et al. Cytochrome P450-mediated herb-drug interaction potential of Galgeun-tang. Food Chem Toxicol. 2013;51:343–9.
Shimada T, Mimura M, Inoue K, Nakamura S, Oda H, Ohmori S, et al. Cytochrome P450-dependent drug oxidation activities in liver microsomes of various animal species including rats, guinea pigs, dogs, monkeys, and humans. Arch Toxicol. 1997;71:401–8.
Singh AP, Pant MC, Ruwali M, Shah PP, Prasad R, Mathur N, et al. Polymorphism in cytochrome P450 1A2 and their interaction with risk factors in determining risk of squamous cell lung carcinoma in men. Cancer Biomark Sect A Dis Markers. 2010;8:351–9.
Wang B, Zhou SF. Synthetic and natural compounds that interact with human cytochrome P450 1A2 and implications in drug development. Curr Med Chem. 2009;16:4066–218.
Jeong HU, Kong TY, Kwon SS, Hong SW, Yeon SH, Choi JH, et al. Effect of honokiol on cytochrome P450 and UDP-glucuronosyltransferase enzyme activities in human liver microsomes. Molecules (Basel, Switzerland). 2013;18:10681–93.
Pandit S, Mukherjee PK, Ponnusankar S, Venkatesh M, Srikanth N. Metabolism mediated interaction of alpha-asarone and Acorus calamus with CYP3A4 and CYP2D6. Fitoterapia. 2011;82:369–74.
Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 2008;9:310–22.
Ericsson T, Sundell J, Torkelsson A, Hoffmann KJ, Ashton M. Effects of artemisinin antimalarials on Cytochrome P450 enzymes in vitro using recombinant enzymes and human liver microsomes: potential implications for combination therapies. Xenobiotica. 2014;44:615–26.
Ito K, Iwatsubo T, Kanamitsu S, Nakajima Y, Sugiyama Y. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu Rev Pharmacol Toxicol. 1998;38:461–99.
Shao F, Wang G, Xie H, Zhu X, Sun J, Jiye A. Pharmacokinetic study of triptolide, a constituent of immunosuppressive chinese herb medicine, in rats. Biol Pharm Bull. 2007;30:702–7.
Konig J, Muller F, Fromm MF. Transporters and drug-drug interactions: important determinants of drug disposition and effects. Pharmacol Rev. 2013;65:944–66.
Ekins S, Clark AM, Wright SH. Making transporter models for drug-drug interaction prediction mobile. Drug Metab Dispos. 2015;43:1642–5.
Li CZ, Xing GZ, Maeda K, Wu CY, Gong LK, Sugiyama Y, et al. The role of breast cancer resistance protein (Bcrp/Abcg2) in triptolide-induced testis toxicity. Toxicol Res. 2015;36:1260–8.
Acknowledgments
This study was supported by “The Pilot Project of Fujian Province Technology Hall (2015Y0013)”.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
HZ, GY and HR have declared no conflict of interest.
Additional information
H. Zhang and G. Ya equally contributed to this work.
Rights and permissions
About this article

Cite this article
Zhang, H., Ya, G. & Rui, H. Inhibitory Effects of Triptolide on Human Liver Cytochrome P450 Enzymes and P-Glycoprotein. Eur J Drug Metab Pharmacokinet 42, 89–98 (2017). https://doi.org/10.1007/s13318-016-0323-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-016-0323-8