
Abstract
Sphingosine 1-phosphate receptor (S1PR) modulators possess a unique mechanism of action in the treatment of multiple sclerosis (MS). Subtype 1 of the S1PR is expressed on the surface of lymphocytes and is important in regulating egression from lymph nodes. The S1PR modulators indirectly antagonize the receptor’s function leading to sequestration of lymphocytes in the lymph nodes. Fingolimod was the first S1PR modulator to receive regulatory approval for relapsing-remitting MS after 2 phase III trials demonstrated potent efficacy, safety, and tolerability. Fingolimod can cause undesirable effects as a result of its interaction with other S1PR subtypes, which are expressed in diverse tissues, including cardiac myocytes. As such, agents that more selectively target subtype 1 of the S1PR are of interest and are at various stages of development. These include ponesimod (ACT128800), siponimod (BAF312), ozanimod (RPC1063), ceralifimod (ONO-4641), GSK2018682, and MT-1303. Data from phase II trials and early results from phase III studies have been promising and will be presented in this review. Of special interest are results from the EXPAND study of siponimod, which suggest a potential role for S1PR modulators in secondary progressive MS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Sphingosine 1-phosphate is a soluble signaling molecule that is involved in immunological, cardiovascular, and neurological processes through interaction with the sphingosine 1-phosphate receptor (S1PR). The S1PR is a G protein-coupled receptor with 5 subtypes: S1PR1, S1PR2, S1PR3, S1PR4, and S1PR5. Owing to the ubiquitous and overlapping expression of S1PR in multiple organs and systems, the S1PR axis has been implicated in several immune-mediated disorders, such as multiple sclerosis (MS), rheumatoid arthritis, and inflammatory bowel diseases. The successful implementation of fingolimod as a treatment for relapsing-remitting MS (RRMS) and the understanding of its mechanisms of action has led to interest in S1PR-targeted therapies for other autoimmune diseases. Clinical trials of S1PR modulators in conditions such as polymyositis and dermatomyositis are ongoing.
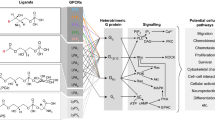
Knowledge of the adverse effects associated with fingolimod (e.g., bradycardia and macular edema) has triggered a search for S1PR modulators with greater selectivity for S1PR1. Agents currently under development include ponesimod, siponimod, ozanimod, ceralifimod, GSK2018682, and MT-1303. Ponesimod is an orally active, reversible, and selective S1PR1 modulator. Siponimod, ozanimod, and ceralifimod are orally active selective modulators of S1PR1 and S1PR5. GSK2018682 is a selective S1PR1 modulator that has some activity at S1PR5, and MT-1303 is known to be a S1PR modulator, but the subtype selectivity is unknown. Figure 1 summarizes the cell types that each S1PR subtype is expressed by and the physiological effects of their activation.

Sphingosine 1-phosphate receptor (S1PR) subtypes, the medications that functionally antagonize them, and the tissues that each subtype is expressed in with their corresponding functions. AV = atrioventricular; CNS = central nervous system; TH17 = T helper 17
Fingolimod
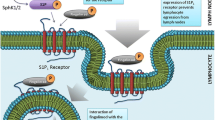
Fingolimod is a lipophilic agent that is phosphorylated by sphingosine kinase 1/2 to become fingolimod phosphate, an S1P analog. Fingolimod phosphate binds to the S1PR, leading to receptor internalization, as illustrated in Figure 2. After fingolimod phosphate dissociation, the receptor returns to the surface [1,2,3]. Fingolimod is nonselective and functions as a modulator of S1PR1, S1PR3, S1PR4, and S1PR5. Its functional antagonism of S1PR1, which is required for lymphocyte egress from the lymph nodes, underlies fingolimod’s principal mechanism of action [4,5,6]. Owing to their inability to respond to the S1P gradient within lymph nodes, B cells and naïve and central memory T cells are sequestered in the lymph nodes. The result is a significant reduction in circulating lymphocytes and reduced inflammatory cell infiltration of the central nervous system. The reduction in circulating lymphocytes is dose dependent, with a 20% to 30% decrease in the first week of treatment [7,8,9]. Fingolimod’s effect is reversible, but is prolonged because of its 7-day elimination half-life. Modulation of S1PR on microglia, oligodendrocytes, astrocytes, and neurons is also postulated to have beneficial effects on the MS disease course [10, 11].

The effect of sphingosine 1-phosphate receptor 1 (S1PR1) internalization in the lymph nodes. After entering the lymph nodes, naïve T cells (TN) and CCR7 expressing central memory T cells (TCM) are sequestered owing to their inability to respond to the sphingosine 1-phosphate gradient after receptor internalization
In the phase III placebo-controlled trial of fingolimod, FREEDOMS (Fingolimod Research Evaluating Effects of Daily Oral therapy in Multiple Sclerosis), 1083 patients with RRMS aged 18 to 55 years were randomized to receive fingolimod 0.5 mg, fingolimod 1.25 mg, or placebo orally once daily [9, 10]. Fingolimod treatment decreased the annualized relapse rate (ARR), the primary endpoint, by 54% and 60% for the 0.5 and 1.25-mg doses, respectively, versus placebo. Results on time to first relapse and confirmed Expanded Disability Status Scale (EDSS) worsening favored fingolimod. Both doses of fingolimod significantly reduced gadolinium-enhancing (GdE) magnetic resonance imaging (MRI) lesions and new or enlarging T2 lesions at 24 months. Moreover, there was significant preservation of brain volume in participants receiving 0.5 mg compared with those receiving placebo (−0.84% vs −1.31%; p = 0.0002) from baseline to 24 months. These results were largely confirmed in a second placebo-controlled phase III trial, FREEDOMS II [12].
A second pivotal phase III trial, TRANSFORMS (TRial Assessing injectable interferon vS. FTY720 Oral in RRMS) was a 12-month, phase III, multicenter, randomized, double-blind, double-dummy, active-comparator trial comparing fingolimod with intramuscular interferon (IFN)-β1a. A total of 1292 patients with RRMS were randomly assigned to receive once-daily fingolimod 0.5 mg or 1.25 mg or once-weekly intramuscular IFN-β1a for 12 months. The primary endpoint was the ARR, which was reduced by 52% in the fingolimod 0.5 mg group, and by 38% in the fingolimod 1.25 mg group relative to intramuscular IFN-β1a. Further, the proportion of relapse-free participants and time to confirmed relapse were greater in both fingolimod groups. MRI endpoints, including the number of GdE lesions, new or enlarging T2 lesions, and brain volume loss all favored the fingolimod groups. No significant differences in time to confirmed disability progression were observed [13, 14].
Given the similar efficacy observed for the 2 doses, fingolimod 0.5 mg was approved under the brand name Gilenya® by the US Food and Drug Administration in 2010 as the first oral disease-modifying therapy for MS. In 2011, it was approved in Canada and by the European Medicines Agency [6, 7].
A double-blind, randomized, multicenter, placebo-controlled, parallel-group study comparing the efficacy and safety of 0.5 mg fingolimod administered orally once daily versus placebo in patients with primary progressive MS (INFORMS) was completed in December 2014. The study used a novel composite endpoint in which worsening beyond a predefined threshold on the EDSS, timed 25-foot walk, or 9-hole peg test, confirmed at 3 months, was considered disability progression. Although the composite outcome performed well, with 69% of participants experiencing confirmed disability progression, no benefit of fingolimod versus placebo was demonstrated [15].
Adverse Effects of Fingolimod
The risk–benefit profile of fingolimod has been demonstrated in a phase II study, 3 phase III studies, a long-term follow-up study [12,13,14,15], and in postmarketing clinical experience. However, use of fingolimod can be complicated by first-dose bradycardia and the occurrence of atrioventricular block in some patients, requiring clinical monitoring of vital signs and electrocardiograms before and after administration of the first dose. Moreover, interaction of fingolimod with S1PR1 and off-target interactions with other S1PR subtypes, particularly S1PR3, S1PR4, and S1PR5, are thought to mediate other risks associated with fingolimod, including QT interval prolongation, hypertension, macular edema, pulmonary toxicity, and possibly hepatotoxicity [16, 17].
The location of S1PR on cardiac myocytes and smooth muscle cells is probably responsible for the adverse cardiovascular effects of fingolimod, which include bradycardia, atrioventricular nodal block, and systemic hypertension. Symptomatic bradycardia occurs in 0.6% of patients treated with fingolimod. It is typically observed 4–5 h after the first dose, with a mean maximum heart rate reduction of 8 beats per minute [17]. The S1PR1 binding by fingolimod activates G protein-coupled inwardly rectifying potassium channels on cardiac myocytes. This leads to an efflux of potassium, thereby hyperpolarizing the cell membrane and temporarily reducing excitability. This effect is transient and resolves with internalization of S1PR1. S1PR3 is activated through the same mechanism and is responsible for the Mobitz type 1 second-degree atrioventricular nodal block observed in 0.2% of patients treated with fingolimod [18, 19].
An increase in blood pressure has also been observed in patients treated with fingolimod [17]. This increase is, on average, +2 mmHg systolic and +1 mmHg diastolic. The blood pressure effect is believed to occur via the presence of S1PR1 and S1PR3 receptors on arterial smooth muscle cells. Activation of S1PR1 in arterial smooth muscle cells causes increased nitric oxide production that leads to vasodilation and an increase in intracellular calcium. The rise in calcium leads to smooth muscle contraction that initially offsets the nitric oxide-induced vasodilation. However, once S1PR1 is internalized, binding shifts to S1PR3, which is also found on arterial smooth muscle cells and smooth muscle contraction becomes the over-riding force. The blood pressure effect peaks at 6 months before stabilizing [20, 21].
Notably, a 48-year-old woman who experienced sudden unexpected death in her fifth month of fingolimod treatment has been reported. Her autopsy results strongly suggested ventricular arrhythmia as the cause of death, and it is unclear whether fingolimod contributed to the arrhythmia [22]. A 20-year-old patient who developed bradycardia and a 7.5-s period of asystole has also been reported. The patient did not require resuscitation and was switched to injectable therapy [23].
Fingolimod is thought to induce macular edema via increased vascular permeability because of its effect on endothelial cells. Sphingolipids are the third most abundant lipid in the retina and it is well recognized that sphingolipid metabolism is involved in retinal cell death and survival. This balance is referred to as the “sphingolipid rheostat”. Ceramide (Cer) is required for sphingolipid production and there are 2 major pathways for Cer production: de novo synthesis and the recycling/degradation of higher-order sphingolipids. Fingolimod can potentially affect sphingolipid metabolism in the retina by inhibiting Cer enzymes, thereby reducing the formation of de novo Cer. However, this is unlikely to be the causative mechanism of fingolimod-related macular edema because fingolimod causes edema exclusively in the macula and not globally across the whole retina [24, 25].
Finally, it should also be noted that 18 patients treated with fingolimod have developed progressive multifocal leukoencephalopathy, including 3 patients without prior exposure to natalizumab [26, 27]. As of now, it is unclear if factors such as JC virus antibody index, a history of immunosuppressive use, or significant lymphopenia increase the risk of progressive multifocal leukoencephalopathy in patients treated with fingolimod.
Siponimod (BAF312)
Siponimod (also known as BAF312) is a novel alkoxyimino derivative and a functional antagonist of S1PR1 and S1PR5 under development for MS by Novartis Pharmaceuticals [24]. Like fingolimod, siponimod induces lymphopenia by preventing lymphocyte egress from lymph nodes. In healthy individuals, siponimod reduces circulating T and B cells within 4 to 6 h. Siponimod has a much shorter half-life than fingolimod and lymphocyte counts recover to baseline levels within a week after treatment discontinuation [28].
Siponimod was found to suppress completely experimental autoimmune encephalomyelitis in a rat model. In another study, siponimod inhibited lysolecithin-induced demyelination in organotypic slice cultures and attenuated lipopolysaccharide or tumor necrosis factor-α/interleukin-17-induced interleukin-6 production in astrocytes and microglia. These findings suggest that siponimod may act directly in the central nervous system in addition to reducing circulating lymphocytes [28,29,30].
A phase II clinical trial of siponimod in RRMS (BOLD) and a subsequent extension study have been successfully completed [31,32,33]. The BOLD trial was a double blind, dose-finding phase II study. The trial employed an adaptive design with 188 patients randomly allocated to cohort 1 in which they received once-daily siponimod 0.5, 2, or 10 mg, or placebo for 6 months. Cohort 2 dosing was based on an interim analysis of cohort 1 and 109 patients were randomized to siponimod 1.25 mg, siponimod 0.25 mg, or placebo once-daily for 3 months. The primary endpoint was the percentage reduction in the number of combined unique active lesions (new or enlarging T2 lesions and GdE lesions) at 3 months for siponimod versus placebo. The results demonstrated a dose–response relationship across the 5 doses of siponimod, with reductions in combined unique active lesions at 3 months versus placebo of 35% for siponimod 0.25 mg, 50% for siponimod 0.5 mg, 66% for siponimod 1.25 mg, 72% for siponimod 2 mg, and 82% for siponimod 10 mg. Adverse events included 1 myocardial infarction in a patient previously treated with siponimod 10 mg and 1 death attributed to acute myocardial insufficiency in a patient who received siponimod 1.25 mg; however, both patients had cardiovascular risk factors and siponimod was washed out before the events occurred [31]. The dose-blinded extension of the BOLD study enrolled 252 patients to assess the safety and efficacy of siponimod for up to 24 months. MS disease activity remained low during the extension and there were no new safety signals [32, 33].
The EXPAND (EXploring the efficacy and safety of siponimod in PAtients with secoNDary progressive multiple sclerosis) trial is the largest double-blind, placebo-controlled phase III study of an agent in secondary progressive MS (SPMS) conducted to date [34]. It enrolled 1651 patients worldwide who were aged 18 to 60 years and had a diagnosis of SPMS. The primary endpoint was time to 3-month-confirmed disability progression as measured by the EDSS. Secondary endpoints included time to 6-month confirmed disability progression, time to 3-month confirmed worsening of at least 20% from baseline on the timed 25-foot walk, and change in T2 lesion volume. Results from the EXPAND trial were presented at the 2016 annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis. The trial met its primary endpoint as siponimod reduced 3-month confirmed disability worsening by 21% versus placebo. In addition, its key secondary endpoint was met with a 26% reduction in 6-month confirmed disability worsening, as well as a significant reduction in the ARR and MRI activity. Subgroup analyses based on the presence of GdE lesions had insufficient power to draw clear conclusions, but trends suggested younger patients with more inflammatory disease activity were the most likely to benefit.
Ozanimod (RPC1063)
Ozanimod is a selective S1PR1/S1PR5 modulator under development by Celgene. Ozanimod also reduces circulating lymphocytes and regulates the immune response. Ozanimod diminished inflammatory markers in experimental autoimmune encephalomyelitis, trinitrobenzene sulfonic acid-induced colitis, and CD4+CD45RBHi T-cell transfer-induced colitis. These findings led to a clinical development program for ozanimod in MS and inflammatory bowel disease [35].
Unlike fingolimod, ozanimod does not need phosphorylation for activation. Ozanimod has a half-life of 19 h, allowing for once-daily dosing. It causes a dose-dependent decrease in circulating lymphocyte counts but with rapid lymphocyte recovery after discontinuation of treatment because of its short half-life. It crosses the blood–brain barrier and has a low peak plasma concentration because of its high volume of distribution and delayed absorption, leading to low systemic exposure that reduces the first-dose effects on heart rate. Ozanimod induces peripheral lymphocyte sequestration and reduces circulating lymphocyte counts in the gastrointestinal tract as well. In a double-blind, placebo-controlled phase II trial in ulcerative colitis, ozanimod induced a significantly higher rate of clinical remission than placebo, which suggests S1PR1 modulation offers a means of treating inflammatory bowel disease, as well as MS [35].
Ozanimod underwent a combined phase II/III trial (RADIANCE) that assessed the safety and efficacy of ozanimod in patients with relapsing MS [36, 37]. The phase II portion of RADIANCE was a randomized, double-blind, placebo-controlled study assessing the efficacy, safety, and tolerability of 2 orally administered doses (0.5 mg and 1 mg) of ozanimod against placebo in 258 patients with RRMS over 24 weeks. The primary endpoint was the cumulative number of total GdE MRI lesions determined by MRI at weeks 12 to 24 after treatment initiation. The trial met its endpoint with both doses significantly reducing the cumulative number of GdE lesions compared with placebo [36]. A 2-year blinded extension study in which all patients received ozanimod was also conducted. The primary endpoint of the placebo-controlled trial was the reduction in the cumulative number of total GdE lesions. At extension week 96, the mean number of GdE lesions was 0.3 for patients on the 0.5 mg dose and 0.1 for the 1 mg dose versus 0.4 and 0.1, respectively, at week 48. The proportion of patients who were free of GdE lesions was 91% for the 0.5 mg dose and 89% for the 1 mg dose. The cumulative number of new or enlarging T2-hyperintense lesions was 1.8 for the 0.5 mg dose and 0.6 for the 1 mg dose versus 1.3 and 0.7, respectively, at week 48 [37]. Ozanimod showed a favorable safety profile with no notable cardiovascular, pulmonary, ophthalmic, infectious, or malignancy-related events occurring. The phase III portion of RADIANCE is ongoing and updated data is expected later in 2017.
A second phase III trial (SUNBEAM) was recently completed. SUNBEAM was a multicenter, randomized, double-blind, double-dummy, active comparator, parallel-group study to evaluate the efficacy and safety of ozanimod against intramuscular IFN-β1a over a minimum 12-month treatment period [38]. It enrolled 1346 patients with RRMS who were randomized to ozanimod 1 mg daily, ozanimod 0.5 mg daily, or intramuscular IFN-β1a. The primary endpoint was the ARR. Secondary endpoints include the number of new or enlarging T2 lesions over 12 months and the number of GdE lesions at month 12. Time to confirmed EDSS worsening will be analyzed as part of a prespecified pooled analysis of SUNBEAM and RADIANCE data. Top-line data showed that both doses of ozanimod demonstrated significant reductions in the ARR, as well as in the number of GdE lesions and the number of new or enlarging T2 lesions at month 12. The overall safety and tolerability profiles were consistent with those from the RADIANCE phase II trial [38].
Ponesimod (ACT-128800)
Ponesimod is an orally active, selective S1PR1 agonist being developed by Actelion. In contrast to the long half-life and slow elimination of fingolimod, ponesimod is eliminated within 1 week of discontinuation, making its pharmacological effects rapidly reversible. The clinical pharmacology of ponesimod has been described in several studies. In lymphocyte-mediated inflammatory diseases, ponesimod reduced several aspects of the inflammatory response, including edema formation, inflammatory cell infiltration, and proinflammatory cytokine levels. In addition, in a nonobese diabetic mouse model of autoimmune diabetes, ponesimod protected against disease development by reducing the number of B and T cells in the blood and spleen [39].
A double-blind, placebo-controlled, dose-finding phase IIb study evaluated the efficacy, safety and tolerability of 3 ponesimod doses in the treatment of RRMS [39]. A total of 464 patients were randomized to receive ponesimod 10, 20, or 40 mg daily, or placebo for 24 weeks. The primary efficacy endpoint, the cumulative number of new GdE lesions from weeks 12 to 24, was significantly reduced in the ponesimod groups. The effect was dose-dependent with reductions of 43%, 83%, and 77% occurring with ponesimod 10, 20, and 40 mg, respectively, versus placebo. Although the study was not designed to show an effect on relapses, the rate of relapses was also reduced by ponesimod treatment. The ARR from baseline to week 24 was 0.33, 0.42, and 0.25 in the 10-, 20-, and 40-mg ponesimod groups, respectively, versus 0.53 in the placebo group. Patients completing the core study were eligible to enter a dose-blinded extension study that ultimately confirmed the above results [40].
The OPTIMUM study, a phase III clinical trial comparing ponesimod to teriflunomide in RRMS is ongoing. This study will randomize approximately 1100 patients in a 1:1 ratio to receive 20 mg ponesimod or 14 mg teriflunomide daily for 2 years. The primary objective of the trial is to assess whether ponesimod is superior to teriflunomide in reducing the ARR over 108 weeks. Secondary endpoints include time to 12-week confirmed disability accumulation, time to first confirmed relapse, cumulative number of combined unique active lesions of the brain, and percent change in brain volume from baseline to week 108 [41].
Ceralifimod (ONO-4641)
Ceralifimod is an oral, selective S1PR1 and S1PR5 modulator that was being jointly developed by Ono Pharmaceuticals and Merck. The phase II DreaMS trial, completed in 2012, randomized 407 participants to 0.05, 0.1, or 0.15 mg ceralifimod or placebo. The primary outcome was the total number of GdE lesions on monthly scans over 26 weeks. The study met its primary endpoint with reductions in GdE lesions versus placebo of 82%, 92%, and 77% for the 0.05, 0.1, and 0.15 mg doses, respectively [41]. The extension study of ceralifimod was prematurely terminated with the companies citing changes in the MS disease-modifying therapy landscape as the reason [42].
GSK2018682
GSK2018682 is a S1PR1 and S1PR5 modulator. In an experimental autoimmune encephalomyelitis mouse model, GSK2018682 and fingolimod exhibited similar efficacy. A phase I clinical trial on GSK2018682 for RRMS was completed in 2011 [43]. The GlaxoSmithKline Product Development Pipeline no longer lists GSK2018682, suggesting that development has been suspended.
Amiselimod (MT-1303)
Amiselimod is a selective S1PR1 modulator developed by Biogen. Amiselimod has been subjected to a phase II study in moderate-to-severe chronic plaque psoriasis, and a phase II study in RRMS with positive results [44]. It has been shown to have low bradycardia risk [43], but Biogen has not revealed plans for a phase III study of amiselimod.
Future of Selective S1PR Modulators in MS
Fingolimod is an attractive treatment because of its efficacy, tolerability, and convenient oral dosing. However, its use has been limited in certain populations, especially those with heart disease and diabetes. Further, some patients are unable to comply with the start-up process, which includes numerous tests and a 6-h observation period.
The advent of second-generation S1PR modulators with greater specificity for S1PR1 will likely expand the population of patients with MS who can benefit from this class of medication. In addition to a lower risk for treatment complications such as bradycardia, the shorter half-lives of the second-generation medications will increase safety, especially if complications such as progressive multifocal leukoencephalopathy arise. The efficacy of the selective S1PR modulators relative to fingolimod has not yet been established, but fingolimod and ozanimod have both shown superior efficacy to a platform disease-modifying therapy.
Of special interest is the potential for treatment of SPMS with S1PR modulators. Although the INFORMS study was negative in the primary progressive population, the EXPAND study suggests a benefit from siponimod in SPMS. Whether this benefit is limited to a subpopulation of patients with SPMS, and whether other S1PR modulators also have a therapeutic effect will need to be addressed with further research. Regardless, as development of second-generation S1PR modulators proceeds, it is likely that more patients with MS will be able to benefit from their unique mechanisms of action. Table 1 summarizes clinical trials of S1PR modulators for MS and Table 2 summarizes key outcome results from those trials.
S1PR Modulators in Other Inflammatory Conditions
While S1PR modulators have demonstrated a promising treatment effect in MS, their value in other inflammatory conditions is currently being explored. Diseases of interest include rheumatoid arthritis, inflammatory bowel disease, systemic lupus erythematosus, polymyositis, and dermatomyositis. Fingolimod has been demonstrated to be effective in animal models of RA, whereas the use of siponimod is currently being explored in clinical trials for the treatment of polymyositis and dermatomyositis. In addition, S1PR modulators are being investigated in psoriasis, ulcerative colitis, autoimmune diabetes, and uveitis. There have also been investigations into their potential usefulness for cancer treatment owing to interference with angiogenesis. Thus, fingolimod and the second-generation S1PR modulators may have wide applications in healthcare outside of MS [45, 46]. Table 3 provides a comprehensive list of S1PR modulators, their current statuses, and the diseases of interest.
Conclusions
The discovery of S1PRs and the subsequent development of fingolimod eventually led to regulatory approval of the first oral disease-modifying therapy for RRMS. The commercial success of fingolimod has stimulated the development of drugs with greater specificity for S1PR1 in the hopes of achieving better efficacy while minimizing start-up burden and reducing safety concerns such as bradycardia, atrioventricular block, and macular edema. These efforts have resulted in multiple drug candidates entering clinical trials, most of which have had promising results. As of this writing, no second-generation S1PR modulators have been approved by regulatory agencies, but several such agents may be available for neurologists and patients with MS in the near future.
References
Radick L, Mehr SR. The latest innovations in the drug pipeline for multiple sclerosis. Am Health Drug Benefits 2015;8;448-53
Gonzalez-Cabrera PJ, Brown S, Studer SM, Rosen H. S1P signaling: new therapies and opportunities. F1000prime Rep 2014;6:109.
Urbano M, Guerrero M, Rosen H, Roberts E. Modulators of the sphingosine 1-phosphate receptor 1. Bioorg Med Chem Lett 2013;23:6377-89.
Aktas O, Kury P, Kieseier B, Hartung HP. Fingolimod is a potential novel therapy for multiple sclerosis. Nat Rev Neurol 2010;6:373-82.
Chun J, Hartung HP. Mechanism of action of oral fingolimod [FTY720] in multiple sclerosis. Clin Neuropharmacol 2010;33:91-101.
Zécri FJ. Natural Product to the First Oral Treatment for Multiple Sclerosis: The Discovery of FTY720 (Gilenya)? Curr Opin Chem Biol. 2016;32:60-66
Mao-Draayer Y, Sarazin J, Fox D, et al. The sphingosine-1-phosphate receptor: A novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin Immunol. 2017;175:10-15
Hunter SF, Bowen JD, Reder AT. The direct effects of fingolimod in the central nervous system: implications for relapsing multiple sclerosis. CNS Drugs 2016;30:135-47.
Subei AM, Cohen JA. Sphingosine 1-phosphate receptor modulators in multiple sclerosis. CNS Drugs 2015;29:565-75.
Kappos L, Antel J, Comi G. et al. Is fingolimod an advancement in the treatment of multiple sclerosis? N Engl J Med 2006;355:1124-40.
Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med 2010;362:387-401.
Calabresi PA, Radue EW, Goodin D, et al. Safety and efficacy of fingolimod in patients with relapsing-remitting multiple sclerosis [FREEDOMS II]: a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Neurol 2014;13:545-56.
Cohen JA, Khatri B, Barkhof F, et al. Long-term [up to 4.5 years] treatment with fingolimod in multiple sclerosis: results from the extension of the randomised TRANSFORMS study. J Neurol Neurosurg Psychiatry 2016;87:468-75.
Khatri B, Barkhof F, Comi G, et al. Comparison of fingolimod with interferon beta-1a in relapsing-remitting multiple sclerosis: a randomised extension of the TRANSFORMS study. Lancet Neurol 2011;10:520-9.
Lublin F, Miller DH, Freedman MS, et al. Oral fingolimod in primary progressive multiple sclerosis [INFORMS]: a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 2016;387:1075-84.
Ziemssen T, Albrecht H, Haas J, et al. 36 months pangaea: a 5-year non-interventional study of safety, efficacy and pharmacoeconomic data for fingolimod patients in daily clinical practice. Value Health 2015;18:A749.
Gold R, Comi G, Palace J, et al. Assessment of cardiac safety during fingolimod treatment initiation in a real-world relapsing multiple sclerosis population: a phase 3b, open-label study. J Neurol 2014;261:267-76.
Ordonez-Boschetti L, Rey R, Cruz A, et al. Safety and tolerability of fingolimod in Latin American patients with relapsing-remitting multiple sclerosis: the open-label FIRST LATAM study. Adv Ther 2015;32:626-35.
Limmroth V, Haverkamp W, Dechend R, et al. Interim analysis of the START study - extensive electrocardiographic monitoring confirms the good cardiac safety profile of fingolimod. Available at: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115132/volker.limmroth.interim.analysis.of.the.start.study.-.extensive.html. Accessed August 6, 2017.
Butzkueven H, Weller B, Giacomini PS, et al. Baseline characteristics and interim analysis results of TRANSITION: a 2-year observational study evaluating the safety profile of patients with multiple sclerosis who switched from natalizumab to fingolimod. Available at: http://onlinelibrary.ectrims-congress.eu/ectrims/2015/31st/115617/helmut.butzkueven.baseline.characteristics.and.interim.analysis.results.of.html. Accessed August 6, 2017.
Ontaneda D, Hara-Cleaver C, Rudick RA, Cohen JA, Bermel RA. Early tolerability and safety of fingolimod in clinical practice. J Neurol Sci 2012;323:167-72.
Lindsey JW, Haden-Pinneri K, Memon NB, Buja LM. Sudden unexpected death on fingolomod. Mult Scler 2012;18:1507-8.
Espinosa PS, Berger JR. Delayed fingolomod-associated asystole. Mult Scler 2011;17:1387-9.
Jain N, Bhatti MT. Fingolimod-associated macular edema: incidence, detection, and management. Neurology 2012;78:672-80
Jeffery DR, Rammohan KW, Hawker K, Fox E. Fingolimod: a review of its mode of action in the context of its efficacy and safety profile in relapsing forms of multiple sclerosis. Expert Rev Neurother 2016;16:31-44.
Pan S, Gray NS, Gao W, et al. Discovery of BAF312 [Siponimod], a potent and selective S1P receptor modulator. ACS Med Chem Lett 2013;4:333-7.
Boudot de la Motte M , Louapre C , Bertrand A, et al. Extensive white matter lesions after 2 years of fingolimod: Progressive multifocal leukoencephalopathy or MS relapse? Mult Scler 2017;23:614-16.
Gergely P, Nuesslein-Hildesheim B, Guerini D, et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol 2012;167:1035-47.
O'Sullivan C, Schubart A, Mir AK, Dev KK. The dual S1PR1/S1PR5 drug BAF312 [Siponimod] attenuates demyelination in organotypic slice cultures. J Neuroinflamm 2016;13:31.
Legangneux E, Gardin A, Johns D. Dose titration of BAF312 attenuates the initial heart rate reducing effect in healthy subjects. Br J Clin Pharmacol 2013;75:831-41.
Selmaj K, Li DK, Hartung HP, et al. Siponimod for patients with relapsing-remitting multiple sclerosis [BOLD]: an adaptive, dose-ranging, randomised, phase 2 study. Lancet Neurol 2013;12:756-67.
Kappos L, Li DK, Stuve O, et al. Safety and efficacy of siponimod [BAF312] in patients with relapsing-remitting multiple sclerosis: dose-blinded, randomized extension of the phase 2 BOLD study. JAMA Neurol 2016;73:1089-98.
Hammond ER. Perspectives on safety and efficacy—the BOLD phase 2 extension study of siponimod in relapsing-remitting multiple sclerosis. JAMA Neurol 2016;73:1052-4.
ClinicalTrials.gov. A multicenter, randomized, double-blind, parallel-group, placebo-controlled variable treatment duration study evaluating the efficacy and safety of siponimod [BAF312] in patients with secondary progressive multiple sclerosis followed by extended treatment with open-label BAF312. Available at: https://clinicaltrials.gov/ct2/show/NCT01665144?term=NCT01665144&rank=1. Accessed August 6, 2017
Scott FL, Clemons B, Brooks J, et al. Ozanimod [RPC1063] is a potent sphingosine-1-phosphate receptor-1 [S1P1] and receptor-5 [S1P5] agonist with autoimmune disease-modifying activity. Br J Pharmacol 2016;173:1778-92.
Cohen JA, Arnold DL, Comi G, et al. Safety and efficacy of the selective sphingosine 1-phosphate receptor modulator ozanimod in relapsing multiple sclerosis [RADIANCE]: a randomised, placebo-controlled, phase 2 trial. Lancet Neurol 2016;15(4):373-81.
ClinicalTrials.gov. A phase 2/3, multi-center, randomized, double-blind, placebo-controlled [Part A] and double-blind, double-dummy, active-controlled [Part B], parallel group study to evaluate the efficacy and safety of RPC1063 administered orally to relapsing multiple sclerosis patients 2017. Available at https://clinicaltrials.gov/ct2/show/NCT02047734?term=NCT02047734&rank=1. Accessed August 6, 2017.
ClinicalTrials.gov. A phase 3, multi-center, randomized, double-blind, double-dummy, active controlled, parallel group study to evaluate the efficacy and safety of RPC1063 administered orally to relapsing multiple sclerosis patients 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT02294058?term=RPC1063&rank=5. Accessed August 6, 2017.
Brossard P, Derendorf H, Xu J, Maatouk H, Halabi A, Dingemanse J. Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br J Clin Pharmacol 2013;76:888-96.
ClinicalTrials.gov. Multicenter, randomized, double-blind, parallel-group extension to study AC-058B201 to investigate the long-term safety, tolerability, and efficacy of three doses of ponesimod, an oral S1P1 receptor agonist, in patients with relapsing-remitting multiple sclerosis 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT01093326?term=Ponesimod&rank=4. Accessed August 6, 2017.
ClinicalTrials.gov. Multicenter, randomized, double-blind, parallel-group, active-controlled, superiority study to compare the efficacy and safety of ponesimod to teriflunomide in subjects with relapsing multiple sclerosis 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT02425644. Accessed August 6, 2017.
ClinicalTrials.gov. A safety and efficacy extension study of ONO-4641 [MSC2430913A] in patients with relapsing-remitting multiple sclerosis 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT01226745?term=ONO-4641&rank=1. Accessed August 6, 2017.
Xu J, Gray F, Henderson A, et al. Safety, pharmacokinetics, pharmacodynamics, and bioavailability of GSK2018682, a sphingosine-1-phosphate receptor modulator, in healthy volunteers. Clin Pharmacol Drug Dev 2014. doi:10.1002/cpdd.98.
ClinicalTrials.gov. A phase II, multicentre, randomised, double-blind,parallel group, placebo-controlled, dose-finding study to evaluate the safety and efficacy of three different oral doses of MT-1303 administered for a period of 24 weeks in subjects with relapsing-remitting multiple sclerosis 2017. Available at: https://clinicaltrials.gov/ct2/show/NCT01742052?term=MT-1303&rank=5. Accessed August 6, 2017.
Tsai HC, Han MH. Sphingosine-1-phosphate [S1P] and S1P signaling pathway: therapeutic targets in autoimmunity and inflammation. Drugs 2016;76:1067-79.
O'Sullivan S, Dev KK. Sphingosine-1-phosphate receptor therapies: advances in clinical trials for CNS-related diseases. Neuropharmacology 2017;113:597-607.
Acknowledgments
Dr. Chaudhry has no disclosures to report. Dr. Cohen has received consulting fees from Adamas, Merck, Mallinckrodt, Novartis, and Receptos, and compensation as Co-Editor of Multiple Sclerosis Journal – Experimental, Translational and Clinical. Dr. Conway has received research support paid to his institution by Novartis Pharmaceuticals.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 281 kb)
Rights and permissions
About this article

Cite this article
Chaudhry, B.Z., Cohen, J.A. & Conway, D.S. Sphingosine 1-Phosphate Receptor Modulators for the Treatment of Multiple Sclerosis. Neurotherapeutics 14, 859–873 (2017). https://doi.org/10.1007/s13311-017-0565-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-017-0565-4