
Abstract
Hypoxia-inducible factor-1α (HIF-1α) has crucial role in adapting cells to hypoxia through expression regulation of many genes. Identification of HIF-1α target genes (HIF-1α-TGs) is important for understanding the adapting mechanism. The aim of the present study was to collect known HIF-1α-TGs and identify their associated pathways. Targets and associated genomics data were retrieved using PubMed, WoS (http://apps.webofknowledge.com/), HGNC (http://www.genenames.org/), NCBI (http://www.ncbi.nlm.nih.gov/), Ensemblv.84 (http://www.ensembl.org/index.html), DAVID Bioinformatics Resources (https://david.ncifcrf.gov/), and Disease Ontology database (http://disease-ontology.org/). From 51 papers, we collected 98 HIF-1α TGs found to be associated with 20 pathways, including metabolism of carbohydrates and pathways in cancer. Reanalysis of genomic coordinates of published HREs (hypoxia response elements) revealed six polymorphisms within HRE sites (HRE-SNPs): ABCG2, ACE, CA9, and CP. Due to large heterogeneity of results presentation in scientific literature, we also propose a first step towards reporting standardization of HIF-1α-target interactions consisting of ten relevant data types. Suggested minimal checklist for reporting will enable faster development of a complete catalog of HIF-1α-TGs, data sharing, bioinformatics analyses, and setting novel more targeted hypotheses. The proposed format for data standardization is not yet complete but presents a baseline for further optimization of the protocol with additional details, for example, regarding the experimental validation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Regulation of gene network is related to changes in environment in order to survive [1, 2]. A eukaryotic genome contains to thousands of genes that have to be expressed at the right time, in the right cells, and on the right level [3]. Gene regulation is performed with transcription factors (TF) through binding on transcriptional binding site (TFBS) [2–4]. DNA-binding proteins bind on promoter regions proximal to gene transcription start sites (TSSs) or to more distant enhancer regions [5]. Transcription factors recognize target’s response elements (RE) very selectively through dynamic conformation ensemble, affinity of TF to RE, and availability of TFBS in the genome and cellular network. The interaction between TF and gene is complex and not always straightforward [6]. However, identification of direct targets of TFs is the key step for understanding complex gene regulatory network [7].
Hypoxia-inducible factor-1 (HIF-1) is a TF with a critical role in adapting cells to lower oxygen level [8]. The HIF-1 protein is a heterodimer and consists of oxygen-regulated subunit (HIF-1α) and constitutively expressed subunit (HIF-1β) [9]. In normoxia, post-translational modification of the α subunit of HIF-1 associate with prolyl hydoxylase (PHD), von Hippel–Lindau protein (pVHL), ubiquitin ligase, and the proteasome promoting oxygen depended degradation [8, 10]. Through binding on hypoxia response elements (HRE) named also as HIF-responsive elements, it regulates many physiological and pathological processes [11]. Natural polymorphisms like single nucleotide polymorphisms (SNPs) in HRE sequence can prevent hypoxic induction [12]. Site-directed mutagenesis experiments that change HRE core motive are applied in experimental validation of targets; HIF-1α binding site mutations alter in decreased luciferase activity in reporter assay [13–15]. It has been reported that only 1–5 % of all human genes are expressed in response to hypoxia in more than one type of cells [16]. HIF-1α regulates the expression of a large number of genes, including genes involved in diseases and carcinogenesis; therefore, known targets involved in regulation mechanism can be applied in therapy [16, 17]. Transcription factor HIF-1α is master regulator turning on or off many protein-coding genes [16] and noncoding RNA genes (ncRNA) including microRNAs (miRNAs) [18]. Additionally, an interaction between HIF-1α and transcribed-ultraconserved regions (T-UCRs) has also been shown in our previous study [19].
The aim of the present study was to collect known HIF-1α targets which will enable further more targeted planning of research and detailed understanding of hypoxia-responsive mechanism and perform pathway enrichment analysis and screening for HRE polymorphisms. Over the course of our work, we reviewed a large number of publications describing HIF-1α target genes. However, we were confronted with data heterogeneity in scientific papers; diverse methodology and presentation of the data. Additionally, an increasing number of new papers are being published in this field; PubMed search for “transcriptomics” filtered to “humans” and “English” currently results more than 36,000 citations. Therefore, we also developed an initiative with standardized format for the results presentation in studies identifying HIF-1α-TG which will facilitate progression of the work towards the complete catalog HIF-1α-TGs and more systematic progress of the field.
Materials and methods
We collected publications on PubMed (http://www.ncbi.nlm.nih.gov/pubmed ) and WoS (http://apps.webofknowledge.com/) between 1992 and May 2016 using the following keywords: HIF-1α, transcription regulation, targets, chromatin immunoprecipitation (ChIP), luciferase assay, and HRE binding site. Obtained scientific papers were reviewed to get information about HIF-1α-TG, and relevant data were collected in the Excel table. From the paper, we extracted the following information: gene name and symbol, species, applied experimental methods, cell lines/tissue, regulation of target gene expression (up- or downregulation), number and location of HRE sites, associated phenotype/disease, and reference’s authors and year of publication. We collected genes, confirmed as HIF-1α targets with the following methods: EMSA (electrophoretic mobility shift assay), ChIP, reporter assay, site directed mutagenesis, HIF1A gene inactivation, overexpression of HIF1A/HIF-1α, methods for detection of mRNA and protein level, and microarrays. Target genes from studies using only high-throughput (HT) screening or only bioinformatics prediction were not included in list of HIF-1α-TGs. Entrez gene ID and the nomenclature of collected genes were obtained and uniformed according to the HUGO Gene Nomenclature Committee (HGNC http://www.genenames.org/). Taxonomy identifiers were obtained in NCBI (http://www.ncbi.nlm.nih.gov/taxonomy). Disease ontology ID (DOID) was extracted in Disease Ontology database (http://disease-ontology.org/). Reference PMID (PubMed ID) number was gained in the NCBI site (http://www.ncbi.nlm.nih.gov/). Enrichment pathway analysis with known HIF-1α downstream targets was employed using DAVID Bioinformatics Resources (Huang da et al. 2009) as described previously [20] from four different pathway databases (KEEG, PANTHER, Reactome, and BioCarta). The result of the enrichment analysis was processed using Bonferroni multiple test correction and a p value significant threshold of 0.01. Genes present in more than one pathway were identified using database KEGG. Reanalysis of genomic locations of previously published HRE locations and analysis of their possible overlap with published genetic variations was performed using Ensembl Release 84 [21].
Results
In the present study, we (1) collected a list of known HIF-1α-TGs, (2) performed pathway enrichment analysis associated with target genes, and (3) reanalyzed genomic location of HRE sites to see if they overlap with previously reported polymorphisms. Extraction of TG results from publications is challenging due to variable studies and heterogeneous results display; therefore, a standardization of the results presentation in scientific literature is required. We propose a simple transparent format that contains relevant data about each HIF-1α target, and we formed initiative guidelines for strengthening and uniform data presentation of results describing HIF-1α downstream target genes.
Overview of the collected data related with HIF-1α target genes
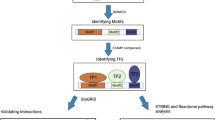
In the present study, we collected papers describing experimentally validated HIF-1α targets. We extracted data from 51 papers. The workflow of the study and main results are presented in the Fig. 1. Collected data associated with 98 HIF-1α TGs are presented in Supplemental Table 1. Gene names of the collected target genes were unified according to HGNC nomenclature. In cases when the gene symbol in the article differed from HGNC nomenclature, we presented synonym in parenthesis. Validation of interactions HIF-1α-target was performed in human, mouse, and rat promoter sequences. The workflows in published studies included analysis of nucleotide sequences for HRE target prediction, followed by experimental validation. For some target genes, their role in hypoxia was confirmed in more than one study: ACAN, ADM, ALDOA, ALDOC, ANGPT2, ANKRD37, BHLHE40, BNIP3, CA9, COL2A1, ENO1, GAPDH, LDHA, LRP1, PFKFB4, PFKL, PGK1, PKM, SLC2A1, SLC2A3, and VEGFA and therefore in our list, HIF-1α-TGs are listed in more than one entry in the Supplemental Table 1. One HIF-1α-target interaction has been validated in two species; target gene ADM has been confirmed in mouse and human [22–25].

Workflow of the study. HIF-1α hypoxia-inducible factor-1α, TG target gene
Researchers used very diverse experimental methods for confirmation of HIF-1α-TG interactions. PCR methods and methods for quantification of protein level were usually used for initial target screening. The most commonly applied assays for identification of HIF-1α target gene interaction were luciferase assay, EMSA, ChIP, site-directed mutagenesis or deletion of promoter’s region, overexpression of HIF1A/HIF-1α, and HIF1A gene inactivation. For 37/98 TGs, researchers used at least two aforementioned assays. Different approaches like RNA interference (RNAi), antisense oligonucleotide, and Cre recombinase were applied for inactivation of HIF1A gene. Overexpression is achieved with VHL-null cells, transient transfection assay, and exposure of cells to inhibitors of hydrolyses activity (CoCl2, desferrioxamine, dimethyloxalylglycine). Aforementioned commonly used assays are suitable for identification DNA–protein interaction [26–30].
Different cell lines and tissues from patients or animals (mouse, rat) were applied for identification of HIF-1α-TG. We collected information of cell lines and/or tissue with confirmed HIF-1α-target interaction. Selection of the cell line for the experimental validation depends on the studied gene, and the most frequently used cell lines were CHO, Hep3B, HeLa, RCC4, and Hek293.
Regulation of target gene expression is also one of the key information in transcription mechanism since TFs can increase or decrease the expression of targets [31]. Based on the detailed review of 98 HIF1A targets, 85 TG have been found to be upregulated and 12 downregulated. Target ANGPT2 has been reported to be up- and downregulated, depending on cell lines [32]; therefore, determining the regulation and cell type is important.
Literature review revealed heterogeneous presentation of HRE genomic coordinates. Some research groups precisely presented location of predicted and experimentally validated HRE sites [33–35]. In most studies, HRE location has been determined relative to the transcription start site (TSS) and in other relative to the translation site. In some cases, researchers did not publish exact genomic location of HRE site, but nucleotide sequence with marked HRE region. In collection of HIF-1α-TG (Supplemental Table 1), we listed all predicted HRE sites on nucleotide sequence from article and underlined HRE site/s which has been reported to interact with HIF-1α. In majority, groups applied 5′-RCGTG-3′ core sequence for a HIF-1α binding site as it has been first reported in Semenza et al. [36]; however; some studies validated different sequences named as HIF-responsive element: CACGT [37] and CACGC [38].
The association between target gene and phenotype/disease presents biological impact of examined interaction and provides data for understanding the connection between gene/protein and phenotype/disease; therefore, this information was extracted from publications. For 18 TGs, association with phenotype has been reported. For example, HIF-1α target AURKA has been shown to be involved in cell proliferation [39], protein BCL2L1 promotes cell growth and inhibits cell death [40], and BSG is involved in metabolic reprogramming of tumor cells under hypoxia and protects tumor cells from apoptosis [41].
Pathway analysis of HIF-1α-TG
Pathway enrichment analysis of collected 98 HIF-1α downstream targets yielded the presence of 20 pathways (Supplemental Table 2). Biological pathways (with numbers of included HIF1A-TGs) most significantly associated with collected HIF-1α-TGs are metabolism of carbohydrates (N = 17), diabetes pathways (N = 14), pathways in cancer (N = 14), and integration of energy metabolism (N = 13). For identification of genes identified in more than one pathway, we used the results of KEGG database. HIF-1α-TGs have been associated with nine biological pathways: glycolysis/gluconeogenesis (N = 12), fructose and mannose metabolism (N = 8), focal adhesion (N = 12), pathways in cancer (N = 14), renal cell carcinoma (N = 7), bladder cancer (N = 5), pancreatic cancer (N = 6), pentose phosphate pathway (N = 4), and galactose metabolism (N = 4). For example, ALDOA and ALODC genes are present in three pathways: glycolysis/gluconeogenesis, fructose and mannose metabolism, and pentose phosphate pathway. Targets PFKL and PFKP are included in all pathways that include metabolism of carbohydrates. Target genes PGK and VEGFA coexist in all pathways associated with cancer and focal adhesion. Genes present in more than one associated pathway have a potential for greater impact in adaption and regulation of hypoxia and deserve further functional experiments.
Reanalysis of genomic location of HRE sites and identification of polymorphic HRE sites
Based on previously reported HRE locations in publications, we used Ensembl genomic browser to acquire genomic HRE locations annotated to the latest genome assembly release. Genomic locations of HRE sites were possible to define for 12 TGs and are presented in Supplemental Table 1. Further, the identified genomic locations of HRE regions were then examined for the presence of polymorphisms in core HRE motive, which we termed HRE-SNPs. Using the genome variation data from Ensembl, we found six polymorphisms located in the HRE regions of four genes (ABCG2, ACE, CA9, and CP), previously reported as HIF-1α-TGs [33, 42–45]. There are three HRE sites in the promoter region of the target gene ACE [42] which includes three polymorphisms: SNP rs746774969 is present in HRE1 and polymorphisms rs769225220; rs776943620 are present in HRE3 (Supplemental Figure 1). Polymorphisms located within ABCG2, CA9, and CP HRE motives are substitutions of single nucleotides. We screened the literature to analyze if those HRE-SNPs have been previously associated with disease in separate studies. Polymorphism rs76656413 located in HRE of the ABCG2 target gene has been shown previously to decrease liver enhancer activity in vivo [46]. For other TGs, the connection between polymorphisms and phenotype has not yet been reported.
Initiative for reporting standardization of format of the results HIF-1α target genes
Based on the literature review and collected and analyzed data regarding the HIF-1α-TGs, we suggest publishing ten basic items for strengthening reports of HIF-1α-TG interactions. Proposed minimal checklist for publishing HIF-target interaction should include the following information: transcription factor, target gene, species, experimental methods, cell lines and/or tissue, regulation of target gene expression, location of HRE sites, HRE polymorphisms, associated phenotype/disease, and reference data (Table 1). Examples of previously published HIF-1α interactions are presented in Table 2 [15, 33, 47] with data presented according to our proposed initiative for reporting standardization.
Discussion
In the present study, we extracted data from publications and collected 98 HIF-1α-TGs, which were found to be associated with 20 biological pathways. Reanalysis of the genomics location of previously reported interaction sites resulted in six polymorphisms located within HRE sites of four TGs. We also proposed a first step towards standardization of the format for presentation the HIF-1α-TG results.
Research groups collected HIF-1α-TGs using different approaches. More than 60 putative HIF-1-regulated genes were collected from literature by Semenza [16]. Benita et al. identified more than 200 HIF-1α-TGs based on integrative genomic approach combining computational and experimental strategies [48]. List of 217 HIF-1α-TGs was predicted based on integration of phylogenetic footprinting and transcription profiling meta-analysis [12]. Database HypoxiaDB contains 3500 hypoxia-regulated proteins from high-throughput experiments [49]. In comparison with gene collections listed above, our list of HIF-1α-TGs includes 98 experimentally validated targets complemented with relevant genomics data. However, the collected gene list is most probably not complete and will have to be updated with discoveries from upcoming publications.
We compared list of pathways associated with HIF-1α-TGs with the results of previous pathway-based studies. The results of the KEGG pathway study in previous report using 21 validated and 60 predicted HIF-1α targets revealed 11 associated pathways [48]. The results of the present study confirmed four enriched pathways identified previously (marked with stars in Supplemental Table 2): glycolysis/gluconeogenesis, fructose and mannose metabolism, renal cell carcinoma, and pancreatic cancer. Our list of validated target genes has been associated with 16 additional enriched pathways: for example, metabolism of carbohydrates, glycolysis, diabetes pathways, pathways in cancer, hemostasis, and bladder cancer. Pathway analysis enables further, more detailed understanding of hypoxia mechanism and predicting novel pathway-based biomarkers and therapeutic targets.
During literature mining, we came across different terminology about hypoxia-responsive elements. HRE was defined as a combination of conserved HIF binding site (HBS) with an RCGTG core motive and highly variable flanking sequence [50]. On the other hand, it has also been suggested that HRE consists of HBS and HAS (HIF-1 ancillary sequence) [51], which has been validated for ABCB1 and CBR1 targets [13, 47]. In the published literature, genomic location of HIF-1α binding site has usually been presented relative to the TSS or in some cases relative to the translation site. In the proposed initiative, we therefore suggest to report HRE locations upstream to TSS. Regarding the location of the HRE site, it would be most suitable to present HRE genomic locations in experimental papers uniformly, in accordance with genomic coordinates of main genomics browsers, like Ensembl, UCSC, or NCBI Map Viewer. For example, we reanalyzed the genomic location HRE1 of the target gene ACE, which is now annotated to the regions 17:63475856–63475860.
Polymorphisms mapping within HRE sequences presents a natural model for studying gain or loss of functional HIF-1α interaction sites and provides novel tool for researching impact of HRE-SNPs on HIF-1α binding and on altered phenotype. These natural polymorphisms in HIF-1α binding core motive could be used in experimental validation of TG in reporter assay instead of site-directed mutagenesis approach. Ortiz-Barahona et al. used computational prediction and identified 146 SNPs in predicted HIF-binding sites. Additionally, they demonstrated that SNP rs1700403 in MIF gene prevents hypoxic induction [12]. In chicken, HRE polymorphism in NOS3 gene has been shown to be associated with pulmonary hypertension syndrome [52]. Analysis of chicken PGK gene sequence revealed SNP within HRE site effecting transcription level of protein PGK in hypoxia [53]. In our study, we identified six polymorphisms within promoter regions of ABCG2, ACE, CA9, and CP genes that present natural examples for experimental validation of functional impact and associated phenotype. Literature mining for HRE-SNPs revealed that polymorphism rs76656413 located within ABCG2 gene has already been associated with decreased liver enhancer activity in vivo in previous study by Eclov [46].
Our reanalysis of genomic locations of HRE sites was more challenging as expected since for only 12 TGs was possible to define genomic coordinates according with the latest genome assembly. For the rest of the targets, obtaining genomic location was not doable due to differences of the nucleotide sequence between genome assemblies. However, the present preliminary analysis raised awareness for the community to standardize reporting the location of the HRE site. In order to determine genomic coordinates for other TGs, a separate study including the collaboration with authors of original reports would be required. Obtaining genomic locations and genetic variations of HRE regions would enable development of bioinformatics algorithms and models for HRE site searching and possible tool for HRE polymorphism identification for medical applications.
Literature reviewing of HIF-1α-TG revealed different and heterogeneous way of presenting results. Different research groups publish results in diverse forms, information need to be extracted from manuscripts and supplemental materials which is time consuming. Thus, proposed format (Table 2) would decrease variability between different research groups. Implementing the proposed format in future studies would enable much faster completion of the catalog of HIF-1α target genes. The value of the present standardized form is not only in the development of catalog of HIF-1α-TG, but it could also be considered for collecting target genes for other TFs. Namely, our proposed format was created based on our literature mining and other existing TF-TG databases: ERGDB, HTRIdb, HypoxiaDB, Myc Target Gene database, and Rel/NF-kappaB target genes [49, 54–57].
Complementing the data with identification numbers, like Entrez ID, DOID, PMID number, and uniform gene nomenclature, will enable obtaining additional relevant genomics information and also further bioinformatics analyses. Explicitly presented data about HIF-1α-TG interaction site, cell lines/tissue, and association with phenotype/disease are important for novel biologic disclosures. Uniform presentations of HIF-1α-TGs data also simplify data extraction for review and meta-analyses papers and bioinformatics studies. The use of this format is recommended in original as well in the review papers.
Diverse assays were applied for elucidating the interaction between HIF-1α-TG, and only combination of various methods can enable reliable results with higher degree of confidence. We acknowledge no gold standard protocol or guidelines recommendation for number and type of methods applied and for results evaluation in case for determination of HIF-1α-TG interaction. For example, Pichiule et al. in 2007 used wide range of methods on multiple levels to determine AGER gene as HIF-1α target [58]; on the other hand, some studies reported potential TG with lower spectrum of assays. The proposed initiative should also motivate experts to define terminology and establish assays for direct/indirect TF-DNA interaction screening. Majority researchers present direct targets as genes controlled with direct binding of TF to target gene’s promoter region and indirect as genes regulated through an indirect cascade [59, 60]. On the other hand, indirect TG can be acknowledged as TF interacted with cofactor or other protein [61, 62]. To enable correctly interpreted results and make some directive for TG identifications, it would be necessary to form appropriate uniform terminology and identify criteria by which direct/indirect type of TF-TG interaction can be determined. Applied methods are not equivalent, so it would be reasonable to divide methods to two groups, for example, to the strong and less strong evidence for interaction, as proposed previously for miRNA-target interactions [63]. Best practice protocol for HIF-1α-TG identification should also consider high-throughput methods, for example, ChIP-chip and ChIP-seq [64–66] and integration HT results with small-scale experimental techniques to meet the criteria for interpretation genes as direct/indirect or as strong/less strongly validated TG.
A role of HIF-1α has been defined in physiological phenotypes (metabolism, pH regulation, oxygen delivery, angiogenesis, motility, mitochondrial function, proliferation, extracellular matrix production, inflammatory cell recruitment) and diseases (ischemic heart disease, pulmonary hypertension, cancer, tumors progression, metastasis, inflammation) [67, 68]. Complete catalog of HIF-1α target genes will enable greater understanding of hypoxia and HIF-1α response with pathway analysis, reveal novel therapeutic targets, and potential biomarkers. It will also enable defining better criteria for planning HIF-1α target experiments. Understanding of mechanism and transcription response to regulator HIF-1α will provide tool for manipulation of genetic regulatory network for treatment for diseases as cancer and ischemia [69, 70].
The number of published papers describing TF-target interactions is increasing; therefore, it is important to make these data easily available and uniform for all researchers. This standardization would save time, and enable correct data interpretations and easier data manipulation for future research. Extracting these key data about TG is crucial for readers and helps researchers to effectively present their results. Special importance of the presented standardized format is for data curators, which would enable faster data extraction from scientific literature and further data manipulation. Because authors would mediate their result in uniform format, one of the advantages is prevention of false interpretations by readers. The situation is similar in other fields of genomic research; initiatives have already been proposed for strengthening the reporting for observational studies in epidemiology [71], genetic association studies [72, 73], immuno-genomic studies [74], genetic risk prediction studies [75], observational studies in molecular epidemiology [76], and genomic methylation [77]. Additionally, in our previous studies, we proposed format for reporting next-generation sequencing data [78] and molecular mechanisms for cryptorchidism development [79]. We acknowledge that no such initiative exists in interactomics area; therefore, we propose format for strengthening the reporting of TF target genes with key data (Fig. 2). Implementation of this initiative would facilitate bioinformatics studies, and experimental studies would help to improve the proposed format with additional details.

Schematic summary of key data related with HIF-1α-target interaction. bp base pair, HIF-1α hypoxia-inducible factor-1α, HRE hypoxia-responsive elements, TF transcription factor
One part of transcription atlas complexity makes attendance special coactivators through interaction with TF to direct transcription activity [80, 81], making standardization for transcriptome, interactome, and regulome more required. For complete overview, regulation mechanism for catalog of HIF-1α-TG and also for other TF-TGs in future would be momentous to also uniformly expose even more data about protein-DNA interaction, like TFBS sequence, cofactors, coactivators, exposure duration in studied terms, fold change, and chromosome location.
Increasing awareness for the integration of the large mass of data also termed integromics [20] contributes to novel biological disclosures; therefore, standardization of genomics data is recommended. Unification of results would make scattered studies easy and more effectively applied in assembling the whole picture and smoother literature mining. The message to all scientific writers is to report their results in explicate and readable form, if possible, in a tabular format, supplemented with available ID numbers, which will enable easier and faster further data analysis.
References
Balazsi G, van Oudenaarden A, Collins JJ. Cellular decision making and biological noise: from microbes to mammals. Cell. United States: Elsevier, 2011. vol 144, pp 910–925.
Latchman DS. Transcription factors as potential targets for therapeutic drugs. Curr Pharm Biotechnol. 2000;1:57–61.
Georges AB, Benayoun BA, Caburet S, Veitia RA. Generic binding sites, generic dna-binding domains: where does specific promoter recognition come from? Faseb J. 2010; 24, pp 346–356.
Wang J, Lu J, Gu G, Liu Y. In vitro dna-binding profile of transcription factors: methods and new insights. J Endocrinol England, 2011 Society for Endocrinology. 2011;210:15–27.
Farnham PJ. Insights from genomic profiling of transcription factors. Nat Rev Genet England. 2009;10:605–16.
Pan Y, Tsai CJ, Ma B, Nussinov R. Mechanisms of transcription factor selectivity. Trends Genet England. 2010;26:75–83.
Kim TM, Park PJ. Advances in analysis of transcriptional regulatory networks. Wiley Interdiscip Rev Syst Biol Med. 2011;3:21–35.
Ke Q, Costa M. Hypoxia-inducible factor-1 (hif-1). Mol Pharmacol. 2006;70:1469–80.
Lee JW, Bae SH, Jeong JW, Kim SH, Kim KW. Hypoxia-inducible factor (hif-1)alpha: its protein stability and biological functions. Experimental and. Mol Med. 2004;36:1–12.
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein vhl targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–5.
Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40:294–309.
Ortiz-Barahona A, Villar D, Pescador N, Amigo J, del Peso L. Genome-wide identification of hypoxia-inducible factor binding sites and target genes by a probabilistic model integrating transcription-profiling data and in silico binding site prediction. Nucleic Acids Res. 2010;38:2332–45.
Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (mdr1) gene. Cancer Res. 2002;62:3387–94.
Zaka R, Dion AS, Kusnierz A, Bohensky J, Srinivas V, Freeman T, Williams CJ. Oxygen tension regulates the expression of ank (progressive ankylosis) in an hif-1-dependent manner in growth plate chondrocytes. J Bone Miner Res. 2009;24:1869–78.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, Bronte V, Chouaib S. Pd-l1 is a novel direct target of hif-1alpha, and its blockade under hypoxia enhanced mdsc-mediated t cell activation. J Exp Med. 2014;211:781–90.
Semenza GL. Targeting hif-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Brocato J, Chervona Y, Costa M. Molecular responses to hypoxia-inducible factor 1alpha and beyond. Mol Pharmacol. 2014;85:651–7.
Gee HE, Ivan C, Calin GA, Ivan M. Hypoxamirs and cancer: from biology to targeted therapy. Antioxid Redox Signal. 2014;21:1220–38.
Ferdin J, Nishida N, Wu X, Nicoloso MS, Shah MY, Devlin C, Ling H, Shimizu M, Kumar K, Cortez MA, Ferracin M, Bi Y, Yang D, Czerniak B, Zhang W, Schmittgen TD, Voorhoeve MP, Reginato MJ, Negrini M, Davuluri RV, Kunej T, Ivan M, Calin GA. Hincuts in cancer: hypoxia-induced noncoding ultraconserved transcripts. Cell Death Differ. 2013;20:1675–87.
Cannistraci CV, Ogorevc J, Zorc M, Ravasi T, Dovc P, Kunej T. Pivotal role of the muscle-contraction pathway in cryptorchidism and evidence for genomic connections with cardiomyopathy pathways in rasopathies. BMC Med Genet. 2013;6:5.
Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, Girón CG, Gordon L, Hourlier T, Hunt SE, Janacek SH, Johnson N, Juettemann T, Keenan S, Lavidas I, Martin FJ, Maurel T, McLaren W, Murphy DN, Nag R, Nuhn M, Parker A, Patricio M, Pignatelli M, Rahtz M, Riat HS, Sheppard D, Taylor K, Thormann A, Vullo A, Wilder SP, Zadissa A, Birney E, Harrow J, Muffato M, Perry E, Ruffier M, Spudich G, Trevanion SJ, Cunningham F, Aken BL, Zerbino DR, Flicek P. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6.
Geiger K, Leiherer A, Muendlein A, Stark N, Geller-Rhomberg S, Saely CH, Wabitsch M, Fraunberger P, Drexel H. Identification of hypoxia-induced genes in human sgbs adipocytes by microarray analysis. PLoS One. 2011;6:e26465.
Garayoa M, Martinez A, Lee S, Pio R, An WG, Neckers L, Trepel J, Montuenga LM, Ryan H, Johnson R, Gassmann M, Cuttitta F. Hypoxia-inducible factor-1 (hif-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: a possible promotion mechanism of carcinogenesis. Mol Endocrinol. 2000;14:848–62.
Cormier-Regard S, Nguyen SV, Claycomb WC. Adrenomedullin gene expression is developmentally regulated and induced by hypoxia in rat ventricular cardiac myocytes. J Biol Chem. 1998;273:17787–92.
Nguyen SV, Claycomb WC. Hypoxia regulates the expression of the adrenomedullin and hif-1 genes in cultured hl-1 cardiomyocytes. Biochem Biophys Res Commun. 1999;265:382–6.
Guille MJ, Kneale GG. Methods for the analysis of dna-protein interactions. Mol Biotechnol. 1997;8:35–52.
Helwa R, Hoheisel JD. Analysis of dna-protein interactions: from nitrocellulose filter binding assays to microarray studies. Anal Bioanal Chem. 2010;398:2551–61.
Remes-Lenicov F, Jacobsen KX, Rogaeva A, Czesak M, Hadjighasem M, Daigle M, Albert PR. Frontiers in neuroscienceidentification of novel transcriptional regulators in the nervous system. In: Chattopadhyay A, editor. Serotonin receptors in neurobiology. Boca Raton: CRC, Taylor & Francis; 2007.
Taverner NV, Smith JC, Wardle FC. Identifying transcriptional targets. Genome Biol. 2004;5:210.
Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-dna interactions. Science. 2007;316:1497–502.
Mitchell PJ, Tjian R. Transcriptional regulation in mammalian cells by sequence-specific dna binding proteins. Science. 1989;245:371–8.
Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–81.
Krishnamurthy P, Ross DD, Nakanishi T, Bailey-Dell K, Zhou S, Mercer KE, Sarkadi B, Sorrentino BP, Schuetz JD. The stem cell marker bcrp/abcg2 enhances hypoxic cell survival through interactions with heme. J Biol Chem. 2004;279:24218–25.
Gimm T, Wiese M, Teschemacher B, Deggerich A, Schodel J, Knaup KX, Hackenbeck T, Hellerbrand C, Amann K, Wiesener MS, Honing S, Eckardt KU, Warnecke C. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J. 2010;24:4443–58.
Wellmann S, Bettkober M, Zelmer A, Seeger K, Faigle M, Eltzschig HK, Buhrer C. Hypoxia upregulates the histone demethylase jmjd1a via hif-1. Biochem Biophys Res Commun. 2008;372:892–7.
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase a, enolase 1, and lactate dehydrogenase a gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–37.
Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, Wilson BC, Teh BT, Park M, Irwin MS, Ohh M. Hypoxia promotes ligand-independent egf receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci U S A. 2012;109:4892–7.
Liao SH, Zhao XY, Han YH, Zhang J, Wang LS, Xia L, Zhao KW, Zheng Y, Guo M, Chen GQ. Proteomics-based identification of two novel direct targets of hypoxia-inducible factor-1 and their potential roles in migration/invasion of cancer cells. Proteomics. 2009;9:3901–12.
Klein A, Flugel D, Kietzmann T. Transcriptional regulation of serine/threonine kinase-15 (stk15) expression by hypoxia and hif-1. Mol Biol Cell. 2008;19:3667–75.
Chen N, Chen X, Huang R, Zeng H, Gong J, Meng W, Lu Y, Zhao F, Wang L, Zhou Q. Bcl-xl is a target gene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem. 2009;284:10004–12.
Ke X, Fei F, Chen Y, Xu L, Zhang Z, Huang Q, Zhang H, Yang H, Chen Z, Xing J. Hypoxia upregulates cd147 through a combined effect of hif-1alpha and sp1 to promote glycolysis and tumor progression in epithelial solid tumors. Carcinogenesis. 2012;33:1598–607.
Zhang R, Wu Y, Zhao M, Liu C, Zhou L, Shen S, Liao S, Yang K, Li Q, Wan H. Role of hif-1alpha in the regulation ace and ace2 expression in hypoxic human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2009;297:L631–40.
Grabmaier K, AdW MC, Verhaegh GW, Schalken JA, Oosterwijk E. Strict regulation of caix(g250/mn) by hif-1alpha in clear cell renal cell carcinoma. Oncogene. 2004;23:5624–31.
Wykoff CC, Beasley NJ, Watson PH, Turner KJ, Pastorek J, Sibtain A, Wilson GD, Turley H, Talks KL, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000;60:7075–83.
Mukhopadhyay CK, Mazumder B, Fox PL. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J Biol Chem. 2000;275:21048–54.
Eclov RJ. Mechanisms regulating the expression and function of mxr: pharmaceutical sciences and pharmacogenomics. Ann Arbor, University of California, San Francisco, 2013, Ph.D., pp 464.
Tak E, Lee S, Lee J, Rashid MA, Kim YW, Park JH, Park WS, Shokat KM, Ha J, Kim SS. Human carbonyl reductase 1 upregulated by hypoxia renders resistance to apoptosis in hepatocellular carcinoma cells. J Hepatol. 2011;54:328–39.
Benita Y, Kikuchi H, Smith AD, Zhang MQ, Chung DC, Xavier RJ. An integrative genomics approach identifies hypoxia inducible factor-1 (hif-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009;37:4587–602.
Khurana P, Sugadev R, Jain J, Singh SB. Hypoxiadb: a database of hypoxia-regulated proteins. Database (Oxford). 2013;2013:bat074.
Kaluz S, Kaluzova M, Stanbridge EJ. Rational design of minimal hypoxia-inducible enhancers. Biochem Biophys Res Commun. 2008;370:613–8.
Kimura H, Weisz A, Ogura T, Hitomi Y, Kurashima Y, Hashimoto K, D'Acquisto F, Makuuchi M, Esumi H. Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J Biol Chem United States. 2001;276:2292–8.
MoncaleanoVega J, Ariza F, Hern¢ndez A. Association of nos3 and hif1¦á gene polymorphisms with the susceptibility of broiler chickens to develop hypoxic pulmonary hypertension. American Journal of Hypertension Research. 2013;1:17–22.
Wang C, Yuan C, Zhang L, Wu C, Li N. Differential gene expression of phosphoglyceric kinase (pgk) and hypoxic adaptation in chicken. Sci China C Life Sci. 2007;50:335–42.
Zeller KI, Jegga AG, Aronow BJ, O'Donnell KA, Dang CV. An integrated database of genes responsive to the myc oncogenic transcription factor: identification of direct genomic targets. Genome Biol. 2003;4:R69.
Bovolenta LA, Acencio ML, Lemke N. Htridb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics. 2012;13:405.
Pahl HL. Activators and target genes of rel/nf-kappab transcription factors. Oncogene. 1999;18:6853–66.
Tang S, Han H, Bajic VB. Ergdb: estrogen responsive genes database. Nucleic Acids Res. 2004;32:D533–6.
Pichiule P, Chavez JC, Schmidt AM, Vannucci SJ. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J Biol Chem. 2007;282:36330–40.
Weinmann AS. Novel chip-based strategies to uncover transcription factor target genes in the immune system. Nat Rev Immunol. 2004;4:381–6.
Shim H, Lewis BC, Dolde C, Li Q, CS W, Chun YS, Dang CV. Myc target genes in neoplastic transformation. Curr Top Microbiol Immunol. 1997;224:181–90.
Stormo GD, Zhao Y. Determining the specificity of protein-dna interactions. Nat Rev Genet. 2010;11:751–60.
Cheng C, Shou C, Yip KY, Gerstein MB. Genome-wide analysis of chromatin features identifies histone modification sensitive and insensitive yeast transcription factors. Genome Biol. 2011;12:R111.
Hsu SD, Lin FM, WY W, Liang C, Huang WC, Chan WL, Tsai WT, Chen GZ, Lee CJ, Chiu CM, Chien CH, MC W, Huang CY, Tsou AP, Huang HD. Mirtarbase: a database curates experimentally validated microrna-target interactions. Nucleic Acids Res. 2011;39:D163–9.
Schodel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of hif-binding sites by chip-seq. Blood. 2011;117:e207–17.
Xia X, Kung AL. Preferential binding of hif-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009;10:R113.
Xia X, Lemieux ME, Li W, Carroll JS, Brown M, Liu XS, Kung AL. Integrative analysis of hif binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci U S A. 2009;106:4260–5.
Cassavaugh J, Lounsbury KM. Hypoxia-mediated biological control. J Cell Biochem. 2011;112:735–44.
Bertout JA, Patel SA, Simon MC. The impact of O2 availability on human cancer. Nat Rev Cancer. 2008;8:967–75.
Li Y, D Y. Cancer therapy by targeting hypoxia-inducible factor-1. Curr Cancer Drug Targets. 2010;10:782–96.
Patiar S, Harris AL. Role of hypoxia-inducible factor-1alpha as a cancer therapy target. Endocr Relat Cancer. 2006;13(Suppl 1):S61–75.
von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. The strengthening the reporting of observational studies in epidemiology (strobe) statement: guidelines for reporting observational studies. Epidemiology. 2007;18:800–4.
Little J, Higgins JP, Ioannidis JP, Moher D, Gagnon F, von Elm E, Khoury MJ, Cohen B, Davey-Smith G, Grimshaw J, Scheet P, Gwinn M, Williamson RE, Zou GY, Hutchings K, Johnson CY, Tait V, Wiens M, Golding J, van Duijn C, McLaughlin J, Paterson A, Wells G, Fortier I, Freedman M, Zecevic M, King R, Infante-Rivard C, Stewart A, Birkett N. Strengthening the reporting of genetic association studies (strega)--an extension of the strobe statement. Eur J Clin Investig. 2009;39:247–66.
Peters SP. Reporting and evaluating genetic association studies. Respir Res. 2009;10:109.
Hollenbach JA, Mack SJ, Gourraud PA, Single RM, Maiers M, Middleton D, Thomson G, Marsh SG, Varney MD. A community standard for immunogenomic data reporting and analysis: proposal for a strengthening the reporting of immunogenomic studies statement. Tissue Antigens. 2011;78:333–44.
Janssens AC, Ioannidis JP, van Duijn CM, Little J, Khoury MJ. Strengthening the reporting of genetic risk prediction studies: the grips statement. Eur J Hum Genet. 2011;19:833–6.
Gallo V, Egger M, McCormack V, Farmer PB, Ioannidis JP, Kirsch-Volders M, Matullo G, Phillips DH, Schoket B, Stromberg U, Vermeulen R, Wild C, Porta M, Vineis P. Strengthening the reporting of observational studies in epidemiology–molecular epidemiology (strobe-me): an extension of the strobe statement. Mutagenesis. 2012;27:17–29.
Saffery R, Gordon L. Time for a standardized system of reporting sites of genomic methylation. Genome Biol. 2015;16:85.
Pipan V, Kunej T. Initiative for standardization of the format of the next-generation sequencing (ngs) results. Discoveries. 2015;3:e44.
Urh K, Kunej T. Molecular mechanisms of cryptorchidism development: update of the database, disease comorbidity, and initiative for standardization of reporting in scientific literature. Andrology 2016;4(5):894–902.
Spiegelman BM, Heinrich R. Biological control through regulated transcriptional coactivators. Cell. 2004;119:157–67.
Luo W, Chang R, Zhong J, Pandey A, Semenza GL. Histone demethylase jmjd2c is a coactivator for hypoxia-inducible factor 1 that is required for breast cancer progression. Proc Natl Acad Sci U S A. 2012;109:E3367–76.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was supported by the Slovenian Research Agency (ARRS) through the Research program (P4-0220).
Electronic supplementary material
Supplemental Figure 1
. Identified polymorphisms within HRE sites of four target genes. Sequence of the target genes: ABCG2 (A), ACE (B), CA9 (C), and CP (D). Biotype of identified polymorphisms within HRE sites (E). (DOCX 1512 kb)
Supplemental Table 1
. Collected data associated with 98 HIF-1α target genes. (XLSX 38 kb)
Supplemental Table 2
. Pathway analysis of HIF-1α downstream targets. (DOCX 17 kb)
Rights and permissions
About this article

Cite this article
Slemc, L., Kunej, T. Transcription factor HIF1A: downstream targets, associated pathways, polymorphic hypoxia response element (HRE) sites, and initiative for standardization of reporting in scientific literature. Tumor Biol. 37, 14851–14861 (2016). https://doi.org/10.1007/s13277-016-5331-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5331-4