
Abstract
Background and Objectives
Curcumin is the major bioactive component of turmeric, but has poor oral bioavailability that limits its clinical applications. To improve the in vitro solubility and alkaline stability, we developed a prodrug of curcumin by succinylation to obtain curcumin diethyl disuccinate, with the goal of improving the oral bioavailability of curcumin.
Methods
The in vivo pharmacokinetic profile of curcumin diethyl disuccinate was compared with that of curcumin in male Wistar rats. Doses of curcumin 20 mg/kg intravenous or 40 mg/kg oral were used as standard regimens for comparison with the prodrug at equivalent doses in healthy adult rats. Blood, tissues, urine, and faeces were collected from time zero to 48 h after dosing to determine the prodrug level, curcumin level and a major metabolite by liquid chromatography-tandem spectrometry.
Results
The absolute oral bioavailability of curcumin diethyl disuccinate was not significantly improved compared with curcumin, with both compounds having oral bioavailability of curcumin less than 1 %. The major metabolic pathway of the prodrug was rapid hydrolysis to obtain curcumin, followed by glucuronidation. Interestingly, curcumin diethyl disuccinate gave superior tissue distribution with higher tissue to plasma ratio of curcumin and curcumin glucuronide in several organs after intravenous dosing at 1 and 4 h. The primary elimination route of curcumin glucuronide occurred via biliary and faecal excretion, with evidence of an entry into the enterohepatic circulation.
Conclusion
Curcumin diethyl disuccinate did not significantly improve the oral bioavailability of curcumin due to first pass metabolism in the gastrointestinal tract. Further studies on reduction of first pass metabolism are required to optimise delivery of curcumin using a prodrug approach.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Curcumin diethyl disuccinate did not significantly improve the oral bioavailability of curcumin due to first pass metabolism in the gastrointestinal tract. |
Curcumin diethyl disuccinate gave superior tissue distribution, with a higher tissue to plasma ratio of curcumin and curcumin glucuronide in several organs after intravenous dosing. |
The major metabolic pathway of curcumin diethyl disuccinate was rapid hydrolysis to give curcumin, followed by glucuronidation, with evidence of clearance through the enterohepatic circulation. |
1 Introduction
There is an increasing trend for use of herbal and oriental medicine worldwide. In Thailand, turmeric is a common herbal product used for gastrointestinal diseases, with an official recommendation in the national drug list [1, 2]. The major bioactive constituents of turmeric are volatile oil and curcuminoids [3, 4], which contain curcumin at not less than 5 % by weight as recommended in the Thai Herbal Pharmacopoeia [5]. However, curcumin has low water solubility (XlogP 3.2) and alkaline instability, which result in very low oral bioavailability and limit clinical applications to diseases of the gastrointestinal tract [6–8]. In addition, some researchers found that tissue levels of curcumin were very limited in heart, liver and kidney after oral administration of curcumin 400 mg in rodents [9]. Biotransformation of curcumin molecules in vivo occurred by three major reactions: phase I reduction, phase II sulphation and glucuronidation [10]. The plasma level of curcumin-o-glucuronide was approximately 10–100-fold higher than curcumin-o-sulphate and tetrahydrocurcumin after oral gavage in mice [11]. Therefore, glucuronide conjugates are likely to be major metabolites of curcumin in vivo [12, 13]. Moreover, curcumin and its glucuronide conjugates are substrates of some efflux transporters in both enterocytes and hepatocytes, which were mainly found in gastrointestinal tracts [14–16]. Interestingly, there are several reports mentioning that glucuronide conjugates of curcumin enter the enterohepatic circulation due to glucuronidase enzymes from normal intestinal flora [9–11]. The major elimination pathway of curcumin and its metabolites is likely to be excretion via bile and faeces, especially after oral administration of curcumin.
Efforts to improve the oral bioavailability of curcumin to expand its clinical applications can be classified into physical [17–19] and chemical [20–22] modifications. Curcumin diethyl disuccinate (CDD) is a prodrug formed by succinylation of curcumin (Fig. 1). CDD has superior lipid solubility and chemical stability in basic conditions, compared with curcumin [23], and these properties may improve the oral bioavailability and pharmacokinetics of CDD. CDD also has improved pharmacodynamics as an antinociceptive agent in a mouse model [24], with the effective daily dose for antinociceptive activity ranging from 20 to 80 mg/kg orally. Given these findings, this study was designed to examine the pharmacokinetic profiles of CDD and metabolites at the effective dose in comparison with curcumin in Wistar rats. Data obtained from the study will be useful for understanding the in vivo pharmacokinetics of CDD and as basic information for further development of curcumin prodrugs.

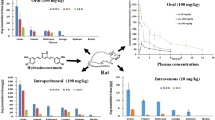
Chemical structures of curcumin and curcumin diethyl disuccinate
2 Materials and Methods
2.1 Chemicals
Curcumin was purchased from Sigma Aldrich Corp., USA. CDD was synthesised as described previously [23]. Both compounds were tested for quality control and had more than 95 % of the active ingredient [23]. β-Glucuronidase from Helix pomatia (Type H-1) was purchased from Sigma Aldrich Corp., USA. 2-Hydroxychalcone used as an internal standard in liquid chromatography-tandem spectrometry (LC-MS/MS) was purchased from Sigma Aldrich Corp., USA.
2.2 Animals
Male Wistar rats were purchased from the National Laboratory Animal Center, Mahidol University, Thailand. The animals were housed for 2 months in a climate- and light-controlled environment with free access to water and food. Rats used in the study had a body weight more than 400 g and normal liver and kidney functions. Rats were moved to a metabolic cage 1 day before experiments and kept there until 72 h afterwards. Animal procedures complied with the guidelines of the Institutional Animal Care and Use Committee of Chulalongkorn University.
2.3 Animal Experiments
Curcumin and CDD were dissolved in dimethylsulfoxide at concentrations equivalent to 100 mg/mL curcumin. Each solution was administered at a dose of 20 mg/kg by an intravenous route via a lateral tail vein or at 40 mg/kg orally. All rats were anaesthetized with 5 % isoflurane by chamber induction method to reduce pain and injury to animals, before administration of drugs or collection of blood via lateral tail vein. Approximately 0.4 mL of blood was collected from lateral tail vein using a heparin-coated syringe at 0, 5, 15, 30 min and 1, 2, 4, 8, 24 h after dosing. The blood was centrifuged at 1500×g for 10 min under refrigerated conditions and the plasma was collected and stored at −80 °C until analysis. Urine and faeces samples from each rat in metabolic cages were measured for weight and volume, and then stored at −80 °C until analysis. For tissue distribution experiments, some rats received curcumin or CDD at a dose of 20 mg/kg by the intravenous route via a lateral tail vein. These rats were killed by decapitation at 1 and 4 h after dosing and major internal organs were collected and rinsed with ice-cold saline, and then stored at −80 °C until analysis. Blood levels of aspartate transaminase (AST), alanine transaminase (ALT) and creatinine in all rats at pre-dose and 24 h post-dose were determined by Professional Laboratory Management Corp. (Bangkok, Thailand).
2.4 Sample Preparations
The concentrations of curcumin and CDD in biological samples were analysed as unchanged and glucuronide-conjugated forms. For unchanged curcumin or CDD, all biological samples were treated by the protein precipitation method. In brief, 50 μL of plasma or urine samples were mixed with 200 μL of methanol containing 10 ng of 2-hydroxychalcone as internal standard for LC/MS/MS analysis. With regard to solid organs and faeces, the biological samples 50 mg were weighed, homogenated in phosphate buffer pH 7.4 with final volume of 200 μL, and mixed with methanol 800 μL containing 40 ng of internal standard. The mixture was then centrifuged at 1500×g for 10 min, and supernatant was collected for further analysis by LC-MS/MS.
For glucuronide conjugates of curcumin or CDD, 50 μL of plasma or urine or homogenated samples were added to 50 μL of 0.2 M acetate buffer (pH 4.5) containing 1000 unit of β-glucuronidase. The mixture was incubated at 37 °C for 15 min, and the reaction was stopped by adding 400 μL of methanol containing 20 ng of 2-hydroxychalcone. The mixture was centrifuged at 1500×g for 10 min, and then supernatant was collected and 10 μL of supernatant were injected into a LC/MS/MS system.
2.5 LC-MS/MS Analysis
LC-MS/MS was performed using an Eksigent ekspert™ UHPLC 100 liquid chromatograph equipped with a QTRAP® 6500 mass spectrometer, controlled by Analyst® software version 1.6 (AB Sciex, USA). The UHPLC system was equipped with a Synergi™ Fusion-RP C18 column as stationary phase (Phenomenex Inc., USA). The mobile phase consisted of 100 % methanol and 0.2 % formic acid in water (pH 2.5). Gradient elution of the mobile phase was started at 10 % methanol for 0.5 min, increased to 90 % methanol at 1.5 min until 3.5 min, and then decreased to 10 % methanol at 4 min until 4.5 min. The retention times of curcumin, CDD and 2-hydroxychalcone were 1.5, 1.6, and 1.7 min, respectively. MS analysis was conducted by negative mode ionisation. Optimised MS conditions are shown in Table 1. Calibration curves of curcumin and CDD showed good correlation coefficients (R 2 > 0.99) over the concentration range from 1 to 100 μg/L for all matrices (100, 50, 25, 12.5, 6.25, 3.12, 1.56 and 0.78 μg/L). The limit of detection was estimated to be 0.1 μg/L with signal to noise ratio of 5 and the percent recovery was more than 50 % for all matrices. The intra-assay accuracy and precision for analysis of both compounds were within ±10 %.
2.6 Data Analysis
Pharmacokinetic parameters were calculated by non-compartmental analysis using PK solution 2.0™ (Summit Research Service, USA). C max was the maximum concentration of curcumin and T max was a time to reach maximum concentration of curcumin after administration of curcumin or CDD, these values were directly observed from the plasma concentration time curve of each animal. AUC was area under plasma concentration time curve of curcumin or curcumin glucuronide from time zero to observed time (0–t) or infinity (0–∞). Vd was volume of distribution of curcumin after administration of curcumin or CDD. MRT was mean resident time of curcumin molecules after administration of curcumin or CDD. T 1/2 was elimination half-life of plasma curcumin. CL was total body clearance curcumin after administration of curcumin or CDD. Absolute oral bioavailability of curcumin was calculated as (AUCpo/DOSEpo) divided by (AUCiv/DOSEiv). Tissue to plasma ratio of curcumin was calculated from actual tissue concentration divided by actual plasma concentration of each rat at the same time point. Percent recovery of curcumin in urine and faeces was determined from glucuronide metabolites found in urine or faeces divided by the dose of curcumin. Comparison of numerical data between groups was performed by independent t test, with p < 0.05 indicating a significance difference. Statistical analysis was performed using SPSS ver. 16 (SPSS Inc., USA).
3 Results
3.1 Animal Tolerability
All rats had a normal appearance from 0 to 24 h after dosing. In liver biochemical profiles, all rats had normal AST and ALT levels pre-dose and post-dose for 24 h. There was a slight increase in AST with intravenous curcumin compared with intravenous CDD, and a slight decrease in ALT with oral curcumin and oral CDD after dosing for 24 h. However, these changes had not exceeded the normal ranges for healthy Wistar rats. In kidney biochemical profiles, blood creatinine showed no significant changes from pre-dose to post-dose for both test compounds given intravenously or orally. All values were within the normal ranges for healthy adult Wistar rats (Table 2).
3.2 Plasma Concentration–Time Profiles
In rats that received curcumin 20 mg/kg intravenously, the plasma concentration of curcumin reached a level of 715 μg/L at 5 min and then rapidly decreased to 10 μg/L at 4 h after dosing. For CDD given intravenously at a dose equivalent to 20 mg/kg curcumin, the plasma curcumin concentration reached a level of 676 μg/L at 5 min and then sharply decreased at 30 min after dosing, but then gradually increased to reach a secondary peak at 2 h (Fig. 2a). Both intravenous groups had high volumes of distribution of curcumin, and CDD group showed significantly higher than that of curcumin group (197 vs. 82 L/kg). Clearance of curcumin between the two intravenous groups did not show any significant difference with approximately 40–50 L/h/kg. Elimination half-lives of curcumin were 1.5–3.0 h and intravenous CDD group exerted slightly longer half-life than intravenous curcumin group (2.81 vs. 1.76 h). CDD given orally at a dose equivalent to 40 mg/kg curcumin had an absolute oral bioavailability of curcumin less than 1 %, similarly to oral curcumin (Table 3). The plasma curcumin level was close to the limit of detection at most sampling time points after oral administration of both compounds (Fig. 2b).

a Plasma concentration–time profiles of curcumin after administrations of curcumin intravenous (filled triangle), CDD intravenous (filled circle). b Plasma concentration–time profiles of curcumin after administrations of curcumin oral (filled square), CDD oral (filled diamond). Data are shown as mean ± SD (n = 4). CDD curcumin diethyl disuccinate
3.3 Tissue Penetration
Intravenous curcumin showed limited tissue distribution of curcumin to most organs, with lower tissue to plasma ratio of curcumin than that of intravenous CDD at 1 h after dosing. Interestingly, intravenous CDD group exerted significantly higher tissue to plasma ratio of curcumin than intravenous curcumin group in most tissues at 1 h and also 4 h in kidney and liver (Fig. 3). We also found these similar patterns of tissue distribution of curcumin glucuronide, a major metabolite of curcumin in rats that received intravenous CDD compared with intravenous curcumin. Administration of intravenous CDD could give superior tissue to plasma ratio of curcumin glucuronide in most internal organ at 1 h, and also in spleen, kidney and liver at 4 h (Fig. 4). The tissue to plasma ratio of curcumin glucuronide was higher than curcumin in most internal organs approximately 10–1000-fold after intravenous dosing of both test compounds for 1 and 4 h (Figs. 3, 4).

Tissue to plasma ratio of curcumin in internal organs after administration of curcumin or CDD 20 mg/kg intravenous injection. Data are shown as mean ± SD (n = 4), * p < 0.05 for curcumin vs. CDD. CDD curcumin diethyl disuccinate

Tissue to plasma ratio of curcumin glucuronide in internal organs after administration of curcumin or CDD 20 mg/kg intravenous injection. Data are shown as mean ± SD (n = 4), *p < 0.05 for curcumin vs. CDD. CDD curcumin diethyl disuccinate
3.4 Metabolic Pathways and Routes of Excretion
CDD was not detectable in samples of plasma, tissues, urine, and faeces in rats that received the prodrug intravenously or orally. However, a high plasma level of curcumin was found after intravenous dosing of the prodrug for 5 min and at other sampling time points. A large amount of curcumin glucuronide was also found in most tissues after intravenous administration of curcumin or CDD. In rats that received curcumin 20 mg/kg intravenously, the plasma concentration of curcumin glucuronide reached a maximum within 15 min after dosing, sharply decreased to 1 h, and then gradually increased to reach a secondary peak at 8 h (Fig. 5). Similarly, intravenous CDD gave a maximum level of curcumin glucuronide at 1 h after dosing, followed by a sharp decrease to 2 h, and then a gradual increase that reached a secondary peak at 8 h. In oral administration, curcumin glucuronide in plasma from rats that received curcumin 40 mg/kg or CDD at equivalent does was below the limit of detection by LC/MS/MS. The ratio of AUCcurcumin glucuronide/AUCcurcumin in intravenous CDD group was approximately 3.45, meanwhile intravenous curcumin group was 2.85 (Table 3). Approximately 30–40 % of oral dose of curcumin or CDD was found as curcumin glucuronides in faeces during 24–48 h after administration. Negligible amounts of curcumin and curcumin glucuronide were found in urine samples from 0 to 48 h after oral or intravenous administration of each compound (Table 4).

Plasma concentration–time profiles of curcumin glucuronide after administration of curcumin intravenous (filled triangle), CDD intravenous (filled circle). Data are shown as mean ± SD (n = 4). CDD curcumin diethyl disuccinate
4 Discussion
Curcumin is a major bioactive component of turmeric, which has been used as a herbal medicine since ancient times [3]. However, curcumin has limited clinical applications due to its low oral bioavailability [12]. Therefore, chemical modification of curcumin was performed by succinylation to improve these properties, including instability under basic conditions and solubility. The resulting prodrug, CDD, showed improved stability in alkaline solution and increased lipid solubility [23]. To determine the effect of these improved properties, the pharmacokinetics of the prodrug were determined in adult male Wistar rats. Curcumin and CDD showed good tolerability profiles in intravenous and oral administration at an effective dose of 20–40 mg/kg, with no major changes in physical appearance, liver biochemical markers, or renal function indicators after dosing for 24 h (Table 2).
In our pharmacokinetic experiments, animal guidelines limited blood collection to within 10 % of the total blood volume of animals. Therefore, we used 8–9 blood sampling times to determine absorption, distribution, metabolism and excretion of the test compounds [25, 26]. One major finding was that CDD still had very low absolute oral bioavailability of curcumin less than 1 %, similar to that for curcumin (Table 3). The plasma curcumin level after oral administration of curcumin or CDD was close to the limit of detection at 0.1 μg/L at most sampling time points (Fig. 2b). The low level of curcumin after oral administration of CDD was due to intestinal and hepatic metabolism, since a large amount of curcumin glucuronide was present in faeces after oral CDD dosing, similarly to oral curcumin. Thus, succinylation at the hydroxyl group of the two phenyl rings in curcumin molecule does not protect against UDP-glucuronosyltransferase (UGT) modification of the hydroxyl group on the alkyl chain linking the two phenyl rings [27, 28]. Enzyme inhibition of intestinal UGTs or substitution of this hydroxyl group are interesting options to decrease first pass metabolism and increase oral bioavailability of curcumin and also CDD [29, 30]. In general, management of first pass metabolism by UGTs in the gastrointestinal tracts may be a critical factor and major challenge in improvement of the oral bioavailability of curcumin.
Intravenous curcumin had a volume of distribution of 82 L/kg. This implies that curcumin has very large volume of distribution with the possibility of accumulation in some tissues. Interestingly, intravenous CDD had a significantly higher volume of distribution (197 L/kg) and this parameter correlated well with its superior lipophilic property. In addition, plasma concentration time profiles of intravenous CDD exerted a sharp decrease of plasma curcumin level in the first hour after dosing. We also found that tissue to plasma ratio of curcumin from intravenous CDD in most internal organs was higher than intravenous curcumin at the first hour after dosing. This superior distribution pattern of curcumin in CDD group was also found for curcumin glucuronide in major internal organs after intravenous CDD (Figs. 3, 4). The superior tissue distribution of curcumin after administration of the succinylated prodrug is an important finding that requires further investigation to understand the molecular mechanism. CDD may passively diffuse through the lipid bilayer of tissue and membranes, and then enter intracellular compartments more effectively than curcumin. This may be because CDD has superior lipid solubility and chemical stability in basic conditions, compared with those of curcumin.
CDD undergoes rapid esterase hydrolysis and a negligible amount of CDD was detected within a few minutes after intravenous administration. The prodrug was hydrolysed and generated high levels of curcumin within 5 min after intravenous dosing. Curcumin has several metabolic pathways in vivo and the major metabolite is curcumin glucuronide [31, 32]. The plasma level of curcumin glucuronide after intravenous administration of curcumin and CDD exerted two peaks (Fig. 5), the first peak of curcumin glucuronide was produced within the first hour after dosing, and the second peak of curcumin glucuronide occurred 2–8 h after dosing. The first peak of curcumin glucuronide might result from abundant UGTs in liver, intestine and other internal organs. The second peak of production of curcumin glucuronide might be due to enterohepatic circulation in the 2–8 h after dosing. Excretion of curcumin glucuronide was likely to occur via the hepatobiliary system, and then secreted with bile into small intestine. This curcumin glucuronide in the intestine could be hydrolysed by glucuronidase enzymes from normal flora of the intestine and reabsorbed back into blood circulation [9]. In addition, curcumin is a substrate of efflux transporters in the hepatobiliary system [14] and these transporters can pump curcumin from the hepatobiliary system into the gut lumen [30, 31]. The physicochemical properties of curcumin molecule include a high partition coefficient (XlogP 3.2) and a molecular weight of 368 Da, which are consistent with substrate patterns for efflux transporters in hepatobiliary system [8, 33]. Therefore, excretion of curcumin and curcumin glucuronide was likely to occur via the hepatobiliary system rather than the urinary system, which prefers small hydrophilic molecules [34]. Consistent with this, most excretion of curcumin occurred in faeces in the form of curcumin glucuronide, with extremely high levels of curcumin glucuronide found in faeces of rats that received oral curcumin or CDD (Table 4). However, percent recovery of curcumin glucuronide after intravenous administration of curcumin or CDD is less than 1 % in both urine and faeces. Intravenous administration of curcumin or CDD could generate very high concentration of curcumin in plasma, and might have possibility to be biotransformed via multiple metabolic pathways such as reduction and sulphation.
5 Conclusion
Clinical applications of curcumin are mainly focused on treatment of gastrointestinal disorders due to limited absorption and low bioavailability. Alkaline degradation and UGT first pass metabolism are major obstacles in improvement of oral bioavailability of curcumin and expansion of its indication for other diseases. Our prodrug, CDD, showed good stability in basic conditions in vitro and had a good tolerability profile in vivo. However, the prodrug still had absolute oral bioavailability of curcumin less than 1 % due to first pass metabolism. Administration of the prodrug resulted in better tissue distribution of curcumin and curcumin glucuronide in major internal organs, compared to those after administration of curcumin itself. Based on the rationale that most sites of drug actions are located intracellularly, CDD may generate higher levels of curcumin more rapidly at sites of actions in cells. Further studies on reduction of first pass metabolism and tissue penetration of curcumin molecules are required to optimise delivery of curcumin using the prodrug approach.
References
Howe E, Keiwkarnka B, Khan MI. Traditional medicine and medicinal plants: utilization, policy and research in Thailand. J Public Health. 2004;2:102.
Satyapan N, Patarakitvanit S, Temboonkiet S, Vudhironarit T, Tankanitlert J. Herbal medicine: affecting factors and prevalence of use among Thai population in Bangkok. J Med Assoc Thai. 2010;93:S139–44.
Prasad S, Aggarwal BB. Turmeric, the golden spice: from traditional medicine to modern medicine. In: Benzie IFF, Wachtel-Galor S, editors. Herbal medicine: biomolecular and clinical aspects. 2nd ed. Boca Raton:CRC Press;2011.
Ammon HP, Wahl MA. Pharmacology of Curcuma longa. Planta Med. 1991;57:1–7.
Department of Medical Sciences. Thai herbal pharmacopoeia, vol. I. Bangkok: Prachachon; 2009.
Anand P, Thomas SG, Kunnumakkara AB, Sundaram C, Harikumar KB, Sung B, et al. Biological activities of curcumin and its analogues (congeners) made by man and mother nature. Biochem Pharmacol. 2008;76:1590–611.
Huang MT, Newmark HL, Frenkel K. Inhibitory effects of curcumin on tumorigenesis in mice. J Cell Biochem. 1997;27:S26–34.
Pubchem Compound. Cited on 8 August 2014 by using search term “Curcumin” https://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=969516&loc=ec_rcs.
Ravindranath V, Chandrasekhara N. Absorption and tissue distribution of curcumin in rats. Toxicology. 1980;16:259–65.
Pan MH, Huang TM, Lin JK. Biotransformation of curcumin through reduction and glucuronidation in mice. Drug Metab Dispos. 1999;27:486–94.
Zhongfa L, Chiu M, Wang J, Chen W, Yen W, Fan-Havard P, et al. Enhancement of curcumin oral absorption and pharmacokinetics of curcuminoids and curcumin metabolites in mice. Cancer Chemother Pharmacol. 2012;69:679–89.
Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:807–18.
Yang KY, Lin LC, Tseng TY, Wang SC, Tsai TH. Oral bioavailability of curcumin in rat and the herbal analysis from Curcuma longa by LC/MS/MS. J Chromatogr B. 2007;853:183–9.
Heger M, van Golen RF, Broekgaarden M, Michel MC. The molecular basis for the pharmacokinetics and pharmacodynamics of curcumin and its metabolites in relation to cancer. Pharmacol Rev. 2014;66:222–307.
Mao Q, Unadkat JD. Role of the breast cancer resistance protein (ABCG2) in drug transport. AAPS J. 2005;7:E118–33.
Ni Z, Bikadi Z, Rosenberg MF, Mao Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr Drug Metab. 2010;11:603–17.
Gupta NK, Dixit VK. Bioavailability enhancement of curcumin by complexation with phosphatidyl choline. J Pharm Sci. 2011;100:1987–95.
Liu A, Lou H, Zhao L, Fan P. Validated LC/MS/MS assay for curcumin and tetrahydrocurcumin in rat plasma and application to pharmacokinetic study of phospholipid complex of curcumin. J Pharm Biomed Anal. 2006;40:720–7.
Zhang W, Tan TM, Lim LY. Impact of curcumin-induced changes in p-glycoprotein and CYP3A expression on the pharmacokinetics of peroral celiprolol and midazolam in rats. Drug Metab Dispos. 2007;35:110–5.
Liang G, Yang S, Zhou H, Shao L, Huang K, Xiao J, et al. Synthesis, crystal structure and anti-inflammatory properties of curcumin analogues. Eur J Med Chem. 2009;44:915–9.
Rinwa P, Kumar A. Piperine potentiates the protective effects of curcumin against chronic unpredictable stress-induced cognitive impairment and oxidative damage in mice. Brain Res. 2012;1488:38–50.
Steward WP, Gescher AJ. Curcumin in cancer management: recent results of analogue design and clinical studies and desirable future research. Mol Nutr Food Res. 2008;52:1005–9.
Wichitnithad W, Nimmannit U, Wacharasindhu S, Rojsitthisak P. Synthesis, characterization and biological evaluation of succinate prodrugs of curcuminoids for colon cancer treatment. Molecules. 2011;16:1888–900.
Wongsrisakul J, Wichitnithad W, Rojsitthisak P, Towiwat P. Antinociceptive effects of CDD in animals models. J Health Res. 2010;24:175–80.
Fan J, Lannoy IAM. Pharmacokinetics. Biochem Pharmacol. 2014;87:93–120.
Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, et al. Curcumin structure–function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther. 2008;326:196–208.
Wahlstrom B, Blennow G. A study on the fate of curcumin in the rat. Acta Pharmacologica et Toxicologica. 1978;43:86–92.
Balant LP, Doelker E, Buri P. Prodrugs for the improvement of drug absorption via different routes of administration. Eur J Drug Metab Pharmacokinet. 1990;15:143–53.
Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas PS. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med. 1998;64:353–6.
Ajazuddin, Alexander A, Qureshi A, Kumari L, Vaishnav P, Sharma M, et al. Role of herbal bioactives as a potential bioavailability enhancer for active pharmaceutical ingredients. Fitoterapia. 2014;97C:1–14.
Asai A, Miyazawa T. Occurrence of orally administered curcuminoid as glucuronide and glucuronide/sulfate conjugates in rat plasma. Life Sci. 2000;67:2785–93.
Ireson CR, Jones DJ, Orr S, Coughtrie MW, Boocock DJ, Williams ML, et al. Metabolism of the cancer chemopreventive agent curcumin in human and rat intestine. Cancer Epidemiol Biomark Prev. 2002;11:105–11.
Limtrakul P. Curcumin as chemosensitizer. Adv Exp Med Biol. 2007;595:269–300.
Parkinson A, Ogilvie BW. Biotransformation of xenobiotics. In: Klaassen CD, editor. Casarette and Doull’s toxicology; the basic sciences of poisons. 7th ed. New York: McGraw-Hill; 2007. p. 161–304.
Acknowledgments
The technical language amendment was supervised by Dr. Ian S. Haworth, Dr. Jessica T. Lin and Dr. Delia Bethell.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was supported by the Ratchadaphiseksomphot Endowment Fund 2013 of Chulalongkorn University, CU-56-329-HR. Dr. Kunan Bangphumi was working under the Postdoctoral Scholarship of Ratchadaphiseksomphot Endowment Fund of Chulalongkorn University.
Conflicts of interest
All authors declare no conflict of interest.
Ethical approval
Animal procedures complied with the guidelines of the Institutional Animal Care and Use Committee of Chulalongkorn University, approval number 13-33-008.
Rights and permissions
About this article

Cite this article
Bangphumi, K., Kittiviriyakul, C., Towiwat, P. et al. Pharmacokinetics of Curcumin Diethyl Disuccinate, a Prodrug of Curcumin, in Wistar Rats. Eur J Drug Metab Pharmacokinet 41, 777–785 (2016). https://doi.org/10.1007/s13318-015-0308-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-015-0308-z