
Abstract
Tanshinone IIA (TSA), an important natural lipophilic diterpene compound from the traditional Chinese herb Salvia miltiorrhiza Bunge, has long been widely used for the prevention and treatment of various diseases because of its anti-inflammatory activities; however, the anti-inflammatory mechanism remains unknown. In the present work, we examined the effects of TSA on experimental autoimmune encephalomyelitis (EAE), a model of autoreactive T/B cell-mediated central nervous system (CNS) autoimmunity. The data showed that TSA significantly attenuates the severity of EAE when administered at the pre-onset and peak of clinical disease. In vivo, the protective effects of TSA on EAE mice are correlated with diminished inflammatory infiltration, demyelination, and GM-CSF-producing CD4+ T cells in the spinal cord and selectively increased regulatory T (Treg) cell frequencies in both the spinal cord and spleen. We further confirm that TSA can promote the polarization of naïve CD4+ T cells into Treg cells both by targeting dendritic cells (DCs) to drive transforming growth factor β1 (TGF-β1) upregulation and by directly targeting naïve CD4+ T cells in vitro. Most importantly, we showed that TSA-induced Treg cells display an effective suppressive activity at a level comparable to TGF-β1-polarized Treg Cells in vitro and in vivo. Taken together, our data provide evidence that TSA can promote Treg cell differentiation, and TSA may have a promising application as a therapeutic agent for the treatment of neuroinflammatory diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is an autoimmune disease that is pathologically characterized by multifocal inflammatory demyelination of the white matter of the central nervous system (CNS) [1, 2]. MS affects an estimated 350,000 people in the USA and 2.5 million people worldwide [3]. Experimental autoimmune encephalomyelitis (EAE) shares many characteristics of clinical symptoms and disease course with MS and has thus become the most commonly used animal model of MS [4,5,6,7].
Increasing evidence suggests that T helper cells with pathogenic potential [mainly interferon-γ (IFN-γ)-producing T helper 1 (Th1) cells and interleukin-17 (IL-17)-producing T helper 17 (Th17) cells] are involved in the pathogenicity of EAE and MS, whereas regulatory T (Treg) cells function to keep the autoimmune response in check [8,9,10]. Treg cells derived from the thymus (natural Treg, nTreg) or induced from naïve CD4+ T cells in the periphery (induced Treg, iTreg) are referred to as suppressor T cells characterized by the expression of the transcription factor forkhead box P3 (FoxP3) [1, 2, 11, 12]. Both types of Treg cells are involved in the maintenance of peripheral tolerance and the prevention of autoimmunity; however, their individual contributions have not yet been closely examined in vivo [11]. Treg cells mediate immune suppression, in part, by secreting the anti-inflammatory cytokines IL-10 and TGF-β [13,14,15].
Tanshinone IIA (TSA) is an important lipophilic diterpene extracted from the traditional Chinese medicine Danshen, which is the dry root of Salvia miltiorrhiza Bunge. Danshen has been commonly used for the prevention and treatment of cardiovascular and cerebrovascular diseases [16,17,18,19,20,21]. Currently, ample evidence supports the observation that TSA has potent immunosuppressive effects on neutrophils and microglia/macrophages [22,23,24,25,26]. TSA can dramatically promote inflammatory resolution in both zebrafish and human systems by accelerating the parallel mechanisms of neutrophil reverse migration and apoptosis [25]. A previous investigation indicated that TSA protects against dextran sulfate sodium–induced colitis in mice by modulating neutrophil infiltration and activation [27]. Studies in microglia/macrophages have confirmed that TSA also suppresses the activation of microglia/macrophages. Encephalomyelitis in the CNS of EAE mice is caused mainly by activated microglia and infiltrating macrophages, which are driven by pathogenic Th1 and Th17 cells [28]. Therefore, we propose the hypothesis that TSA can ameliorate progressive EAE.
In the present study, we focused on exploring the immunomodulatory effects of TSA on EAE and clarifying the underlying mechanisms. We demonstrated that TSA administration significantly inhibited the progression of EAE, and the reduced pathology (inflammation and demyelination) in TSA-treated EAE mice was associated with increased frequencies of Treg subsets. We further establish that TSA enhanced Treg cell differentiation by acting directly and parallelly on naïve CD4+ T cells dependent on T cell antigen receptor (TCR) and TGF-β1 signaling and the TGF-β1 signaling pathway by driving TGF-β1 expression in dendritic cells.
Materials and Methods
Mice
Female C57BL/6 mice (6–8 weeks) were obtained from Beijing HFK Bioscience Co., Ltd. Mice were maintained in air-filtered cages and fed normal mouse chow ad libitum. All procedures were approved by the Committee for Research and Animal Ethics of Shaanxi Normal University and carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Investigators who recorded clinical symptoms and stained and analyzed for pathology were blinded to the experimental groups during the experiments.
Reagents and Antibodies
TSA (purity ≥ 97%), dimethyl sulfoxide (DMSO), mitomycin C, and lipopolysaccharide (LPS) were obtained from Sigma-Aldrich (St. Louis, MO). Carboxyfluorescein diacetate succinimidyl ester (CFSE) proliferation dye was from eBioscience (Santa Clara, CA). Myelin oligodendrocyte glycoprotein (MOG)35–55 peptide (MEVGWYRSPFSRVVHLYRNGK, purity ≥ 98%) was purchased from GenScript (Nanjing, China). Recombinant murine granulocyte–macrophage colony-stimulating factor (rmGM-CSF), recombinant murine IL-4, and recombinant human transforming growth factor β1 (rhTGF-β1) were purchased from PeproTech (Rocky Hill, NJ). TGF-β-neutralizing monoclonal IgG1 (aTGF-β, clone: 1D11.16.8) was from Bio X Cell (West Lebanon, NH). SB431542 (an inhibitor of TGF-β receptor) was from MCE. The antibodies (Abs) PE-anti-CD4 (clone: RM4-5), allophycocyanin-anti-CD4 (clone: RM4-5), Alexa Flour 488-anti-CD11b (clone: M1170), PE-anti-CD11c (clone: N418), FITC-anti-IFN-γ (clone: XMG1.2), PE-anti-IL-17A (clone: TC11-18H10.1), PE-cy7-anti-GM-CSF (clone: MP1-22E9), allophycocyanin-anti-FoxP3 (clone: MF-14), purified anti-CD3 (αCD3, clone: 17A2), and purified anti-CD28 (αCD3, clone: 37.51) were purchased from BioLegend (San Diego, CA). The Abs allophycocyanin-anti-CD45 (clone: 30-F11), FITC-anti-CD25 (clone: PC61), PE-cy7-anti-CD25 (clone: PC61), and allophycocyanin-anti-TGF-β1 (LAP, clone: TW7-16B4) were obtained from eBioscience (Santa Clara, CA). Anti-CD45 (clone: 30-F11) and anti-CD31 (clone: MEC 13.3) were obtained from BD Biosciences (San Jose, CA). Anti-ionized calcium-binding adapter molecule 1 (Iba-1) was obtained from Wako (Osaka, Japan). Anti-ZO-1 (clone: ZO1-1A12) and anti-Claudin-5 (clone: 4C3C2) were purchased from Invitrogen (Carlsbad, CA). Alexa Fluor 594 donkey anti-rat IgG, Alexa Fluor 488 donkey anti-rabbit IgG, and Alexa Fluor 488 donkey anti-mouse IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA).
Active EAE Induction and Treatment
The animals were divided into the TSA-treated group and the control (vehicle-treated) group. The mice were immunized subcutaneously (s.c.) to actively induce EAE as described previously [29]. Briefly, immunization was performed with a mixture containing 200 μg MOG35–55 peptide in complete Freund’s adjuvant (Sigma-Aldrich, St. Louis, MO) containing 5 mg/mL Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI). A total of 250 ng pertussis toxin (List Biologic, Campbell, CA) was administered intraperitoneally (i.p.) on days 0 and 2 post immunization (p.i.). To test the therapeutic effects of TSA (purity ≥ 97%; Sigma-Aldrich, St. Louis, MO) on EAE, 736 μg/kg TSA (200 μL/d) or the vehicle [2% DMSO (Sigma-Aldrich, St. Louis, MO) in phosphate-buffered saline (PBS)] was administered i.p. starting at the day of disease pre-onset (13 days p.i.) or disease peak (19 days p.i.). Based on the previous reports [25], we measured the dose–effect relationship of TSA and obtained the optimal dose of TSA for EAE mice treatment (Fig. S1). The mice were scored daily for the appearance of clinical signs of EAE on a scale from 0 to 5 as previously described [30]: 0, no clinical signs; 1, fully limp tail; 2, paralysis of 1 hind limb; 3, paralysis of both hind limbs; 4, paralysis of trunk; and 5, death.
Histopathology
At the end of the experiments (22 days p.i.), mice were transcardially perfused with ice-cold 4% paraformaldehyde (PFA) (Solarbio, Beijing, China) while deeply anesthetized. Lumbar spinal cords were postfixed in the same fixative at 4 °C overnight and then processed into paraffin-embedded sections. The paraffin-embedded transverse sections (6 μm) were stained with hematoxylin and eosin (H&E) and Luxol fast blue (LFB) and then examined by light microscopy (Leica DM6000B; Wetzlar, German). H&E and LFB staining was performed to assess inflammatory cell infiltration and demyelination, respectively. The pathological examination of the spinal cords was performed in a blinded fashion. The scale used to evaluate inflammation was as follows [31]: 0, no inflammatory cells; 1, a few scattered inflammatory cells; 2, organization of inflammatory infiltrates around blood vessels; and 3, extensive perivascular cuffing with extension into the adjacent parenchyma or parenchymal infiltration without obvious cuffing. Demyelination of the spinal cords was scored as previously described [32]: 0, none; 1, rare foci; 2, a few areas of demyelination; and 3, large (confluent) areas of demyelination. The CNS histopathology was quantified in 5 sections per mouse, and 3–5 mice per group were analyzed.
Immunofluorescence
For immunofluorescent staining, the lumbar spinal cords postfixed with 4% PFA were dehydrated in 30% sucrose (Solarbio, Beijing, China) at 4 °C overnight. The tissue was subsequently embedded in optimal cutting temperature (OCT) compound (Sakura, Torrance, CA) and placed at − 80 °C at least overnight. Frozen transverse sections (6 μm) were permeabilized with ice-cold acetone for 7 min. After blocking with 5% BSA (Sigma-Aldrich, St. Louis, MO) in PBS, the sections were incubated with anti-CD45 (1:100), anti-Iba-1 (1:500), and anti-CD31 (1:100) together with anti-ZO-1 (1:200) or anti-Claudin-5 (1:200) at 4 °C overnight, respectively. After being washed 3 times with PBS, sections were incubated with the corresponding secondary Abs at room temperature for 60 min. Alexa Fluor 594 donkey anti-rat IgG, Alexa Fluor 488 donkey anti-rabbit IgG, and Alexa Fluor 488 donkey anti-mouse IgG (Jackson ImmunoResearch Laboratories, West Grove, PA) were used as secondary antibody. Finally, the slides were covered with Fluoroshield mounting medium with DAPI (Abcam, Cambridge, UK). The slides were visualized by a Leica DM6000B fluorescence microscope.
Isolation of Leukocytes from the Spleen and CNS
Cells were isolated as previously described [33]. Briefly, the spinal cords or spleens from MOG35–55-immunized mice were homogenized into a single cell suspension with a 70-μm cell strainer (Corning, Durham, NC). For the isolation of infiltrating leukocytes from the spinal cord, spinal cord suspensions were centrifuged at 400×g for 5 min at 4 °C. After removal of the supernatants, pelleted cells were resuspended in 5 mL of 30% Percoll (GE Healthcare), overlaid onto an equal volume of 70% Percoll, and centrifuged at 800×g for 30 min at 4 °C. Then, cells at the gradient interface were collected, washed twice with PBS, and treated as described below.
For the mononuclear cell (MNC) isolation from the spleen, suspensions were spun at 400×g for 5 min at 4 °C, and the supernatants were discarded. The remaining red blood cells (RBCs) in each supernatant were removed using RBC lysis buffer (Solarbio, Beijing, China) for 10 min at room temperature. Cells were then washed twice in PBS.
Flow Cytometry
For intracellular cytokine staining, the cells from the spleen or spinal cord were plated in 24-well plates at a density of 1 × 106 cells/mL in RPMI 1640 complete medium [10% heat-inactivated FBS, 1 mM sodium pyruvate, 10 mM HEPES, 100 U/mL penicillin, 100 μg/mL streptomycin, 0.1 mM nonessential amino acids (NEAAs), and 0.055 mM β-mercaptoethanol (Gibco BRL, Gaithersburg, MD)] and were stimulated with 50 ng/mL phorbol-12-myristate-13-acetate (PMA), 500 ng/mL ionomycin, and 5 μg/mL brefeldin A (BFA) (Sigma-Aldrich, St. Louis, MO) for 5 h. The cells were harvested and washed twice in staining buffer containing 1% BSA and 0.02% sodium azide (NaN3) in PBS. Then, the cells were surface stained with fluorescent Abs against surface markers for 20 min at room temperature. After washing, the cells were fixed and permeabilized with fixation/permeabilization buffer (eBioscience, San Diego, CA), and intracellular cytokines were stained with fluorescently labeled Abs. Foxp3 staining was carried out using a commercial kit according to the manufacturer’s instructions (eBioscience, San Diego, CA). After the last wash, the cells were acquired by using a NovoCyte flow cytometer (ACEA). Data were analyzed using FlowJo software (Tree Star, Ashland, OR).
Magnetic-Activated Cell Sorting for Naïve CD4+ T Cells
Single cell suspensions derived from the spleens of normal female C57BL/6 mice (6–8 weeks) were purified by negative selection with a mouse naïve CD4+ T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). For the magnetic depletion of non-CD4+ T cells and memory CD4+ T cells, the splenocytes were incubated with a cocktail of biotinylated Abs against CD8a, CD11b, CD11c, CD19, CD25, CD45R (B220), CD49b (DX5), CD105, MHC class II, Ter-119, TCR-γ/δ, and CD44. Isolation of highly purified, naïve CD4+ T cells was achieved by depleting magnetically labeled cells. A small portion of the sorted cells was analyzed using a NovoCyte flow cytometer to confirm the efficiency of the sorting. The cells were then used for polarization assays and coculture assays.
In Vitro Treg Cell Polarization
For Treg cell polarization, sorted naïve CD4+ T cells were cultured for 72 h with plate-bound αCD3 (5 μg/mL) and soluble αCD28 (2 μg/mL) Abs under Treg cell–polarizing conditions (5 ng/mL rhTGF-β1). To investigate the influence of TSA on T cell differentiation, TSA (5 μM) or the vehicle (0.08% DMSO) was added to the cultures. All cells were analyzed by flow cytometry analysis using PE-anti-CD4, FITC-anti-CD25, and allophycocyanin-anti-FoxP3 Abs.
Generation of BMDCs
Bone marrow cells were flushed from the femurs and tibias of female C57BL/6 mice (6–8 weeks) with precooled PBS and depleted of red blood cells. The cells were cultured at a density of 2 × 106 cells/mL in 100-mm dishes in RPMI 1640 complete medium supplemented with 20 μg/mL rmGM-CSF plus 10 ng/mL IL-4. Nonadherent cells were removed, and fresh medium was added every 2 days. On day 8, nonadherent cells (bone marrow–derived dendritic cells, BMDCs) released spontaneously from the proliferating cell clusters were collected for subsequent experiments. The purity of the DCs was confirmed by flow cytometry using a PE-anti-CD11c Ab.
Naïve CD4+ T Cells Cocultured with BMDCs
In brief, BMDCs were incubated with mitomycin C (60 μg/mL) in fresh RPMI 1640 complete medium without rmGM-CSF for 30 min and then treated with 100 ng/mL LPS for 16 h in the presence of TSA (5 μM) or the vehicle (0.08% DMSO). After being washed 3 times with precooled PBS, these cells were cocultured in 24-well plates with the purified naïve CD4+ T cells described above at a ratio of 1:1 (2.5 × 105 BMDCs:2.5 × 105 naïve CD4+ T cells) in the presence of plate-bound αCD3 (5 μg/mL) and soluble αCD28 (2 μg/mL) Abs, as well as aTGF-β (10 μg/mL) or SB431542 (5 μM) [34]. Treg cell frequencies were determined 72 h later via flow cytometry analysis.
Cytokine Measurement
For cytokine detection, supernatants were harvested from the cultures at the indicated time points. The levels of IL-10 and TGF-β1 in supernatants were measured by ELISA kits (eBioscience, Santa Clara, CA) according to the manufacturer’s instructions.
In Vitro Treg Suppression Assay
To evaluate the suppressive capacity of TSA-induced Treg cultures, a proliferation assay was performed [35, 36]. Naive CD4+CD25− T effector cells (Teffs) and CD4+CD25+ Treg cells were isolated with the mouse CD4+CD25+ regulatory T cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Untouched resting B cells were sorted by negative selection with the mouse B cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Untouched resting B cells and CD4+CD25− Teffs were sorted from the spleens and peripheral lymph nodes of allogeneic mice. CD4+CD25+ Treg cells were purified from polarized naïve CD4+ T cells in the presence of TSA (5 μM) or the vehicle (0.08% DMSO) as previously described. Briefly, freshly isolated CD4+CD25− Teffs were used as responder cells upon CFSE (5 μM) labeling. Responder cells (1 × 105) were activated in the presence of soluble αCD3 (1 μg/mL) and mitomycin C (60 μg/mL)–treated B cells (APC, 0.2 × 105) alone or in the presence of Treg cells (suppressors, 0.2 × 105) for 3 days. CFSE dilution of CD4+CD25− Teffs was analyzed by flow cytometry.
Adoptive Transfer of Treg Cells
CD4+CD25+ Treg cells were isolated from polarized naïve CD4+ T cells and resuspended in PBS for injection into recipient mice (6–8-week female C57BL/6 mice). Mice were adoptively transferred by i.v. injection with CD4+CD25+ Treg cells (2 × 105 cells in 100 μL PBS) from allogeneic mice. As negative controls, mice were injected i.v. with PBS (100 μL). One day later, all recipients were immunized with 200 μg MOG35–55 peptide in CFA. Pertussis toxin (250 mg) per mouse in PBS was injected i.p. at the time of immunization and 48 h later [29, 37]. The clinical score of mice was monitored daily until 23 days p.i. Spleens were harvested at the end of the experiments for flow cytometry analyses of the levels of Th1 and Th17.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 5.0 software. All data were presented as mean ± standard error of mean (SEM). For the EAE clinical score, 2-way ANOVA followed by Bonferroni posttests were used. When 2 groups were compared, a 2-tailed unpaired Student’s t test or Mann–Whitney test was used. One-way analysis of variance (ANOVA) followed by Turkey posttests or Newman–Keuls posttests was performed to analyze experiments with more than 2 groups. A P value < 0.05 was deemed statistically significant.
Results
TSA Effectively Inhibits the Progression of EAE
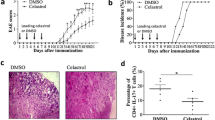
To investigate the role of TSA in EAE, we examined the in vivo effects of TSA on an actively induced EAE model. For the disease pre-onset treatment regiment, TSA (736 μg/kg/day) or the vehicle (2% DMSO in PBS) was administered i.p. daily to MOG35–55-induced mice starting from 13 days p.i., and TSA-treated mice exhibited a significant decrease in the severity of EAE compared with the vehicle control (Fig. 1a). After the fifth injection (17 days p.i.), the TSA recipients went into the peak phase, whereas the vehicle-treated mice showed continued disease progression. The clinical effects became obvious at the time of 20 days and 21 days p.i. (Fig. 1a). For the disease peak treatment protocol, TSA or the vehicle was administered i.p. daily to EAE mice starting from day 19 p.i. (Fig. 1b). Mice treated with TSA effectively displayed a suppressed secondary course of disease from 33 days p.i. to 41 days p.i. compared with the vehicle control, but it took more time for the same dose of TSA to work than it was administrated starting from EAE pre-onset (15 days for disease peak treatment vs 8 days for disease pre-onset treatment; Fig. 1a, b). The protective effect of TSA was associated with markedly reduced inflammatory foci (Fig. 1cupper, d left) and demyelination (Fig. 1clower, d right) in the TSA-treated mice compared with the vehicle-treated mice. Taken together, these data suggest that TSA has an effective therapeutic effect on EAE.

TSA ameliorates EAE and inhibits CNS inflammation. Mice were injected i.p. every day with TSA (736 μg/kg) or the vehicle (2% DMSO in PBS) starting at the day of disease onset (13 days p.i.) or disease peak (19 days p.i.), which is indicated by the black arrow. (a) Mean clinical scores for TSA (n = 14) or the vehicle (n = 10)–treated mice are shown. *P < 0.05, 2-way ANOVA followed by Bonferroni posttests. (b) Mean clinical scores for mice in the disease peak treatment experiment (n = 7 in the TSA-treated group vs n = 7 in the vehicle-treated group) are shown. *P < 0.05, ***P < 0.001, 2-way ANOVA followed by Bonferroni posttests. (c) The lumbar spinal cords from EAE mice treated with TSA or the vehicle were obtained at 21 days p.i. and stained with H&E and LFB. Scale bars = 100 μm. (d) Pathological scores for inflammation and demyelination (n = 3 in the TSA-treated group vs n = 5 in the vehicle-treated group) are shown. Data are representative of 3 independent experiments. *P < 0.05, Mann–Whitney test. Error bars denote mean ± SEM.
TSA Treatment Dramatically Reduces the Levels of Leukocytes and Microglia/Macrophages in the CNS
MNCs from the spleen or spinal cord of EAE mice were analyzed by flow cytometry for surface expression of CD45 and CD11b to differentiate between leukocytes and microglial/macrophage infiltrates. TSA treatment caused significant reductions in the percentages of leukocytes (CD45+; Fig. 2a, b), microglia (CD45intCD11b+; Fig. 2c, d left), and infiltrating macrophages (CD45hiCD11b+; Fig. 2c, d right) in the spinal cord. Histological analysis also confirmed the same results, with the numbers of both CD45+ leukocytes (Fig. 2e) and Iba-1+ microglia/macrophages (Fig. 2f) being reduced in the spinal cord of mice treated with TSA. These findings suggest that TSA inhibits the infiltration of macrophage and other leukocytes into the CNS.

TSA dramatically reduces the levels of leukocytes and activated microglia/macrophages in the CNS. (a–f) MNCs were isolated from the spinal cord of TSA or vehicle-treated EAE mice 21 days after EAE induction. Representative flow cytometry plots for leukocytes (a, CD45+), microglia (c, CD45intCD11b+), and macrophages (c, CD45hiCD11b+) gated on all MNCs are shown. The percentages of leukocytes (b) and microglia/macrophages (d) are shown (n = 3 in the TSA-treated group vs n = 5 in the vehicle-treated group). (e, f) The spinal cords were obtained from EAE mice treated with TSA or the vehicle at 21 days p.i. and immunofluorescently stained with Abs against CD45 (e, red, bar = 50 μm) and Iba-1 (f, green, bar = 50 μm). Data are representative of 3 independent experiments. *P < 0.05, ***P < 0.001, 2-tailed unpaired Student’s t test. Error bars denote mean ± SEM.
The infiltration of inflammatory cells in autoimmune inflammatory diseases of the CNS such as EAE/MS is closely related to the BBB leakage and the release of chemokines [38]. Maintaining the integrity of the BBB on a molecular level, tight junction proteins such as ZO-1 and Claudin-5 form a complex network between endothelial cells to ensure BBB integrity [39]. To elucidate the mechanism by which TSA can suppress the infiltration of inflammatory cells (leukocyte and macrophage) into the CNS, we therefore investigated whether TSA can maintain tight junction protein levels. We observed that the expression levels of ZO-1 and Claudin-5 colocalized with CD31 in the spinal cord were dramatically downregulated in vehicle-treated EAE mice on EAE peak (21 days p.i.) compared with TSA-administrated EAE mice (Fig. S2). In addition, we found that the frequencies of GM-CSF-producing CD4+ T cells from the spinal cord were also reduced (Fig. 3a, b). Collectively, these findings imply that the reduction of neuroinflammation in EAE mice administrated with TSA may be related to the maintenance of BBB integrity and the reduced release of GM-CSF.

Treg cell subsets in the spinal cord are selectively enhanced by TSA in vivo. MNCs were isolated from the spinal cord of TSA or vehicle-treated EAE mice at 21 days p.i. (a) Subpopulations of CD4+ T cells in the CNS were analyzed by flow cytometry. The subsets of Th1, Th17, and GM-CSF+ Th cells in the CD4+ T cell gate were analyzed by combining surface staining for CD4 with intracellular staining for IFN-γ, IL-17A, and GM-CSF following stimulation with 50 ng/mL PMA and 500 ng/mL ionomycin in the presence of 5 μg/mL BFA for 5 h at a density of 1 × 106 cells/mL in RPMI 1640 complete medium. Treg cells were analyzed by staining with Abs against CD4/FoxP3 and assessing the cells in the CD4+ T cell gate. (b, c) The percentages of CD4+ T (b); Th1, Th17, and GM-CSF+ Th cells; and Treg cells (c) are shown (n = 3 in the TSA-treated group vs n = 5 in the vehicle-treated group). Data are representative of 3 independent experiments. *P < 0.05, 2-tailed unpaired Student’s t test. Error bars denote mean ± SEM.
TSA Selectively Enhances the Proportion of Treg Cells in the CNS and Periphery
To investigate whether the CD4+ T cell subsets (Th1, Th17, and Treg) in TSA-treated mice are altered, surface marker (CD4) and intracellular cytokine (IFN-γ, IL-17A, and GM-CSF) or transcription factor (FoxP3) staining were performed on MNCs from the spinal cord (Fig. 3) and spleen (Fig. 4) of EAE mice. It was interesting that the frequencies of Treg cells were dramatically elevated in TSA-treated mice versus vehicle-treated EAE mice in both the spinal cord (19.90% ± 1.31% vs 13.00% ± 1.21%, respectively, P = 0.0357, Mann–Whitney test; Fig. 3a, c) and spleen (16.30% ± 0.85% vs 8.71% ± 0.82%, respectively, P < 0.0001, 2-tailed unpaired t test; Fig. 4a, c), whereas the percentages of total CD4+ T cells (spinal cord, Fig. 3a, b; spleen, Fig. 4a, b), Th1 cells, and Th17 cells (spinal cord, Fig. 3a, c; spleen, Fig. 4a, c) were not affected. In addition, the frequencies of GM-CSF-producing CD4+ T cells were reduced in the spinal cord (Fig. 3a, b) but were not changed in the spleen (Fig. 4a, b). Hence, these results indicate that the therapeutic effect of TSA may be a result of selectively promoting Treg cell differentiation in vivo.

Treg cell subsets in the spleen are elevated by TSA. (a) The subsets of CD4+ T cells in the spleen were assessed by flow cytometry. The Treg cell subsets were analyzed by staining with Abs against CD4/FoxP3 and assessing the cells in the CD4+ T cell gate. Th1, Th17, and GM-CSF+ Th cells gated on the CD4+ T cell in the spleen were analyzed by intracellular staining for IFN-γ and IL-17A following stimulation with 50 ng/mL PMA and 500 ng/mL ionomycin in the presence of 5 μg/mL BFA for 5 h at a density of 1 × 106 cells/mL in RPMI 1640 complete medium. (b, c) The percentages of CD4+ T (b); Th1, Th17, and GM-CSF+ Th cells; and Treg cells (c) are presented (n = 7 in the TSA-treated group vs n = 9 in the vehicle-treated group). Data are representative of 3 independent experiments. ***P < 0.001, 2-tailed unpaired Student’s t test. Error bars denote mean ± SEM.
In addition, to determine whether TSA is capable of driving increased MOG-reactive Treg cell generation, splenocytes obtained from EAE mice on day 8 p.i. were subjected to ex vivo experiments. Briefly, the splenocytes were restimulated with 30 μg/mL MOG35–55 peptide in the presence or absence of TSA at concentrations ranging from 1 to 10 μM for 60 h. Then, the cultured cells were analyzed by flow cytometry for CD4+CD25+FoxP3+ Treg cells, and the supernatants were assessed by ELISA for the concentrations of IL-10 secreted by the splenocytes. As shown in Fig. 5a–c, TSA (5 μM and 10 μM) treatment led to an increase in MOG-specific Treg cell generation compared with the vehicle treatment (P < 0.001, n = 5; one-way ANOVA with Tukey’s multiple comparison test; Fig. 5a, b). Similarly, the production of anti-inflammatory IL-10 was also upregulated in the supernatants of TSA-treated splenocytes (P = 0.0055, n = 5; unpaired Student’s t test; Fig. 5c). These results suggest that TSA can accelerate MOG-reactive Treg cell production.

TSA induces an increase in MOG-specific Treg cell generation in vitro. Splenocytes isolated from EAE mice at 8 days p.i. were reactivated with MOG35–55 (30 μg/mL) in the presence or absence of TSA (1 μM, 5 μM, 10 μM) or the vehicle (0.08% DMSO) for 60 h. (a) CD25+FoxP3+ Treg cells were analyzed in the CD4+ T cell gate by flow cytometry. (b) The frequencies of Treg cells were shown (n = 5, ***P < 0.001, one-way ANOVA followed by Tukey posttests). (c) Supernatants from a culture stimulated in the absence of BFA were analyzed by ELISA for the production of IL-10 (n = 5, **P < 0.01 vs vehicle, 2-tailed unpaired Student’s t test). Data are representative of 3 independent experiments. Error bars denote mean ± SEM.
TSA Significantly Enhances Treg Cell Polarization In Vitro
To further substantiate that TSA can enhance Treg cell differentiation, naïve CD4+ T cells were purified from the splenocytes of female C57BL/6 mice (6–8 weeks) and stimulated with αCD3/αCD28 Abs in the presence or absence of TSA and/or rhTGF-β1 for 72 h. We determined that the frequencies of CD25+FoxP3+ Treg cells were slightly increased by treatment with only 5 μM TSA (from 0.60% ± 0.10% in the absence of TSA to 1.75% ± 0.29% with 5 μM TSA, P > 0.05; Fig. 6a, b) but significantly improved by treatment with exogenous rhTGF-β1 (from 0.60% ± 0.10% in the absence of TSA to 8.18% ± 0.63% with exogenous rhTGF-β1, P < 0.001; Fig. 6a, b). This is precisely because Treg polarization is absolutely dependent on both TCR and TGF-β1 signaling. Moreover, the enhancement of Treg cell polarization is even more obvious when naïve CD4+ T cells are simultaneously treated with exogenous rhTGF-β1 plus 5 μM TSA (from 9.02% with rhTGF-β1 to 15.70% with rhTGF-β1 plus 5 μM TSA, P < 0.001; Fig. 6a, b). These results indicate that TGF-β1 signals are necessary for the development of Treg cells in vitro and that TSA enhances naïve CD4+ T cell polarization toward Treg cells under Treg cell–polarizing conditions.

TSA induces naïve CD4+ T cell polarization toward Treg cells in vitro by enhancing BMDC TGF-β1 production. (a, b) Sorted naïve CD4+ T cells (CD3+CD4+CD62L+CD25−CD44low) from normal mice (6–8 weeks) were cultured with TSA or the vehicle (0.08% DMSO) in the presence or absence of rhTGF-β1 following stimulation with αCD3/αCD28 Abs for 72 h. (a) Representative plots for CD25+Foxp3+ Treg cells among the CD4+ T cells were shown. (b) The percentages of CD25+Foxp3+ Treg cells among the CD4+ T cells were shown. ***P < 0.001, ns, not significant, one-way ANOVA with Newman–Keuls posttests. (c) BMDCs were prepared as previously described and analyzed by flow cytometry with a PE-anti-CD11c Ab staining to assess purity. The shaded gray histogram depicts the fluorescence-minus-one (FMO) control, the solid red line represents the anti-CD11c Ab staining, and the numbers indicate the purity of the CD11c+ DCs. (d–f) BMDCs were treated with 100 ng/mL LPS for 16 h in the presence of TSA or the vehicle in fresh RPMI 1640 complete medium without rmGM-CSF. Then, these cells were analyzed by flow cytometry using PE-CD11c/allophycocyanin-TGF-β1 Abs staining to detect the production of LAP-TGF-β1 in DCs. In addition, the culture supernatants were used to determine the TGF-β1 secretion via ELISA. (d) The production of LAP-TGF-β1 in DCs. (e) The mean fluorescence intensity (MFI) of the LAP-TGF-β1-expressing DCs was shown (n = 4). ***P < 0.001, 2-tailed unpaired Student’s t test. (f) The concentrations of TGF-β1 in the culture supernatants were shown. **P < 0.01, 2-tailed unpaired Student’s t test. (g–h) Naïve CD4+ T cells were cocultured with TSA or the vehicle-pretreated BMDCs for 72 h in the presence of αCD3/αCD28 Abs, with or without TGF-β-neutralizing Ab or SB431542 (an inhibitor of TGF-β receptor). (g) Representative plots for CD25+Foxp3+ Treg cells in CD4+ gate were shown. (h) The frequencies of CD25+Foxp3+ Treg cells among the CD4+ T cells were presented. ***P < 0.001, one-way ANOVA followed by Tukey posttests. Error bars denote mean ± SEM.
TSA Promotes Treg Cell Differentiation by Elevating the Production of BMDC-Derived TGF-β1
To identify whether APCs expressing TGF-β1 are involved in the Treg cell polarization induced by TSA, we isolated BMDCs from C57BL/6 mice (6–8 weeks) and carried out in vitro assays. The purity of the BMDCs was 95.3%, as determined by flow cytometry with an anti-CD11c Ab (Fig. 6c). We found that the expression of LAP-TGF-β1 was dramatically elevated in mitomycin C-preconditioned BMDCs by treatment for 16 h with TSA compared with the vehicle (Fig. 6d, e). Meanwhile, the concentration of TGF-β1 in the culture supernatants was also significantly enhanced by TSA treatment (from 633.1 pg/mL ± 35.8 pg/mL with the vehicle to 829.6 pg/mL ± 10.7 pg/mL with 5 μM TSA, P < 0.01; Fig. 6f). Then, these treated DCs were cocultured with freshly isolated naïve CD4+ T cells for 3 days in the presence of αCD3/αCD28 Abs. As shown in Fig. 6 g and h, TSA-pretreated BMDCs efficiently accelerated CD4+CD25+Foxp3+ Treg generation in comparison with the vehicle-pretreated BMDCs. Meanwhile, this phenomenon was almost completely eliminated by αTGF-β or SB431542 (Fig. 6g, h). Taken together, these findings demonstrate that TSA indirectly promotes the differentiation of naïve CD4+ T cells toward Treg cells by inducing TGF-β1 secretion from DCs.
TSA-Induced Treg Cells Display an Effective Suppressive Activity at a Level Comparable to TGF-β1-Polarized Treg Cells
The above experimental results indicate that TSA can induce Treg differentiation; however, we have no direct evidence to fully confirm whether TSA-induced Treg differentiation is responsible for EAE suppression. Therefore, we evaluated the suppressive capacity of TSA-induced Treg cells next. In in vitro Treg cell suppression assay, Treg cells induced with TGF-β1 alone or plus TSA significantly reduced the frequencies of proliferating Teffs (from 55.94% ± 2.73% to 35.43% ± 2.98% for Treg cells induced with TGF-β1 alone vs from 55.94% ± 2.73% to 34.53% ± 1.11% for Treg cells induced with TGF-β1 plus TSA; Fig. 7a, b); however, the suppressive activity of TSA-induced Treg cells fail to be improved compared with TGF-β1-polarized Treg cells (Fig. 7a, b). This suggests that TSA-induced Treg cells display suppressive capacity in vitro, but TSA does not alter the suppressive function of Treg cells.

TSA-induced Treg cells display suppressive activity at a level comparable to TGF-β1-polarized Tregs in vitro and in vivo. (a, b) CD4+CD25− Teffs were isolated via MACS sorting from spleens and peripheral lymph nodes of normal mice. CD4+CD25+ Treg cells were purified via MACS sorting from polarized naïve CD4+ T cells in the presence of TSA (5 μM) or the vehicle (0.08% DMSO) as previously described. CFSE-labeled CD4+CD25− Teffs (responder cells, 1 × 105) were plated in a 96-well round-bottomed plate together with CD4+CD25+ Treg cells (suppressor cells, 0.2 × 105) in the presence of mitomycin (60 μg/mL)-treated untouched rest B cells (0.2 × 105) and plate-bound αCD3 (1 μg/mL) for 72 h. Then, CFSE dilution of CD4+CD25− Teffs was analyzed by FACS. (a) Representative plots for CFSE progressive dilution of cells in the CD4+CD25− gate were shown. Bars indicate the gate of proliferated cells. (b) The percentages of proliferating CD4+CD25− Teffs were shown. n = 6, ***P < 0.001, ns, not significant, one-way ANOVA with Tukey’s posttests. (c–e) Mice were transferred by i.v. injection with CD4+CD25+ Treg cells (2 × 105 cells in 100 μL PBS) at the time of − 1 days p.i., which is indicated by the black arrow. Treg cells were purified from polarized naïve CD4+ T cells in the presence of TSA (5 μM) or the vehicle (0.08% DMSO) plus rhTGF-β1 in vitro. As negative controls, mice were injected i.v. with the same amount of PBS. After 24 h, the MOG35–55-induced EAE model was performed for all mice. The severity of mice was recorded daily until 23 days p.i. Splenocytes were harvested at the end of the experiments for flow cytometry analyses of the levels of Th1 and Th17 as described above in Fig. 4. (c) Mean clinical scores for Treg cell–transferred mice and PBS-treated mice are shown. n = 6, *P < 0.05, **P < 0.01, ***P < 0.001, for the rhTGF-β1-polarized Treg cell group versus the PBS-treated group; #P < 0.05, ##P < 0.01, ###P < 0.001, the for rhTGF-β1 plus TSA-induced Treg cell group versus the PBS-treated group; 2-way ANOVA followed by Bonferroni posttests. (d) Representative plots for Th1 and Th17 cells in the spleen (n = 6). *P < 0.05, **P < 0.01, ns, not significant, 2-tailed unpaired Student’s t test. (e) The percentages of Th1 and Th17 cells are shown (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant, 2-tailed unpaired Student’s t test. Error bars denote mean ± SEM.
Next, we preformed Treg cell adoptive transfer experiment. Similarity, we observed that the EAE progression was ameliorated in mice transferred with Treg cells compared with control mice (PBS treatment) (Fig. 7c). Moreover, Treg cell–injected mice also display delayed disease onset (Fig. 7c, d). Both the severity and onset of EAE did not display a difference between Treg cell–transferred mice (Fig. 7c, d). We also found that the frequencies of Th1 and Th17 in mice transferred with Treg cells were reduced in the spleen compared with control mice, but there were no difference between Treg cell–transferred mice (Fig. 7e, f). Collectively, these results fully confirm that TSA-induced Treg cells possess suppressive capacity at a level comparable to TGF-β1-polarized Treg cells in vitro and in vivo.
TSA Is not Capable of Protective Effects on Oligodendrocyte Progenitor Cells In Vitro
To promote the clinical translation of TSA, it is necessary to evaluate the neuroprotective effects of this small molecular compound during CNS autoimmunity. Considering that the disruption of myelin repair and myelination in the CNS is one of the main features of neuropathy in MS/EAE, oligodendrocyte progenitor cells (OPCs) were selected for neuroprotection experiment in vitro. We induced the conditioned media which derived from MOG35–55 peptide with or without TSA-stimulated splenocytes from EAE mice at 8 days p.i. This media can simulate the environment in which OPCs are exposed to the CNS autoimmunity. We observed that all kinds of conditioned media resulted in reduced proliferation, increased apoptosis, and limited differentiation of OPC in vitro (Fig. S3). TSA did not effectively improve or reverse the situation (Fig. S3). These results indicate that TSA fail to displayed protective influences on OPCs in vitro.
Discussion
Here, we are the first to discover and confirm a previously unrecognized immunoregulatory effect of TSA on Treg cell differentiation in therapeutic EAE. We identified 2 possible mechanisms by which TSA promoted Treg cell differentiation; TSA promotes Treg cell differentiation not only by acting directly on naïve CD4+ T cells dependent on TCR and TGF-β1 signaling but also through the TGF-β1 signaling pathway by driving TGF-β1 expression in APCs (such as DCs).
Treg cells are a suppressive subset of CD4+ T cells that play major roles in the maintenance of peripheral tolerance and the prevention of autoimmunity [10, 11]. It has been reported that Treg cell defects have been discovered in both MS patients and EAE mice [36, 40, 41]. Treg cells might influence disease susceptibility or course and might be associated with the relapsing–remitting MS (RR-MS) nature of this disease in some cases. Therefore, Treg therapy may have the prospect of becoming a clinical application in MS [11, 42,43,44].
Treg cell polarization is strictly dependent on TGF-β1 signaling. TGF-β1, a regulatory cytokine, has pleiotropic roles in T cell development, tolerance, and homeostasis [45,46,47]. T cell-specific deletion of the mouse Tgfb1 gene results in lethal immunopathology in multiple organs [46]. Local administration of a neutralizing TGF-β1 Ab can inhibit Th17 cell differentiation and the induction of EAE [48]. It is well documented that there are defects in the Treg cells of MS patients; thus, the modulation of TGF-β signaling may be used to rescue this deficiency [49]. Although TGF-β is produced by multiple lineages of cells, including leukocytes and stromal cells, the major source of TGF-β for iTreg cell generation in the peripheral lymphoid organs has not been fully clarified [50]. TGF-β1 is indispensable for peripheral Treg cell homeostasis; however, TGF-β1 produced by T cells is not necessary for the maintenance of peripheral Treg cells [45, 51]. Thus, TGF-β1 derived from other cellular sources (not T cells), such as APCs, may be responsible for Treg cell maintenance [29, 46]. Indeed, TGF-β signals from DCs are required for proper Treg cell development, because a DC-specific disruption of TGF-βRII produces abnormal Treg cell phenotypes and spontaneous multiorgan autoimmunity [52].
Our early results suggest that the therapeutic effect of TSA may be a result of driving Treg cell differentiation in vivo. In addition, TSA can enhance MOG-reactive Treg cell production in vitro. To test whether APCs are involved in the promotive effect of TSA on Treg cell differentiation, polarization assays and coculture assays were performed. In the coculture assay, we observed that TSA induced a tolerogenic phenotype with high TGF-β1 expression in BMDCs. Consequently, TSA-pretreated BMDCs efficiently promoted naïve CD4+ T cell differentiation into CD25+FoxP3+ Treg cells when cocultured with naïve CD4+ T cells, which was almost completely eliminated by blocking TGF-β1 pathway with αTGF-β or SB431542. These results demonstrated that TSA promotes Treg cell differentiation in vitro by targeting BMDCs to drive TGF-β1 production. In the polarization assay, we found that TSA can directly induce Treg cell polarization from naïve CD4+ T cells in the presence of αCD3/αCD28 Abs in a TGF-β1-dependent manner.
To further confirm whether TSA-mediated Treg cell differentiation is responsible for the benefit effect of TSA on EAE mice, the suppressive activity of Treg cells was evaluated. Both Treg cell suppressive assay in vitro and Treg cell transfer experiment showed that rhTGF-β1 plus TSA-induced Treg cells possessed a suppressive function at a level comparable to rhTGF-β1-polarized Treg cells, but TSA fails to alter the suppressive capacity of Treg cells. These results fully demonstrate that TSA-mediated Treg cell differentiation is directly responsible for EAE suppression.
Given the clinical transformation and safety of TSA, we also evaluated the protective activity of the small molecular compound on primary OPCs under neuroinflammation. TSA did not display benefit protective effect on OPCs under CNS autoimmunity in vitro.
In conclusion, we found that TSA potently inhibited the progression of EAE by driving Treg differentiation through the parallel mechanisms of triggering TGF-β1 production by DCs and acting directly on naïve CD4+ T cells. It is important to realize, moreover, that the precise molecular mechanisms involved are not yet fully understood. Therefore, it is worth further study to identify which receptor binds specifically to TSA and which downstream signaling molecules are potentially involved in the positive influence of TSA on Treg cell differentiation. The present study provides insights into the role of TSA as a unique therapeutic agent in the treatment of autoimmune diseases.
References
Compston A, Coles A. Multiple sclerosis. Lancet 2008; 372: 1502–1517.
Lu Y, Chen B, Song JH, et al. Eriocalyxin B ameliorates experimental autoimmune encephalomyelitis by suppressing Th1 and Th17 cells. Proc Natl Acad Sci, 2013; 110: 2258–2263.
Frohman EM. Multiple sclerosis. Med Clin North Am 2003; 87: 867–897, viii–ix.
Lassmann H. Models of multiple sclerosis: new insights into pathophysiology and repair. Curr Opin Neurol 2008; 21: 242–247.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell 2010; 140: 918–934.
Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 2011; 164: 1079–1106.
Nylander A, Hafler DA. Multiple sclerosis. J Clin Invest 2012; 122: 1180–1188.
Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006; 441: 235–238.
Jager A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol 2009; 183: 7169–7177.
Kleinewietfeld M, Hafler DA. Regulatory T cells in autoimmune neuroinflammation. Immunol Rev 2014; 259: 231–244.
Chen W, Jin W, Hardegen N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003; 198: 1875–1886.
Qin X, Guo BT, Wan B, et al. Regulation of Th1 and Th17 cell differentiation and amelioration of experimental autoimmune encephalomyelitis by natural product compound berberine. J Immunol 2010; 185: 1855–1863.
Zhang X, Koldzic DN, Izikson L, et al. IL-10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol 2004; 16: 249–256.
Taylor A, Verhagen J, Blaser K, Akdis M, Akdis CA. Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 2006; 117: 433–442.
Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature 2008; 453: 1051–1057.
Lam BY, Lo AC, Sun X, Luo HW, Chung SK, Sucher NJ. Neuroprotective effects of tanshinones in transient focal cerebral ischemia in mice. Phytomedicine 2003; 10: 286–291.
Fu J, Huang H, Liu J, Pi R, Chen J, Liu P. Tanshinone IIA protects cardiac myocytes against oxidative stress-triggered damage and apoptosis. Eur J Pharmacol 2007; 568: 213–221.
Yang R, Liu A, Ma X, Li L, Su D, Liu J. Sodium tanshinone IIA sulfonate protects cardiomyocytes against oxidative stress-mediated apoptosis through inhibiting JNK activation. J Cardiovasc Pharmacol 2008; 51: 396–401.
Yang L, Zhang B, Yin L, et al. Tanshinone IIA prevented brain iron dyshomeostasis in cerebral ischemic rats. Cell Physiol Biochem 2011; 27: 23–30.
Li S, Jiao Y, Wang H, et al. Sodium tanshinone IIA sulfate adjunct therapy reduces high-sensitivity C-reactive protein level in coronary artery disease patients: a randomized controlled trial. Sci Rep 2017; 7: 17451.
Zhou ZY, Huang B, Li S, et al. Sodium tanshinone IIA sulfonate promotes endothelial integrity via regulating VE-cadherin dynamics and RhoA/ROCK-mediated cellular contractility and prevents atorvastatin-induced intracerebral hemorrhage in zebrafish. Toxicol Appl Pharmacol 2018; 350: 32–42.
Chen Y, Wu X, Yu S, et al. Neuroprotection of tanshinone IIA against cerebral ischemia/reperfusion injury through inhibition of macrophage migration inhibitory factor in rats. PLoS One 2012; 7:e40165.
Yin X, Yin Y, Cao FL, et al. Tanshinone IIA attenuates the inflammatory response and apoptosis after traumatic injury of the spinal cord in adult rats. PLoS One 2012; 7: e38381.
Liu X, Ye M, An C, Pan L, Ji L. The effect of cationic albumin-conjugated PEGylated tanshinone IIA nanoparticles on neuronal signal pathways and neuroprotection in cerebral ischemia. Biomaterials 2013; 34: 6893–6905.
Robertson AL, Holmes GR, Bojarczuk AN, et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Sci Transl Med 2014; 6: 225ra229.
Li L, Zhang YG, Tan YF, et al. Tanshinone II is a potent candidate for treatment of lipopolysaccharide-induced acute lung injury in rat model. Oncol Lett 2018; 15: 2550–2554.
Liu X, He H, Huang T, et al. Tanshinone IIA Protects against Dextran Sulfate Sodium- (DSS-) Induced Colitis in Mice by Modulation of Neutrophil Infiltration and Activation. Oxid Med Cell Longev 2016; 2016: 7916763.
Lassmann H, van Horssen J. The molecular basis of neurodegeneration in multiple sclerosis. FEBS Lett 2011; 585: 3715–3723.
Yan Y, Zhang GX, Gran B, et al. IDO upregulates regulatory T cells via tryptophan catabolite and suppresses encephalitogenic T cell responses in experimental autoimmune encephalomyelitis. J Immunol 2010, 185: 5953–5961.
Fitzgerald DC, Zhang GX, El-Behi M, et al. Suppression of autoimmune inflammation of the central nervous system by interleukin 10 secreted by interleukin 27-stimulated T cells. Nat Immunol 2007, 8: 1372–1379.
O’Neill EJ, Day MJ, Wraith DC. IL-10 is essential for disease protection following intranasal peptide administration in the C57BL/6 model of EAE. J Neuroimmunol 2006; 178: 1–8.
Yang J, Jiang Z, Fitzgerald DC, et al. Adult neural stem cells expressing IL-10 confer potent immunomodulation and remyelination in experimental autoimmune encephalitis. J Clin Invest 2009; 119: 3678–3691.
Kang Z, Wang C, Zepp J, et al. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci 2013; 16: 1401–1408.
Zhang D, Chia C, Jiao X, et al. D-mannose induces regulatory T cells and suppresses immunopathology. Nat Med 2017; 23: 1036–1045.
Fantini MC, Dominitzki S, Rizzo A, et al. In vitro generation of CD4+ CD25+ regulatory cells from murine naive T cells. Nat Protoc 2007; 2: 1789–1794.
McGeachy MJ, Stephens LA, Anderton SM. Natural recovery and protection from autoimmune encephalomyelitis: contribution of CD4+CD25+ regulatory cells within the central nervous system. J Immunol 2005; 175: 3025–3032.
Duffy SS, Keating BA, Perera CJ, et al. Regulatory T Cells and Their Derived Cytokine, Interleukin-35, Reduce Pain in Experimental Autoimmune Encephalomyelitis. J Neurosci 2019; 39: 2326–2346.
Takeshita Y, Ransohoff RM. Inflammatory cell trafficking across the blood-brain barrier: chemokine regulation and in vitro models. Immuno Rev 2012; 248: 228–239.
Coisne C, Engelhardt B. Tight junctions in brain barriers during central nervous system inflammation. Antioxid Redox Signal 2011; 15:1285–1303.
Yu P, Gregg RK, Bell JJ, et al. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol 2005; 174: 6772–6780.
Zozulya AL, Wiendl H. The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol 2008; 4: 384–398.
Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: take a billion or so and call me in the morning. Immunity 2009; 30: 656–665.
Fransson M, Piras E, Burman J, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation 2012; 9: 112.
Keeler GD, Kumar S, Palaschak B, et al. Gene Therapy-Induced Antigen-Specific Tregs Inhibit Neuro-inflammation and Reverse Disease in a Mouse Model of Multiple Sclerosis. Mol Ther 2018; 26: 173–183.
Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006; 25:455–471.
Li MO, Wan YY, Flavell RA: T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 2007; 26: 579–591.
Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-beta: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol 2017; 9: 1–33.
Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol 2006; 7: 1151–1156.
Lee PW, Severin ME, Lovett-Racke AE. TGF-beta regulation of encephalitogenic and regulatory T cells in multiple sclerosis. Eur J Immunol 2017; 47: 446–453.
Kashiwagi I, Morita R, Schichita T, et al. Smad2 and Smad3 Inversely Regulate TGF-beta Autoinduction in Clostridium butyricum-Activated Dendritic Cells. Immunity 2015; 43: 65–79.
Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med 2005; 201: 1061–1067.
Ramalingam R, Larmonier CB, Thurston RD, et al. Dendritic cell-specific disruption of TGF-beta receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol 2012; 189: 3878–3893.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81571596, 81601044, and 81771279) and the Fundamental Research Funds for the Central Universities (2018TS088, GK201701009, and GK201603110).
Required Author Forms Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Contributions
Langjun C and Yaping Y designed the research; YG, Huanhuan S, and Yuanchu L performed the research; YG, Xiaoli D, and YF analyzed the data; and YG, YF, Langjun C, and Yaping Y wrote the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Gong, Y., Liu, YC., Ding, XL. et al. Tanshinone IIA Ameliorates CNS Autoimmunity by Promoting the Differentiation of Regulatory T Cells. Neurotherapeutics 17, 690–703 (2020). https://doi.org/10.1007/s13311-019-00789-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-019-00789-2