
Abstract
Insufficient supply of selenium to antioxidant enzymes in the brain may contribute to Alzheimer’s disease (AD) pathophysiology; therefore, oral supplementation may potentially slow neurodegeneration. We examined selenium and selenoproteins in serum and cerebrospinal fluid (CSF) from a dual-dose 24-week randomized controlled trial of sodium selenate in AD patients, to assess tolerability, and efficacy of selenate in modulating selenium concentration in the central nervous system (CNS). A pilot study of 40 AD cases was randomized to placebo, nutritional (0.32 mg sodium selenate, 3 times daily), or supranutritional (10 mg, 3 times daily) groups. We measured total selenium, selenoproteins, and inorganic selenium levels, in serum and CSF, and compared against cognitive outcomes. Supranutritional selenium supplementation was well tolerated and yielded a significant (p < 0.001) but variable (95% CI = 13.4–24.8 μg/L) increase in CSF selenium, distributed across selenoproteins and inorganic species. Reclassifying subjects as either responsive or non-responsive based on elevation in CSF selenium concentrations revealed that responsive group did not deteriorate in Mini-Mental Status Examination (MMSE) as non-responsive group (p = 0.03). Pooled analysis of all samples revealed that CSF selenium could predict change in MMSE performance (Spearman’s rho = 0.403; p = 0.023). High-dose sodium selenate supplementation is well tolerated and can modulate CNS selenium concentration, although individual variation in selenium metabolism must be considered to optimize potential benefits in AD. The Vel002 study is listed on the Australian and New Zealand Clinical Trials Registry (http://www.anzctr.org.au/), ID: ACTRN12611001200976.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Selenium is essential for normal neurological function [1]. Insufficient selenium intake produces inactive selenoproteins, which increases vulnerability to oxidative stress. In the brain, this has been associated with cognitive decline [2, 3]. This may contribute to the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease (AD).
Oxidative damage is a biochemical hallmark of AD [4], and therapies designed to reduce cellular oxidative load may have therapeutic potential [5]. The clinical trials of selenium supplementation in AD patients are small in number and power, and have produced inconclusive data (reviewed by Loef et al. [6]). Nevertheless, reported associations between selenium status and cognition from human post mortem findings, as well as animal studies, indicate a potential role for selenium deficiency in AD.
There is no consensus yet regarding changes in circulating selenium levels in AD [7], although several large cohort studies reported lowered levels [3, 8, 9]. We recently reported that post mortem temporal cortex samples from AD cases contain ≈ 14% less total selenium than age-matched healthy controls [10]. The apolipoprotein-E (APOE) ε4 allele, which is the major genetic risk factor for AD, was also associated with a decrease in selenium in these samples, and a redistribution of selenium in the tissue from the membrane-bound and insoluble fractions to the soluble fraction [10].
A small-scale randomized pilot trial of subjects with mild cognitive impairment (MCI) [11], supplemented daily with selenomethionine (SeMet)-rich Brazil nuts (Bertholletia excelsa; ≈ 290 μg selenium/day; ~75% as SeMet [12]) found improved verbal fluency and constructional praxis after 6 months compared to controls [13]. In contrast, the large-scale primary prevention PREADViSE study (ClinicalTrials.gov identifier NCT00040378) found that long-term (5.4 ± 1.2 years of supplement use) daily supplementation with SeMet (200 μg/day) did not decrease the risk for dementia in cognitively healthy males over 60 years of age [14].
Selenium supplementation can also be achieved using inorganic forms, such as selenate (SeO42−) and selenite (SeO32−). Selenate reduces pathological tau hyperphosphorylation common to AD via activation of protein phosphatases 2A both in vitro and in animal models of tauopathies [15,16,17]. It also downregulates the expression of BACE1, a key enzyme involved in the AD-associated amyloid deposition [18], and reduces levels of amyloid and markers of nucleic acid oxidation in APP/PS1 transgenic mice [19]. Our group recently reported results of a Phase IIa exploratory trial of selenate in AD (Vel002) [20], and showed that despite a significant amelioration of brain structural deterioration, there were no significant effects on cognitive performance outcomes. However, it is not proven whether selenate effectively delivers selenium into the central nervous system (CNS). In this study, as exploratory analysis, we examined the selenium concentration in serum and cerebrospinal fluid (CSF) taken from patients participating in this Phase IIa trial to assess the degree to which 24-week sodium selenate (Na2SeO4) supplementation at the doses used increased serum and CSF selenium concentration, the latter indicative of selenium uptake by the CNS.
Subjects and Methods
Study Participants
The Vel002 study (Australian and New Zealand Clinical Trials Registry ID: ACTRN12611001200976) recruited patients in 4 centers in Melbourne, Australia, who were diagnosed with probable AD according to NINCDS-ADRDA criteria [21]. Eligible subjects were ≥ 55 years old; presented with a modified Hachinski score ≤ 4 and a “mild” to “moderate” degree of dementia, as defined by a Mini-Mental Status Examination (MMSE) score of between 14 and 26 at screening; were under treatment with an acetylcholine esterase inhibitor at a stable dose for at least 4 months; and had a documented volumetric MRI brain scan performed within 14 days of baseline that revealed no gross structural abnormality. Exclusion criteria were contraindication for lumbar puncture; history of alcohol and/or other substance abuse; known sensitivity to selenium; presence of any other dementia syndrome or other neurological or psychiatric illness; significant medical disease not adequately controlled; history of epilepsy, diabetes, impaired renal, hepatic, or hematological function; known history of familial AD; current or recent (within 6 weeks of screening) treatment with lithium, NMDA receptor antagonists, steroids, or injectable non-steroidal anti-inflammatory drugs; current treatment with carbamazepine, digoxin, phenobarbitone, phenytoin, or warfarin; and consumption of dietary supplements containing more than 26 μg selenium/day [20].
Informed consent was obtained before the interview from all participants or their legally authorized representative, and the participant’s caregiver. The study was approved by the Melbourne Health Institutional Ethics Committee.
Study Protocol
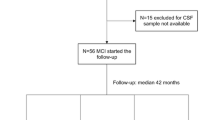
Full details of the trial protocol can be found in Malpas et al. [20]. This was a double-blind, randomized, placebo-controlled pilot study. An initial screening visit was performed to confirm AD diagnosis by an experienced clinical neurologist or neuropsychiatrist. Following screening, 40 participants were randomly assigned to one of 3 study groups for 24 weeks’ treatment: placebo, “nutritional” (0.32 mg of sodium selenate, 3 times per day); and “supranutritional” (10 mg of sodium selenate, 3 times daily). The randomization sequence was a 1:1:2 ratio, and this sample size was determined based on the early stage of investigation, i.e. Stage IIa as reported by Malpas et al. [20]. All investigators, participants, and caregivers remained blinded to randomization status until the conclusion of the trial [20]. A total of 36 participants completed the study. The participant CONSORT flow chart is shown in Fig. 1. A list of the biofluid samples for this study is provided in Supplementary Table 1.

Participant CONSORT flow chart. “Analyzed” refers to subjects who completed the protocol, where cognitive testing was completed at baseline and week 24, and where at least 1 biofluid sample was measured for selenium content
Determination of Total Selenium Concentration in Serum and CSF
Selenium concentration in serum and CSF was measured using inductively coupled plasma-mass spectrometry (ICP-MS). Neat serum and CSF were diluted in 1% nitric acid (1:20 and 1:3 respectively, to 300 μL final volume). Selenium was measured on mass at m/z = 78 (78Se; natural abundance = 23.8%) using an Agilent Technologies 7700× ICP-MS system (Agilent Technologies, Australia) fitted with “cs” lenses and platinum cones. Hydrogen (4 mL/min) was used as a reaction gas to remove polyatomic interferences at m/z = 78. Values were the average of 4 technical replicates. Selenium concentrations were calculated by external calibration using multi-element standards (AccuStandard, USA) containing of 0, 0.1, 0.5, 1, 5, 10, 50, and 100 μg/L of selenium. An internal standard solution containing 200 μg/L of yttrium (89Y) was introduced online via a Teflon T-piece. Analytical validity was assessed using reconstituted lyophilized Seronorm™ Trace Elements in Serum (Sero AS, Norway) standard reference materials, which was prepared using the same protocol for serum samples. The measured analytical recovery of selenium in the Seronorm™ standard was within the acceptable range, per manufacturer’s guidelines (measured serum = 153.89 ± 6.48 μg/L, n = 4; certified range = 95–176 μg/L).
Chromatographic Analysis of Selenoproteins
Frozen aliquots of serum and CSF samples from the supranutritional group (baseline and 24 weeks) were brought to 4 °C and transferred into standard glass chromatography vials with polypropylene low-volume inserts. A 20-μL injection of neat serum/CSF was resolved using a BioSEC3 150 Å, 4.6 × 300 mm size exclusion chromatography (SEC) column (Agilent Technologies) with a molecular weight range of 500 to 150,000 Da on an Agilent Technologies 1200 Series liquid chromatography (LC) system equipped with a Peltier-cooled (4 °C) autosampler. A 200-mM ammonium nitrate buffer containing 10 μg/L cesium and antimony as online internal standards (see Lothian and Roberts [22]) was adjusted to pH 7.5–7.7 with 28% ammonium hydroxide and used as the isocratic mobile phase (0.4 mL/min flow rate) for all separations. The column was calibrated for molecular mass estimation using a standard mix of heteroatom and metal-containing proteins [23], and injections of sodium selenate (1.88 ppb) prepared in the chromatography buffer were used to estimate selenate retention time in serum and CSF samples. Selenium was measured with the same instrument configuration described above. The size exclusion chromatography (SEC) eluent was directly connected to the concentric nebulizer (Glass Expansion, Australia) of the ICP-MS via polyethyl ether ketone tubing. The LC and ICP-MS systems were controlled using Mass Hunter (Agilent Technologies) and all SEC-ICP-MS chromatographic traces were measured in time resolved analysis mode.
Cognitive Testing
Cognitive testing was performed at baseline and week 24. Conventional “pencil-and-paper” tests were administered, including the Alzheimer’s Disease Assessment Scale cognitive subscale (ADAS-Cog), MMSE, controlled oral word association test (COWAT), and the category fluency test (CFT). Three tests were also administered from the CogState computerized battery (CogState Ltd., Melbourne, Australia). These included the 1-card learning memory task (OCL), identification reaction time task (IDN), and the detection reaction time task (DET).
Statistical Analysis
All statistical analyses were conducted on the intention to treat population. Data were included for all participants enrolled in the study who had complete data for this exploratory analysis. Baseline characteristics were compared between groups using ANOVA for continuous data and using Fisher’s exact test when data were categorical. Longitudinal changes were assessed by Wilcoxon Signed Rank or paired Student’s t tests. Correlation analyses were performed using Pearson’s correlation coefficient or Spearman’s rho, depending on the presence or absence of normal data distribution as assessed by Kolmogorov-Smirnov. When correlation analysis was performed using change variables, log transformed data was included instead of raw numbers. For SEC-ICP-MS chromatograms, peaks areas were derived using the standard width-at-half-height method in Prism Version 6 h (GraphPad, USA), and compared against peaks with corresponding molecular mass using a paired Student’s t test.
Changes in selenium biomarkers (i.e. total selenium and chromatographically-separated selenoproteins) and cognitive tests were calculated as the difference between values at 24 weeks and baseline. Participants were classified as responsive or non-responsive to selenate treatment according to the difference in measured indicators of selenium status, with the responsive group presenting an increment in serum or CSF post treatment at least 3 times above the highest value at baseline. Comparisons between responsive and non-responsive groups were performed using a Mann-Whitney U test. The Kruskall-Wallis followed by Dunn’s post hoc test was conducted on the comparison of the changes in selenium levels in CSF normalized by change in serum in the 3 treatment groups. All statistical analyses were carried out using the Statistical Package for the Social Sciences software, version 22.0 (SPSS; IBM, USA), and figures were constructed in Prism version 6 h (GraphPad, USA) and Adobe Illustrator CC 2018 (Adobe Systems, USA). Statistical significance was set at p < 0.05.
Results
Studied groups were similar for age, sex, MMSE score, and APOE ε4 allele frequency (Table 1), as well as for selenium biomarkers (Table 2). All but 1 participant were selenium sufficient at baseline according to the plasma selenium reference range of 84–100 μg/L recommended by Thomson [24] required to maintain adequate glutathione peroxidase (GPx) activity and selenoprotein P concentration (Table 2). Both experimental groups receiving sodium selenate treatment showed significant increases in selenium concentration in serum (nutritional + 45%, p < 0.01, Student’s t test; supranutritional + 504%, p < 0.001, Wilcoxon Signed Rank test) and CSF (nutritional + 69.3%, p < 0.05; supranutritional + 1680%, p < 0.001, Student’s t test; Table 2; Fig. 2) from baseline. One individual in the nutritional group had unchanged serum selenium concentrations and decreased levels in CSF, which we believe was due to poor compliance with treatment regime. As expected, there was a dose-dependent effect on serum selenium concentration, with the magnitude of change in serum selenium concentration in the supranutritional group showing an approximately tenfold increase in serum concentration compared to the nutritional group at the study conclusion. CSF selenium was only moderately increased by nutritional supplementation (+ 56%, p < 0.05 vs baseline), whereas supranutritional intake produced a more marked change (+ 1395%, p < 0.001 vs baseline) with high variance between participants.

Selenium concentrations in serum (A, B, and C) and cerebrospinal fluid (D, E, and F) at baseline and after 24 weeks. (A and D) Placebo; (B and E) Nutritional group; (E and F) Supranutritional group. (A, B, D, E, F) Student’s paired t test; (C) Wilcoxon Signed Rank test. *p < 0.05; ***p < 0.001
As CSF represents the main export pathway from the brain, increased selenium concentration is indicative that more has entered the CNS. We therefore examined the correlation between serum and CSF selenium concentration to assess the neuro-bioavailability of selenate. At baseline, no correlation was observed between serum and CSF concentration (Fig. 3a). Post-treatment, there was also no correlation between serum and CSF selenium concentrations for either the placebo or nutritional groups, although these sample sizes were small (Fig. 3b, c). Post-treatment, only the high-dose supranutritional group showed a correlation between serum and CSF selenium (r = 0.653, p < 0.05, Spearman’s rho; Fig. 3d). We examined whether the dimension of the change in CSF selenium content after 24 weeks’ treatment was commensurate with the change in serum selenium content in our 3 treatment groups. We analyzed the change in serum selenium matched to the change in CSF selenium in subjects where both samples were available and assayed at baseline and 24 weeks of treatment. The mean changes in each group indicated that the boost in serum selenium following supplementation was matched by a muted increase in CSF selenium. Of the increase in serum selenium in the 2 supplementation group, only ≈ 3% of the increase was transduced into the CSF (Fig. 3e). The change in CSF selenium was approximately proportional to the change in serum in both the nutritional and the supranutritional selenate supplementation groups. In the supranutritional group, the increase in CSF selenium as a proportion of the change in serum selenium was significantly more than in the placebo group (Fig. 3f, ≈ 3%, Dunn’s test p = 0.0019), consistent with a small proportion of the boost in serum selenium surmounting the blood brain barrier with this high dosage regimen.

Correlation between selenium concentration in serum and CSF. (A) Baseline (n = 32). (B) Placebo group, post treatment (n = 6). (C) Nutritional group, post treatment (n = 7). (D) Supranutritional group, post treatment (n = 14). (E) Changes in serum selenium (x axis) and CSF selenium (y axis) in matched subjects (where both serum and CSF samples were available and assayed at both baseline and 24 weeks of treatment). Data are means ± SD, n = 6, 7 and 13 for placebo, nutritional and supranutritional groups, respectively. The axes are in log units to capture the large shifts in values as the doses are increased. (F) Individual data points from each subject (with box and whisker analysis) expressed as the change in CSF selenium per unit change in serum selenium for each matched subject. The supranutritional group exhibited a small boost in CSF selenium when normalized to the change in serum selenium (≈ + 3%, or ≈ 30 mg/g). P value is from Dunn’s multicomparison’s test
According to the original trial data reported [20], the diffusion tensor imaging (DTI) analysis revealed that the supranutritional group had less of a reduction in white matter organization, evidenced by decreased mean, axial and radial diffusivity. This may suggest a measurable clinical benefit, as sodium selenate may have a direct effect on slowing white matter atrophy in the human brain. We analyzed subsets of serum and CSF (n = 11, samples with sufficient volume left after other measurements) from the supranutritional-dose group by SEC-ICP-MS to assess treatment-induced changes in selenium-containing macromolecules and low molecular weight inorganic selenium species. Selenate supplementation clearly increased binding of selenium into serum proteins (Fig. 4a), with a large increase (+ 656%; p < 0.001, Student’s t test) in the earliest eluting peak (Peak #1, Fig. 4a). We have previously characterized the selenium content in this peak by SEC-ICP-MS and MS/MS bottom up proteomics, and confirmed the presence of both selenoprotein P (comprising ≈ 50% of this peak) and albumin that could not be discriminated by chromatography at this resolution [25]. Selenoprotein P has a molecular mass of 43 kDa, but is highly glycosylated. The molecular mass of albumin is 67 kDa, and Peak #1 has an apparent Mr of 75 kDa against size standards. Using this approach, we cannot discriminate between selenium incorporated as selenocysteine and that being transported via transient binding to free thiol groups on serum albumin [26]. Chemically inert buffers at physiological pH used for SEC-ICP-MS preserve the integrity of selenium thiol ligands, with the compromise being relatively low separation efficiency [27]. As such, we suspect that Peak #1 contains highly abundant serum albumin followed closely (without resolution) by selenoprotein P. Differentiating the proportional increase in selenium directly attributable to both proteins would require higher resolution chromatographic methods [28], though the effects of the denaturing conditions typically employed on albumin-selenium binding have not been characterized.

Selenium trace of supranutritional group samples at baseline (blue) and post treatment (red) on LC-ICP-MS. Dashed line = sodium selenate (1.88 ppb). Selenium-containing peaks identified based on mass alone and should be considered approximations. (A) Serum: Peak #1: co-eluting albumin-associated selenium and selenoprotein P; Peak #2: selenate; Peak #3: inorganic selenium; (B) CSF: Peak #1: albumin-associated selenium; Peak #2: selenoprotein P; Peaks #3 and #4: inorganic selenium. Data are average ± SEM, n = 11 matched serum or CSF samples from the same subjects; ***p < 0.001, Student’s t test of area under curve
Two additional resolved peaks were also significantly increased (p < 0.001, Student’s t test) in serum from the supranutritional group, representing inorganic selenate and an unidentified selenium-containing compound, both eluting below the lower molecular weight limit of the SEC column. Interactions between the silica column and negatively charged inorganic selenium compounds are known to influence retention time in SEC-ICP-MS [29]; thus, we suspect that the peak following the selenate standard (serum Peak #3, CSF Peak #4, Fig. 4) might be selenite (SeO32−). A previous analytical study of the stability of selenocompounds in human serum suggested selenite is not present in freshly-drawn serum and is an artifact of storage, but the report examined serum from only 2 donors, with total serum selenium at levels not commensurate with our supranutritional group [30]. Regardless of its chemical species, the appearance of this selenium-containing peak at 24-weeks is indicative of a specific response to supranutritional selenium supplementation.
In CSF samples, we again identified a marked increase in the low mass peaks, selenate (Peak #3 provisionally, +29.7%, p < 0.001) plus selenite (suspected for Peak #4), following supranutritional supplementation. The lower total selenium concentration in CSF reduced the effect of peak tailing, allowing selenoprotein P (likely to be Peak #2), which we have previously confirmed in CSF samples using a targeted proteomics approach [25], to be resolved from albumin (likely to be Peak #1), which is also present in CSF [31]. Selenium bound to albumin was markedly increased in the supranutritional group (Peak #1 in Fig. 4b, + 3022%, p < 0.001), as was the selenoprotein P peak following selenate treatment (Peak #2 in Fig. 4b, p < 0.001; Table 3). Both selenium-binding proteins were below the limit of detection in untreated CSF, consistent with previous findings [32]. It was also apparent from the ratios of the selenium in the chromatographic protein peaks to the low mass peaks (Table 3) that selenate supplementation boosted selenoprotein production far more in the periphery (serum. Fig. 4a) than in the brain (CSF, Fig. 4b). Thus, to augment brain levels of selenoprotein P (and potentially other selenoproteins) may require higher levels of selenate precursor than peripheral organs.
In both the nutritional and supranutritional groups, there was considerable variance in the response in serum and CSF selenium concentration to selenium supplementation. By stratifying the data into responsive (defined as change in serum and CSF 3 times above the highest baseline value) or non-responsive (all remaining samples), regardless of dose, we were able to reassess the effects of selenate supplementation in cognitive performance per the original trial outcomes report [20]. For the MMSE readout, according to the CSF marker, the unresponsive group deteriorated during the trial by − 3.1 ± 3.5 points (p < 0.0049; paired Student’s t test), but the responsive group did not significantly deteriorate (− 0.4 ± 3.0 points; p = 0.646; paired Student’s t test), with paired analysis indicating that the difference between these 2 groups was significant (p = 0.03; Mann-Whitney U test; Fig. 5a). No differences were observed in any other measures of cognition (Fig. 5b–g). Pooled CSF selenium concentration from all groups correlated with change in MMSE performance (r = 0.403; p < 0.05, Spearman’s rho; Fig. 5h), indicating that change in CSF selenium may be associated with improved cognitive performance following supplementation.

Cognitive performance changes in subjects categorized as either responsive or non-responsive to sodium selenate treatment, according to serum (n = 17 and 18 for responsive and non-responsive, respectively) and CSF (n = 12 and 14 for responsive and non-responsive, respectively) changes. Changes (Δ) were calculated as [post-treatment − baseline] scores. (A) MMSE: Mini-Mental State Examination (*p < 0.005, Mann-Whitney U test). (B) ADAS-Cog: Alzheimer’s Disease assessment scale—cognitive subscale. (C) CFT: Category fluency test. (D) COWAT: Controlled oral word association test. (E) DET: Detection reaction time task. (F) OCL: 1-card learning memory task. (G) IDN: Identification reaction time task. (H) Correlation between changes in CSF selenium concentration and in MMSE score in combined responsive and non-responsive groups (r = 0.403; p < 0.05, Spearman’s rho)
Treatment emergent adverse events (TEAEs) were reported previously according to treatment group, with 90% reporting at least 1 TEAE [20]. To summarize, all TEAEs were reported as mild. The most common solicited TEAEs (incidence ≥ 20% in both placebo/nutritional and supranutritional groups) were fatigue, headache, and lethargy; with nausea, muscle spasms, and dizziness reported in the supranutritional group. One participant experienced a pre-syncopal serious adverse event that was resolved in 24 h and continued in the study. Two supranutritional group participants withdrew due to TEAEs; 1 due to appearance of a skin rash of uncertain cause, and the other resulting from dysmorphic changes in toenails and fingernails. In the latter case, the participant had the 24-week observation and was not excluded from further analysis. On application of our responsive/non-responsive stratification, no relationship between the number of TEAEs and level of selenium uptake was observed (p = 0.976 for serum; p = 0.900 for CSF; Fisher’s exact test).
Discussion
While the readouts of this pilot study of selenate supplementation in AD reported no significant benefit on cognitive performance [20], there was no stratification by biofluid selenium biomarkers in the initial report. Here, we find that the retention of selenium in serum and CSF was highly variable in this study. When the participants were stratified according to response in biofluid to the selenate supplementation, a significant arrest in cognitive deterioration on MMSE was noted. This was not corroborated by the other performance tests used. However, this pilot study was underpowered to detect cognitive changes. Our findings inform future trial design of selenium supplementation, supporting stratification of outcome measures by biofluid selenium changes.
Due to concern about the potential toxicity of inorganic selenium [33], intervention studies have tended to focus on organic compounds. For instance, a double-blind, randomized, placebo-controlled trial of selenium-enriched yeast supplementation [34], which contained SeMet at 54–60% of the total selenium (including inorganic species) found that a dose of 300 μg of selenium-enriched yeast per day over 5 years was well tolerated by older adults (mean age 66.1 years) during the 5 years of dosing. However, at a 10-year follow-up, this dose group exhibited increased all-cause of mortality (hazard ratio = 1.59, 95% CI = 1.02–2.46), especially for those who had baseline plasma selenium ≥ 82 μg/L (hazard ratio = 2.20, 95% CI = 1.16–4.17) [35]. While such studies do raise concerns regarding the possible long-term toxic effects of the dose used in our trial (equivalent to 12.5 mg of elemental selenium per day, much higher than Rayman et al. [35]), our 6-month intervention did not report any life-threatening TEAEs. In contrast to this long-term supplementation study of healthy adults, we tested high-dose selenate treatment as a chemotherapeutic disease-modification intervention.
While significantly more subjects in the Supranutritional group (35%) experienced TEAEs compared to the placebo group (10%) [20], the adverse effect profile was similar to that associated with toxicity resulting from consumption of a misformulated nutritional supplement in the US in the late 2000s that contained selenium at ≈ 400-fold the recommended daily allowance (RDA) [36, 37]. In this incident, an unknown number of consumers inadvertently received a daily selenium dose in the order of 30 mg equivalent, as well as ≈ 30 mg of chromium and other substances. We note that the misformulated supplement exposed subjects to an average of ≈ 30 mg of elemental selenium per day. The highest dose we tested was 30 mg of sodium selenate per day in divided doses, which is equivalent to 12.5 mg of elemental selenium per day, i.e. less than half the dose of selenium in the misformulated supplement. Furthermore, the chromium in the misformulated supplement (17-fold the RDA), as well as many other bioactive ingredients in the mixture, may have potentiated the toxicological burden. In any case, since our trial of sodium selenate at 30 mg per day was completed under controlled conditions, the data are important to examine whether this drug can be used safely in this disease context. Our observations that 30 mg of sodium selenate per day for 24 weeks, being less than half that of the misformulated supplement, without serious adverse events is reassuring for the use of this dose as a chemotherapeutic, where the benefits may exceed the risk of mild TEAEs.
Our data indicate that selenate supplementation promotes protein incorporation, supporting the possibility that inorganic selenium toxicity in the brain is mitigated by endogenous selenoprotein production [38]. Furthermore, it has been shown that while both SeMet and selenate are readily bioavailable (both > 90%), the half-life of selenate is considerably shorter [39]. It has been suggested that the dose-response for selenium intake and benefits to human health follows a U-shaped relationship, indicating that selenium supplementation to populations with adequate or high selenium status could cause adverse effects [40]. Further studies to clarify risk-benefit profiles for different selenocompounds are needed, particularly when being used as a treatment for a terminal condition such as AD.
To our knowledge, this is the first study to determine selenium concentration in paired serum and CSF samples before and after supplementation, which allowed us to evaluate the bioavailability of sodium selenate to the CNS and its ability to promote selenoprotein synthesis. Following supplementation, the distribution of selenium into selenoproteins as a proportion of total selenium in the biofluid was much greater for serum than for CSF (Fig. 4), which confirms that the blood-brain barrier may limit the entry of selenium into the brain.
Our findings that small molecular weight selenium species are the dominant forms in the CSF are at variance with those of Solovyev et al. [32], who found selenoprotein P was the major selenocompound in CSF of healthy individuals, followed by selenomethionine bound to albumin. Both Solovyev and colleagues’ study and the present work used archived frozen samples; thus, the discrepancy between the studies is unlikely to be due to selenite artifact from storage [30].
Our observations are consistent with 2 potential fates for sodium selenate: (i) reduction to selenite, according to a pathway previously identified in the gut [41]; and (ii) direct incorporation into selenoproteins that cross the blood-brain barrier, which would support efficiency of this compound in promoting antioxidant activity in the CNS. While selenoprotein P is the master regulator of selenium delivery to the CNS [42], glutathione peroxidase 4 (GPx4) is the most abundant selenoprotein in brain, and has recently garnered attention as an important regulator of ferroptosis [43], a newly identified form of iron-dependent programmed cell death that causes aggressive lipid peroxidation [44, 45] thought to play a major role in AD pathology [46]. Since AD-affected brain tissue has lower levels of selenium [6, 10] and selenium supplementation has been shown to directly interdict amyloid and iron neurotoxicity by modulating GPx activity [47], GPx4 expression and activity may suffer from insufficient selenium supply, and on this basis, supplementation trials are worth exploring.
Conclusions
This pilot trial showed that sodium selenate supplementation at a high or supranutritional dose induced an increase in selenium uptake into the CNS. The elevation in CSF selenium induced by treatment varied considerably among participants, indicating that factors, such as genetics, influence selenium delivery to the brain. Analysis of selenoproteins in CSF suggested inorganic selenium could increase expression and incorporation of selenium into biomolecules. When stratifying the study groups as either responsive or non-responsive to selenate supplementation, we found subtle but significant, improvement in MMSE score was associated with selenium CSF. Although 24 weeks of treatment was well tolerated, the potential benefits of selenium supplementation for AD must be weighed against recent data reporting increased mortality in healthy elderly subjects after long term supplementation.
Abbreviations
- AD:
-
Alzheimer’s disease
- ApoE:
-
Apolipoprotein E
- CNS:
-
Central nervous system
- CSF:
-
Cerebrospinal fluid
- GPx:
-
Glutathione peroxidase
- ICP-MS:
-
Inductively coupled plasma-mass spectrometry
- MMSE:
-
Mini-Mental Status Examination
- MRI:
-
Magnetic resonance imaging
- Se:
-
Selenium
- SeMet:
-
Selenomethionine
- SeO4 2− :
-
Selenate
- SeO3 2− :
-
Selenite
- SAE:
-
Serious adverse event
- SEC:
-
Size exclusion chromatography
- TEAE:
-
Treatment emergent adverse event
References
Schweizer U, Bräuer AU, Köhrle J, Nitsch R, Savaskan NE. Selenium and brain function: a poorly recognized liaison. Brain Res Rev. 2004;45(3):164–78.
Berr C, Balansard B, Arnaud J et al. Cognitive Decline Is Associated with Systemic Oxidative Stress: The EVA Study. J Am Geriatr Soc. 2000;48(10):1285–91.
Cardoso BR, Ong TP, Jacob-Filho W et al. Nutritional status of selenium in Alzheimer's disease patients. Br J Nutr. 2010;103(6):803–6.
Lin MT, Beal FM. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95.
Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3(3):205–14.
Loef M, Schrauzer GN, Walach H. Selenium and Alzheimer's disease: a systematic review. J Alzheimers Dis. 2011;26:81–104.
da Silva S, Vellas B, Elemans S et al. Plasma nutrient status of patients with Alzheimer's disease: Systematic review and meta-analysis. Alzheimers Dement. 2014;10(4):485–502.
Gao S, Jin Y, Hall KS et al. Selenium level and cognitive function in rural elderly Chinese. Am J Epidemiol. 2007;165(8):955–65.
Reddy VS, Bukke S, Dutt N, Rana P, Pandey AK. A systematic review and meta-analysis of the circulatory, erythrocellular and CSF selenium levels in Alzheimer's disease: A metal meta-analysis (AMMA study-I). J Trace Elem Med Biol. 2017;42:68–75.
Cardoso BR, Hare DJ, Lind M et al. The APOE ε4 Allele Is Associated with Lower Selenium Levels in the Brain: Implications for Alzheimer’s Disease. ACS Chem Neurosci. 2017;8:1459–64.
Portet F, Ousset PJ, Visser PJ et al. Mild cognitive impairment (MCI) in medical practice: a critical review of the concept and new diagnostic procedure. Report of the MCI Working Group of the European Consortium on Alzheimer’s Disease. J Neurol Neurosurg Psychiatry. 2006;77(6):714–8.
da Silva E, Mataveli L, Arruda M. Speciation analysis of selenium in plankton, Brazil nut and human urine samples by HPLC–ICP-MS. Talanta. 2013;110:53–7.
Cardoso BR, Apolinario D, da Silva Bandeira V et al. Effects of Brazil nut consumption on selenium status and cognitive performance in older adults with mild cognitive impairment: a randomized controlled pilot trial. Eur J Nutr. 2016;55(1):107–16.
Kryscio RJ, Abner EL, Caban-Holt A et al. Association of antioxidant supplement use and dementia in the prevention of alzheimer’s disease by vitamin e and selenium trial (preadvise). JAMA Neurol. 2017;74(5):567–573.
van Eersel J, Ke YD, Liu X et al. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer's disease models. Proc Natl Acad Sci USA. 2010;107(31):13888–93.
Corcoran NM, Martin D, Hutter-Paier B et al. Sodium selenate specifically activates PP2A phosphatase, dephosphorylates tau and reverses memory deficits in an Alzheimer's disease model. J Clin Neurosci. 2010;17(8):1025–33.
Shultz SR, Wright DK, Zheng P et al. Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain. 2015;138(Pt 5):1297–313.
Jin N, Zhu H, Liang X et al. Sodium selenate activated Wnt/beta-catenin signaling and repressed amyloid-beta formation in a triple transgenic mouse model of Alzheimer's disease. Exp Neurol. 2017;297:36–49.
Lovell MA, Xiong S, Lyubartseva G, Markesbery WR. Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic Biol Med. 2009;46(11):1527–33.
Malpas CB, Vivash L, Genc S et al. A Phase IIa Randomized Control Trial of VEL015 (Sodium Selenate) in Mild-Moderate Alzheimer's Disease. J Alzheimers Dis. 2016;54(1):223–32.
McKhann G, Drachman D, Folstein M et al. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34(7):939–44.
Lothian A, Roberts BR. Standards for Quantitative Metalloproteomic Analysis Using Size Exclusion ICP-MS. J Vis Exp. 2016; https://doi.org/10.3791/53737(110):53737.
Hare DJ, Grubman A, Ryan TM et al. Profiling the iron, copper and zinc content in primary neuron and astrocyte cultures by rapid online quantitative size exclusion chromatography-inductively coupled plasma-mass spectrometry. Metallomics. 2013;5(12):1656–62.
Thomson CD. Assessment of requirements for selenium and adequacy of selenium status: a review. Eur J Clin Nutr. 2004;58(3):391–402.
Kipp AP, Strohm D, Brigelius-Flohé R et al. Revised reference values for selenium intake. J Trace Elem Med Biol. 2015;32:195–9.
Xia Y, Hill KE, Li P et al. Optimization of selenoprotein P and other plasma selenium biomarkers for the assessment of the selenium nutritional requirement: a placebo-controlled, double-blind study of selenomethionine supplementation in selenium-deficient Chinese subjects. Am J Clin Nutr. 2010;92(3):525–31.
Cardoso BR, Hare DJ, Bush AI et al. Selenium Levels in Serum, Red Blood Cells, and Cerebrospinal Fluid of Alzheimer's Disease Patients: A Report from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL). J Alzheimers Dis. 2017;57(1):183–93.
Haratake M, Hongoh M, Miyauchi M et al. Albumin-mediated selenium transfer by a selenotrisulfide relay mechanism. Inorg Chem. 2008;47(14):6273–80.
Bishop DP, Hare DJ, Clases D, Doble PA. Applications of liquid chromatography-inductively coupled plasma-mass spectrometry in the biosciences: a tutorial review and recent developments. TrAC Trends in Analytical Chemistry. 2018;104:11–21.
Delafiori J, Ring G, Furey A. Clinical applications of HPLC–ICP-MS element speciation: A review. Talanta. 2016;153:306–31.
Onning G, Bergdahl IA. Fractionation of soluble selenium compounds from fish using size-exclusion chromatography with on-line detection by inductively coupled plasma mass spectrometry. Analyst. 1999;124(10):1435–8.
Palacios Ò, Lobinski R. Investigation of the stability of selenoproteins during storage of human serum by size-exclusion LC–ICP-MS. Talanta. 2007;71(4):1813–6.
Ganrot K, Laurell CB. Measurement of IgG and albumin content of cerebrospinal fluid, and its interpretation. Clin Chem. 1974;20(5):571–3.
Solovyev N, Berthele A, Michalke B. Selenium speciation in paired serum and cerebrospinal fluid samples. Anal Bioanal Chem. 2013;405(6):1875–84.
Vinceti M, Mandrioli J, Borella P et al. Selenium neurotoxicity in humans: Bridging laboratory and epidemiologic studies. Toxicol Lett. 2014;230(2):295–303.
Cold F, Winther KH, Pastor-Barriuso R et al. Randomised controlled trial of the effect of long-term selenium supplementation on plasma cholesterol in an elderly Danish population. Br J Nutr. 2015;114(11):1807–18.
Rayman M, Winther KH, Pastor-Barriuso R et al. Effect of long-term selenium supplementation on mortality: results from a multiple-dose, RCT. The 11st International Symposium on Selenium in Biology and Medicine and The 5th International Conference on Selenium in the Environment and Human Health (Se2017); Stockholm 2017. p.p. 44. Abstract.
Morris J, Crane SB. Selenium Toxicity from a Misformulated Dietary Supplement, Adverse Health Effects, and the Temporal Response in the Nail Biologic Monitor. Nutrients. 2013;5(4):1024–57.
MacFarquhar JK, Broussard DL, Melstrom P et al. Acute selenium toxicity associated with a dietary supplement. Arch Intern Med. 2010;170(3):256–61.
Jäger T, Drexler H, Göen T. Human metabolism and renal excretion of selenium compounds after oral ingestion of sodium selenate dependent on trimethylselenium ion (TMSe) status. Arch Toxicol. 2016;90(1):149–58.
Rayman MP. Selenium and human health. Lancet. 2012;379(9822):1256–68.
Cardoso B, Roberts BR, Bush AI, Hare DJ. Selenium, selenoproteins and neurodegenerative diseases. Metallomics 2015;7(8):1213–28.
Cardoso B, Hare D, Bush A, Roberts B. Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry. 2017;22(3).
Dixon SJ, Lemberg KM, Lamprecht MR et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–72.
Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10(1):9–17.
Stockwell BR, Angeli J, Bayir H et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171(2):273–85.
Xiong S, Markesbery WR, Shao C, Lovell MA. Seleno-L-Methionine Protects Against β-Amyloid and Iron/Hydrogen Peroxide-Mediated Neuron Death. Antioxid Redox Signal. 2007;9(4):457–67.
Acknowledgments
The Florey Institute of Neuroscience and Mental Health wishes to acknowledge the Victorian Government’s Operational Infrastructure Support Program, as well as the assistance of the Cooperative Research Centre for Mental Health and the Neuroproteomics Facility.
Funding
Funded by Fellowships from the Brazilian Government Science Without Borders program (Ciência sem Fronteiras; BRC), Deakin University (BRC), and the Australian National Health and Medical Research Council (GNT1138673, BRR; GNT1105784, SC; GNT1122981, DJH; GNT1103703, AIB); a Program Grant from the Australian National Health and Medical Research Council (GNT1132604, AIB); and by Velacor Therapeutics. Agilent Technologies provided material and research support (BRR, DJH).
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Cardoso, B.R., Roberts, B.R., Malpas, C.B. et al. Supranutritional Sodium Selenate Supplementation Delivers Selenium to the Central Nervous System: Results from a Randomized Controlled Pilot Trial in Alzheimer’s Disease. Neurotherapeutics 16, 192–202 (2019). https://doi.org/10.1007/s13311-018-0662-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-018-0662-z