
Abstract
C-C chemokine receptor type 1 (CCR1) and chemokine C-C motif receptor-like 2 (CCRL2) have not yet been sufficiently investigated for their role in colorectal cancer (CRC). Here, we investigated their expression in rat and human CRC samples, their modulation of expression in a rat liver metastasis model, as well as the effects on cellular properties resulting from their knockdown. One rat and five human colorectal cancer cell lines were used. CC531 rat colorectal cells were injected via the portal vein into rats and re-isolated from rat livers after defined periods. Following mRNA isolation, the gene expression was investigated by microarray. In addition, all cell lines were screened for mRNA expression of CCR1 and CCRL2 by reverse transcription polymerase chain reaction (RT-PCR). Cell lines with detectable expression were used for knockdown experiments; and the respective influence was determined on the cells’ proliferation, scratch closure, and colony formation. Finally, specimens from the primaries of 50 patients with CRC were monitored by quantitative RT-PCR for CCR1 and CCRL2 expression levels. The microarray studies showed peak increases of CCR1 and CCRL2 in the early phase of liver colonization. Knockdown was sufficient at mRNA but only moderate at protein levels and resulted in modest but significant inhibition of proliferation (p < 0.05), scratch closure, and colony formation (p < 0.05). All human CRC samples were positive for CCR1 and CCRL2 and showed a significant pairwise correlation (p < 0.0004), but there was no correlation with tumor stage or age of patients. In summary, the data point to an important role of CCR1 and CCRL2 under conditions of organ colonization and both chemokine receptors qualify as targets of treatment during early colorectal cancer liver metastasis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer (CRC) is one of the major causes of death from malignant diseases and the fourth leading cause of cancer death in the world [1]. The treatment of CRC is based on surgical resection, but about half of the colorectal carcinoma patients will relapse within 5 years after primary surgery and succumb to the disease. The prognosis depends on the histopathologic stage at the time of diagnosis and despite improved radio-therapeutic and chemotherapeutic regimens will worsen progressively from stage I to stage IV. Of note, the outcome of individual patients cannot be predicted reliably from the disease stage alone [2], as the shedding of disseminated tumor cells and the subsequent initiation of early microscopic metastatic spread cannot be reliably assessed. Macroscopic metastatic spread of colorectal cancer, as indicated by stage IV, is associated with the worst prognosis [3]. Cancer progression is based on continued cell proliferation and the acquisition of other properties like invasion (loss of cell to cell adhesion, increased cell motility, and basement membrane degradation), intravasation of tumor cells, their distribution with the blood stream, and subsequent extravasation, which can lead to establishing a metastatic colony, and then will also induce angiogenesis [4–6]. Therefore, the effective inhibition of cancer progression is a key problem to improve the outcome of cancer patients. The identification of new biomarkers enabling the early detection of colorectal cancer will be critical in developing more successful therapies and will likely have an important positive effect on the prognosis of CRC patients.
Tumor invasion and metastasis share many similarities with leukocyte trafficking, which is critically regulated by chemokines and their receptors [7]. Chemokines are associated with a multitude of diseases including asthma, HIV infection, rheumatoid arthritis, multiple sclerosis, and cancer. An increasing amount of evidence highlights the importance of chemokines and their receptors in the metastasis process of many cancers, including colon cancer [8–10]. Chemokines play a pivotal role in cancer progression because they trigger numerous important cellular responses such as growth, adhesion, migration, and metastasis of tumor cells [11–15]. Chemokines are a family of small (8–11 kDa) secreted proteins, which initially were identified as mediators of leukocyte trafficking and homing. They also participate in the growth and lymphatic or hematogenic spread of malignant tumors [16–18]. C-C chemokine receptor type 1 (CCR1, CD191) is a chemokine receptor that is expressed on neutrophils, monocytes, eosinophils, dendritic cells, activated T lymphocytes, and B lymphocytes [19–21]. CCR1 binds to three chemokines including MIP-1α, RANTES, and monocyte chemoattractant protein (MCP)-1 [22]. The role of CCR1 has been well characterized for several inflammatory conditions, such as arthritis [23, 24], chronic kidney disease [25–27], immune-mediated hepatitis [28], tumor angiogenesis [29], ischemia-reperfusion injury [30], sepsis [31, 32], and transplant rejection [33, 34]. The chemokine C-C motif receptor-like 2 (CCRL2) is a heptahelical transmembrane receptor, expression of which has been shown on almost all human hematopoietic cells [35]. CCRL2 shows the highest degree of homology with the inflammatory chemokine receptor CCR1 [36], as indicated by more than 40 % identity in amino acid sequence [35]. CCR1 and CCRL2 were previously considered to be orphan receptors without any known biological roles; however, recent studies identified ligands for these receptors and their functions have begun to be unveiled [37].
In preceding experiments we investigated the mRNA expression of rat colorectal CC531 cells following their re-isolation from the liver of animals, in which they had grown for different periods of time [38, 39]. Interestingly, claudins and other gene groups were among those mRNAs, which were most intensively modulated after intraportal CC531 cell implantation and subsequent liver colonization by these cells. In the present study we focused on the modulation of the chemokine receptors CCR1 and CCRL2 in re-isolated rat colorectal CC531 cells and compared these results with the expression profiles found in patient samples. In addition, five human colorectal cancer cell lines as well as rat colorectal CC531 cells were used for studying the effects on cellular functions as proliferation, scratch healing, and colony formation in response to knockdown of CCR1 and CCRL2 by using small interfering RNA (siRNA) for unraveling the effects of these receptors in tumor cells.
Materials and methods
Cell lines and culture conditions
The colon adenocarcinoma cell lines SW480, SW620, Caco2, HT29, LS174T (human), and CC531 (rat) were maintained under standard culture conditions (37 °C, humidified atmosphere with 5 % CO2) in RPMI-1640 medium (Invitrogen, Karlsruhe, Germany). The medium was supplemented with 10 % fetal calf serum (FCS), l-glutamine (2 mM), penicillin (100 IU/ml), and streptomycin (100 μg/ml; Invitrogen). For isolation and propagation, the cells were washed with phosphate buffered saline (PBS), trypsinized (0.25 % trypsin/EDTA), pelleted at 1500 rpm for 5 min, and suspended at the desired concentration in RPMI-1640 medium (Invitrogen).
RNA isolation and reverse transcriptase polymerase chain reaction
For RNA isolation from cells of the above cell lines, the RNeasy Mini Kit (Qiagen, Hilden, Germany) was used. The amount and purity of isolated RNA were measured in a spectrophotometer using the 260/280 ratio.
To produce cDNA from the isolated RNA for PCR reaction, the Maxima first strand cDNA synthesis kit (Thermo Fisher Scientific, Karlsruhe, Germany) was used. In brief, a mixture of 100 ng RNA, buffer (1×), deoxynucleotide (dNTPs) (5 μM), oligo-dT-primers (1 μM), RNAse inhibitor (10 units), and reverse transcriptase enzyme (4 units) was incubated in a total volume of 20 μl at 37 °C for 1 h. Three microliters of cDNA from the last step was mixed with MgCl2 (1.75 mM), dNTPs (200 μM), buffer (1×), red Taq DNA polymerase (2.5 units), and the selected primer pair (each 0.5 μM) (for primer sequences of target and household genes, see suppl. Table 1). The reaction mixture was incubated in a thermal cycler (DNA engine, PTC200 Peltier) using a program with the following profile: Initial heating (95 °C, 3 min) was followed by 35 cycles of denaturation (95 °C, 40 s), annealing (60 °C, 1 min), and elongation (72 °C, 40 s), which was followed by a final elongation step (72 °C, 10 min) before the sample was kept at 4 °C until it was removed from the thermal cycler. Thereafter, the PCR products were analyzed either by polyacrylamide gel or by capillary electrophoresis.
Patients and tissue samples
For determining the levels of CCR1 and CCRL2 in samples from colorectal cancer patients by real-time reverse transcription polymerase chain reaction (RT-PCR) analysis, samples from 50 sporadic CRC patients (33 males, 17 females) were selected, who were admitted and underwent surgery in the time (between January 1998 and July 2001) at the Municipal Hospital in Nürnberg, Germany (Department of Abdominal, Thorax, and Endocrine Surgery). The samples included in this study were used based on the patients’ informed consent and approved by the responsible Ethics Committee. The Light Cycler 480 real-time PCR system with the LC480 RNA Master hydrolysis probes and the human Universal Probe Library kit (Roche Diagnostics, Mannheim, Germany) were used following the manufacturer’s protocol. One microliter containing cDNA derived from the RT reaction with 1 μg RNA was pipetted in triplicate into 384-well plates and amplified at 60 °C for 50 cycles. The cDNA input was normalized to the expression of the housekeeping gene GAPDH. As positive control, a mixture of 11 normal mucosa samples was used.
Small interfering RNA knockdown experiments
Small interfering RNA duplexes designed against rat Ccr1, as well as human CCR1 and CCRL2, were purchased from Invitrogen (Table 2). The tumor cells cultured in 6-well plates were transfected with different nanomolar concentrations of small interfering RNA (siRNA) or negative siRNA control using X-tremeGENE siRNA transfection reagent (Roche), following the manufacturer’s instructions. At 24, 48, and 72 h after treatment, the cells were harvested for RT-PCR analysis.
Cell proliferation assay (MTT)
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to assess the effect of gene knockdown on the proliferation of tumor cells. Briefly, 4 × 103 cells of the cell lines CC531, SW480, SW620, HT29, LS174T, and Caco2 were seeded per well of 96-well plates and treated with specific or control siRNA as described above. After defined periods (24, 48, or 72 h), 10 μl MTT solution (10 mg/ml, Sigma, Munich, Germany; dissolved in PBS, pH 7.4) was added to each well and incubated for 3 h at 37 °C. Subsequently, the medium was removed and formazan crystals were solubilized with 0.04 N HCl-isopropanol. The optical density was measured at 540 nm wavelength (690 nm reference wavelength) using an ELISA plate reader (Anthos Mikrosysteme, Krefeld, Germany).
Wound healing assay (scratch assay)
Tumor cells were seeded into 24-well plates and treated after 24 h with nonsense or specific siRNA as detailed before. At 48 h after transfection, straight scratches (wounds) were created in confluent cells using a sterile pipette tip. The cells were then carefully rinsed with culture medium to remove free-floating cells and debris. Fetal calf serum-free medium was then added, and culture plates were incubated at 37 °C. The migration path of cells was tracked at 0, 12, 24, 36, and 48 h using the Axio Observer.Z1 microscope (Carl Zeiss AG, Germany), and representative scratch zones for each cell line were photographed. Each experiment was repeated twice, and the duplicate wells of each condition were examined for each experiment.
Colony formation assay
To determine the effect of siRNA knockdown of CCR1 and CCRL2 on the ability of colorectal cancer cells to form colonies, the procedure previously detailed was performed [30]. The following criteria were taken into account for evaluating the results after 1 week: clusters of ≥30 cells were counted as colony, and clusters of ≥60 cells were considered as large colony.
Western blotting
The cell pellets were thoroughly suspended, lysed, heated at 99 °C for 5 min, and then centrifuged at 13,000 rpm for 10 min at 4 °C. In order to load the same amount of protein from each sample, the protein concentration of lysates was determined using BCA protein assay kit (Pierce, USA). A volume containing 30–40 μg protein was separated electrophoretically (X-cell Sure Lock system, Invitrogen, Karlsruhe, Germany) in SDS running buffer at 150 V for 1 h. For size orientation, 15 μl of full-range rainbow marker (Fermentas, Germany) was run with the lysate. The separated proteins were transferred onto PVDF membranes (Roche, Mannheim, Germany) at room temperature using a semi-dry system based on Whatman papers wetted with transfer buffer (3 g Tris-base + 14.4 g glycine + 200 ml methanol) for 60 min at a current of 0.8 mA/cm2 and a voltage of ∼30 V. The membranes were blocked for unspecific binding (CCR1–3 h and CCRL2–1 h) in blocking solution (TBS 0.05 % Tween plus 3 % albumin for CCR1 and TBS plus 1 % western blocking solution for CCRL2) and then incubated with the first antibody (diluted 1:500 in TSB 0.05 % Tween pH 7.4 plus 1.5 % albumin for CCR1 and in TSB 0.5 % Tween pH 7.4 for CCRL2) on a shaker at 4 °C overnight. The specific antibodies (goat polyclonal sc-6125 and rabbit polyclonal sc-102422) as well as the respective secondary antibodies (sc-2020 and sc-2004) were obtained from Santa Cruz Biotechnology, Heidelberg. The blots were washed three times followed by an incubation step with the appropriate secondary antibody (1:5000 dilutions) for 45 min for CCR1 and 1 h for CCRL2 on a shaker at room temperature. A second washing step was repeated four times before developing the membranes with ECL (Amersham Bioscience). For re-probing, the PVDF membranes were incubated in stripping solution (200 mM glycine pH 2.5, 0.05 % Tween) at 80 °C for 20–30 min. Then the membranes were washed three times for 10 min followed by the same procedure as mentioned above.
Preparation of CC531 tumor cells for injection
The stably transfected CC531RFP cells were washed with PBS, trypsinized, pelleted, and suspended at a concentration of 4 × 106 cells/500 μl (350 μl PBS + 150 μl Biomatrix EHC; Serva Electrophoresis, Heidelberg).
CC531 tumor cell injection and re-isolation
Six to eight-week-old male WAG/Rij rats (Charles River Laboratories, Germany) were used for the experiments. They were fed a standard diet ad libitum and given an adaptation period of 1 week prior to any experimental procedures. All animal experiments were approved by the responsible governmental animal ethics committee (RP Karlsruhe, Germany). Tumor cell re-isolation was performed as described before [38, 39]. In brief, liver tissue with tumor nodules was transferred into a single cell suspension and the tumor cells were separated from the liver cells by Ficoll gradient centrifugation followed by FACS sorting for the marker RFP. The resulting purity of the tumor cells was >99 %.
Microarray analysis
For RNA isolation from CC531 cells, the RNeasy mini-kit (Qiagen, Hilden, Germany) was used. RNA was eluted in water. The quality of total RNA was checked by gel analysis using the total RNA nanochip assay on an Agilent 2100 bioanalyzer (Agilent Technologies GmbH, Berlin, Germany). Only samples with RNA index values >8.5 were selected for expression profiling. RNA concentrations were determined using the NanoDrop spectrophotometer (NanoDrop Technologies, Wilmington, DE). Microarray probe labeling and Illumina Sentrix BeadChip array hybridization were performed as described in [38, 39]. In brief, biotin-labeled cRNA samples for hybridization on Illumina Rat Sentrix-12 BeadChip arrays (Illumina, San Diego, CA) were prepared according to Illumina’s sample labeling procedure based on the modified Eberwine protocol. Following hybridization, microarray scanning was performed on a BeadStation array scanner. Data extraction was done for all beads individually and outliers were removed. All remaining data points were used for the calculation of the average signal for a given probe, and the standard deviation for each probe was calculated. Data analysis was performed by normalization of the signals using the quantile normalization algorithm without background subtraction, and differentially regulated genes were defined by calculating the standard deviation differences of a given probe in one-by-one comparisons of samples or groups. Gene expression was considered significantly changed for p values ≤0.005, a fold change of at least ±2 and a greater than 12-fold difference between the bead standard error of treatment and control samples.
Statistical analysis
The T-test was used to test the significance of proliferation, migration, and colony formation inhibition in cells treated with specific vs. nonsense siRNA. Calculations were carried out using the ADAM statistical software package (DKFZ, Germany). For mRNA expression values, quantile-normalized mRNA data were log2 transformed. Spearman’s correlation coefficient was used to assess pairwise correlation of genes and correlation of expression levels with age. Kruskal-Wallis test was used to test for association of expression levels with tumor stage. Overall survival (OS) was defined as time from surgery to time of death. Univariate Cox regression was used to test for association of expression levels with OS. The interquartile hazard ratio is given, i.e., the change in risk between the upper 25 % of samples (upper quartile, Q3) and the lower 25 % (lower quartile, Q1). All p values were adjusted for multiple testing using Benjamini-Hochberg correction in order to control the false discovery rate. All p values are two-sided. All analyses were carried out using software R 3.0 [40]. The p values below 0.05 were considered statistically significant.
Results
Regulation of Ccr1 and Ccrl2 genes during CC531 liver colonization
The microarray data showed that during propagation in vitro, the expression levels of Ccr1 and Ccrl2 were low in rat colorectal CC531 cells, with the former showing a twofold higher expression than the latter, which showed basal expression only. After intraportal injection, Ccr1 mRNA levels increased threefold during initial liver colonization before returning to normal levels after 1 week and thereafter rising to a maximally fivefold higher concentration than in vitro (Fig. 1). In contrast, Ccrl2 levels showed initially a 27-fold increase and subsequently a decrease to levels, which were only 1.5-fold higher than that found for cells growing in vitro.

Modulation of Ccr1 and Ccrl2 mRNA levels as shown by microarray analysis in CC531 rat colorectal cells during liver colonization. The values represent the gene expression in isolated metastasizing cells in comparison to the expression in cells growing in vitro. The x-axis gives the time (in days) after intraportal implantation of CC531 rat colorectal cells to syngeneic rats. Each dot corresponds to one time point, at which the cells were re-isolated. The y-axis gives the mRNA expression of Ccr1 and Ccrl2 relative to that level found in CC531 cells growing in vitro
Spontaneous expression of CCR1 and CCRL2 in colorectal cancer cell lines
In addition to rat CC531 cells, five human colorectal cancer cell lines were examined for the expression of CCR1 and CCRL2 mRNAs by conventional RT-PCR using specifically designed primers (suppl. Table 1). The results show that the CCR1 gene was expressed in rat CC531 and human SW480, SW620, and HT29 cell lines, whereas the CCRL2 gene was expressed in human SW480, SW620, LS174T, and Caco2 cell lines (Table 1). The sensitivity of this assay was too low for detecting the basal expression of Ccrl2 in CC531 cells.
Knockdown of CCR1 and CCRL2 by specific siRNA species
Cells of the colorectal cancer cell lines, which had been found to express CCR1 or CCRL2 by conventional RT-PCR, were transfected at 50–500 nM concentrations with specific rat or human siRNAs, as shown in suppl. Table 2. An mRNA knockdown of more than 75 % was observed, except for CCRL2 in human SW480 cells (Table 2). Consequently, this cell line was excluded from the investigation of CCRL2 knockdown effects. In addition, the effect on CCR1 and CCRL2 was examined at protein levels. By Western blot, bands of approximately 35 and 40 kDa were detected, corresponding to the molecular weight of these proteins (Fig. 2). Overall, no significant knockdown was observed; except for SW620 cells, the CCR1 levels decreased at least slightly or temporarily in response to CCR1siRNA, as did CCRL2 levels in response to CCRL2siRNA, except for Caco2 cells.

Western blot analysis of CCR1/Ccr1 and CCRL2/Ccrl2 protein levels in a series of colorectal cancer cell lines (CCR1 in SW620, SW480, HT29, Ccr1 in CC531 and CCRL2 in SW620, LS174T, Caco2) in response to siRNA knockdown. The relative concentrations for CCR1 (left part, top) and CCRL2 (left part, bottom) were normalized to actin as loading control (middle part) and evaluated by the program ImageJ. The values resulting from ImageJ analysis are plotted by GraphPadPrism 6 software to show the respective relative protein levels (right part). T siRNA-treated cells, U untreated cells; the numbers denote the period after transfection of cells
Effect on cell proliferation
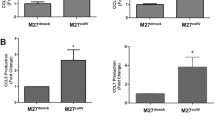
The downregulation of CCR1 and CCRL2 was further analyzed regarding a potential effect on the proliferation of the six colorectal cancer cell lines mentioned above. The data show that there was reduced proliferation in cells from all cell lines as compared to respective untreated cells (see Fig. 3). The inhibition of proliferation was most pronounced in CC531 cells in response to CCR1siRNA (79 % at 24 h, p < 0.05) and in LS174T cells in response to CCRL2siRNA (32 % at 72 h, p < 0.05).

Proliferation of colorectal cancer cells in response to knockdown by CCR1siRNA/Ccr1siRNA (top) and CCRL2siRNA (bottom). The x-axis of this grouped bar chart denotes the period (24, 48, and 72 h) following transfection of cells, after which the proliferation was determined by MTT assay. The y-axis gives the proliferation of cells with knockdown as percentage of respective control cells. Significant differences from comparing control and treated cells (n = 48 samples) by T-test are indicated by an asterisk
Effect on scratch closure
The scratch closure assay revealed effects of CCR1 and CCRL2 knockdown on migratory properties of colorectal cancer cells. Overall, downregulation of CCR1 and CCRL2 by siRNA resulted in a reduced potential of CRC cells to fill in the gap (Fig. 4a, b).

Scratch closure (migration) of colorectal cancer cells in response to knockdown by CCR1siRNA/Ccr1siRNA (a) and CCRL2siRNA (b). The x-axis indicates the various cell lines investigated and for each cell line a pairwise comparison of pictures is given showing control and siRNA-treated cells. The y-axis gives the time after exposure of colorectal cancer cells to siRNA
Effect on colony formation
The colony formation assay revealed further effects of CCR1 and CCRL2 knockdown (see Fig. 5): Exposure to CCR1siRNA uniformly caused a mild reduction of colony formation in the four colorectal cancer cell lines investigated; a significant inhibition was observed only for large colonies formed by SW620 and SW480 cells (p < 0.05). Knockdown of CCRL2, however, showed equivocal results, as demonstrated from more than 50 % reduction of colony formation in LS174T cells (p < 0.05), whereas SW620 and Caco2 cells showed a slightly increased colony formation.

Colony formation of colorectal cancer cells in response to knockdown by CCR1siRNA/Ccr1siRNA (top) and CCRL2siRNA (bottom). Colonies were counted after 7 days, and a comparison was made between the colony-forming ability of control and siRNA-treated cells, differentiating between small (>30 cells) and large (>60 cells) colonies. The x-axis of this grouped bar chart denotes small or large colonies for each cell line; the y-axis indicates the number of colonies as percentage of respective controls. Significant differences from comparing control and treated cells (n = 3 samples) by T-test are indicated by an asterisk
CCR1 and CCRL2 expression in colorectal cancer patient samples
Tumor samples from 50 colorectal cancer patients (33 males and 17 females; 10 International Union Against Cancer (UICC) stage 1, 15 UICC stage 2, 15 UICC stage 3, and 10 UICC stage 4) were screened by real-time RT-PCR for the presence of CCR1 and CCRL2 levels. The results are shown in Fig. 6. The mean expression level from 11 control mucosa samples was set to unity. Based on this, the mean expression of CCR1 was 1.41 (SD 1.62) and that of CCRL2 was 0.31 (SD 0.8). There was neither significant correlation of CCR1 and CCRL2 levels with the stage of colorectal carcinomas (Fig. 6a) nor with the age of the patients (Fig. 6b); there was, however, a significant correlation between CCR1 and CCRL2 expression, when using log2-transformed mRNA expression values (Fig. 6c, p < 0.0004, Spearman’s correlation coefficient rho = 0.5)

Expression of CCR1 and CCRL2 mRNA in human primary colorectal cancers. Raw values of mRNA expression were related to the mean expression of 11 samples from normal mucosa (control), which was set to unity. Then, the relative expression of tumor samples was log2 transformed and plotted as single dots. Box plots indicating the median and 25 % and 75 % quantiles are shown in relation to tumor stages 1–4 (a) and to age of patients (b). Finally, the correlation between CCR1 and CCRL2 expression levels was assessed by pairwise scatterplot (c) and evaluated by Spearman’s correlation coefficient (rho = 0.5) as well as moderated t statistics for significance (p = 0.00037)
Discussion
Chemokines have been shown to participate in tumor growth, angiogenesis, lymphatic and hematogenic spread of malignant tumors [41–46], and regulate leukocyte traffic [18]. The biological effects of chemokines are triggered by interacting with seven-span transmembrane domain receptors coupled to trimeric G proteins that are selectively found on the surface of respective target cells. Consequently, chemokines and their receptors can exert a variety of effects including those, which are supportive of organ colonization.
CCR1 and other chemokine receptor genes, including CCR2, CCRL2, CCR3, CCR5, and CCXCR1, form a gene cluster on chromosome 3p and are expressed in various cells [47–50]. One of the most recent additions to the atypical chemokine receptor family is CCRL2, also known as CRAM. CCRL2 is reported to act as a receptor for CCL19 and CCL5, as well as the nonchemokine chemoattractant chemerin, which also binds to the chemokine-like receptor (CMKLR)-1, alternatively known as ChemR23.
CCR1 expression has been detected in some types of cancer, for example, in prostate cancer cells [51], multiple myeloma cells [52], and in hepatocellular carcinoma cells [53]. CCRL2 expression at the mRNA level has been described in murine macrophages [54], glial cells, astrocytes, microglia stimulated with LPS [55, 56], and in mast cells [57]. CCRL2 was also reported to be upregulated in lung macrophages and epithelial cells after in vivo sensitization [58]. In our experiments, CCR1 and CCRL2 were detected by conventional RT-PCR in four of six colorectal carcinoma cell lines, but there was concordant expression in only one third of the cell lines. The basal Ccrl2 expression found in CC531 cells by microarray was presumably below the detection limit of our conventional RT-PCR. It is interesting, therefore, to note that there were at least basal expression levels of the two chemokine receptors in 50 of 50 patient samples as determined by real-time RT-PCR, although the levels of CCRL2 were on average five times lower than that of CCR1. In addition, with the exception of some outliers, all primary carcinoma expression levels were similar to those found in normal mucosa samples.
These low levels contrast with our results from the experiment, in which rat colorectal CC531 cells had been implanted via intraportal injection into the liver of syngeneic rats to mimic colorectal cancer liver metastasis. Thereafter, CC531 cells were re-isolated at regular intervals and changes in mRNA expression were detected by mRNA microarray.
We chose this syngeneic model for its intact tumor-host interaction, whereas xenograft models lack important components of the host microenvironment, such as immune cell functions. Our model, which focuses on colorectal cancer cells, is complementary to a model used by Kitamura et al. [59] and Hirai et al. [60] who could show that bone marrow cells upregulate Ccr1 in a mouse model and thus contribute to colorectal cancer liver metastasis.
Remarkably, several groups of genes were found modulated in our model, among them claudins [39], IGF-BPs [38], and chemokines like Ccr1 and Ccrl2 (see Fig. 1). These results indicate that Ccr1 levels peak at the initial stage of liver colonization, whereas Ccrl2 levels rose in the more advanced stages. Therefore, it seemed promising to further investigate the role of these chemokines regarding cellular properties, which are essential for organ colonization.
First, we investigated by siRNA technique the effect of CCR1 and CCRL2 knockdown on cell proliferation. Despite a 90 % reduction in CCR1 mRNA levels of CC531 cells, the associated decrease in protein expression did not exceed 30 %. Nevertheless, the proliferation of these cells was significantly inhibited by more than 50 %. In the other cell lines, which exhibited even less intensive reductions in CCR1 protein expression, little but still significant inhibition in proliferation was observed. A similar relationship was observed for CCRL2 expression and related inhibition in proliferation: LS174T cells showed 70 % reduced mRNA level concomitant with the least intensive knockdown of CCRL2 at protein level, but still a significant inhibition of proliferation and colony formation was observed. Altogether, however, inhibition of proliferation in response to CCR1 and CCRL2 knockdown was limited. This is in agreement with data from Wang et al., who found only a minor effect of CCR1 knockdown on cell proliferation in two NSCLC cell lines [7], and of Wu et al., who found a minor effect of a decreased CCR1 level on cell proliferation in a hepatoma cell line [53]. Similarly, there was an only modest effect of CCR1 and CCRL2 knockdown regarding the inhibition of wound healing as surrogate for migration and colony formation, respectively. Knockdown of CCR1 was, however, instrumental in reducing the invasive abilities of hepatoma and NSCLC cells as well as inhibiting the secretion of MMP-2 and MMP-9, respectively [7, 53]. The limited knockdown at protein level could well be explained by the basal mRNA expression of colorectal cancer cells growing in vitro. As a consequence, the mRNA knockdown was less efficacious in reducing the respective protein levels within our observation period of 3 days. A similar observation was made for bone sialoprotein (BSP), the level of which was not sufficiently affected by siRNA exposure within 3 days. Concomitantly, the proliferation of breast cancer cells exposed to siRNA against BSP was not reduced [61]. However, a prolonged knockdown of 6 days was instrumental in reducing the protein concentration of BSP significantly, and concomitantly the proliferation of breast cancer cells was inhibited [62].
Our animal data show that Ccr1 and Ccrl2 are highly expressed during the dynamic phase of metastasis, such as early liver colonization, whereas in primary human carcinomas, only a minority of samples was found to express the two receptors at increased levels. In line with these observations, there was no discernible relation of the mRNA levels of the two chemokine receptors to tumor stage in patients with colorectal carcinoma, which indicates that they do not significantly contribute to the progression of the primary cancer. The only significant correlation was found between the levels of the two chemokines, thus pointing to a common factor, which is responsible for their expression. As increased levels were found related to liver metastasis, future experiments will focus on augmenting CCR1 and CCRL2 levels by overexpressing these genes and analyzing the resulting cellular properties.
Taken together, our data indicate for the first time that the two chemokine receptors Ccr1 and Ccrl2 are increased in rat colorectal carcinoma cells during colonization of the liver and thus can be considered a target during this phase. In primary human colorectal cancers, however, they are seemingly less important and their downregulation will have only modest consequences on proliferation, migration, and colony formation. Future experiments will show whether small molecule drugs with affinity to these receptors will have activity against certain aspects of human colorectal cancer progression, such as liver colonization.
References
Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–502.
Kattan MW, Wheeler TM, Scardino PT. Postoperative nomogram for disease recurrence after radical prostatectomy for prostate cancer. J Clin Oncol Off J Am Soc Clin Oncol. 1999;17:1499–507.
Gallagher DJ, Kemeny N. Metastatic colorectal cancer: from improved survival to potential cure. Oncology. 2010;78:237–48.
Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72.
Duffy MJ, McGowan PM, Gallagher WM. Cancer invasion and metastasis: changing views. J Pathol. 2008;214:283–93.
Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34.
Wang CL, Sun BS, Tang Y, Zhuang HQ, Cao WZ. CCR1 knockdown suppresses human non-small cell lung cancer cell invasion. J Cancer Res Clin Oncol. 2009;135:695–701.
Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights into cancer-related inflammation. Trends Mol Med. 2010;16:133–44.
Kruizinga RC, Bestebroer J, Berghuis P, de Haas CJ, Links TP, de Vries EG, et al. Role of chemokines and their receptors in cancer. Curr Pharm Des. 2009;15:3396–416.
Erreni M, Bianchi P, Laghi L, Mirolo M, Fabbri M, Locati M, et al. Expression of chemokines and chemokine receptors in human colon cancer. Methods Enzymol. 2009;460:105–21.
Horuk R. Chemokine receptor antagonists: overcoming developmental hurdles. Nat Rev Drug Discov. 2009;8:23–33.
Pease JE, Horuk R. Chemokine receptor antagonists: part 1. Exp Opin Ther Patents. 2009;19:39–58.
Pease JE, Horuk R. Chemokine receptor antagonists: part 2. Exp Opin Ther Patents. 2009;19:199–221.
Strieter RM. Chemokines: not just leukocyte chemoattractants in the promotion of cancer. Nat Immunol. 2001;2:285–6.
Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer Lett. 2008;267:226–44.
Luster AD. Chemokines–chemotactic cytokines that mediate inflammation. N Engl J Med. 1998;338:436–45.
Wang JM, Deng X, Gong W, Su S. Chemokines and their role in tumor growth and metastasis. J Immunol Methods. 1998;220:1–17.
Zlotnik A, Yoshie O. Chemokines: a new classification system and their role in immunity. Immunity. 2000;12:121–7.
D’Ambrosio D, Panina-Bordignon P, Sinigaglia F. Chemokine receptors in inflammation: an overview. J Immunol Methods. 2003;273:3–13.
Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, et al. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood. 2001;97:1144–6.
Fioretti F, Fradelizi D, Stoppacciaro A, Ramponi S, Ruco L, Minty A, et al. Reduced tumorigenicity and augmented leukocyte infiltration after monocyte chemotactic protein-3 (MCP-3) gene transfer: perivascular accumulation of dendritic cells in peritumoral tissue and neutrophil recruitment within the tumor. J Immunol. 1998;161:342–6.
Gao JL, Wynn TA, Chang Y, Lee EJ, Broxmeyer HE, Cooper S, et al. Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J Exp Med. 1997;185:1959–68.
Revesz L, Bollbuck B, Buhl T, Eder J, Esser R, Feifel R, et al. Novel CCR1 antagonists with oral activity in the mouse collagen induced arthritis. Bioorg Med Chem Lett. 2005;15:5160–4.
Shahrara S, Proudfoot AE, Woods JM, Ruth JH, Amin MA, Park CC, et al. Amelioration of rat adjuvant-induced arthritis by Met-RANTES. Arthritis Rheum. 2005;52:1907–19.
Anders HJ, Ninichuk V, Schlondorff D. Progression of kidney disease: blocking leukocyte recruitment with chemokine receptor CCR1 antagonists. Kidney Int. 2006;69:29–32.
Ninichuk V, Anders HJ. Chemokine receptor CCR1: a new target for progressive kidney disease. Am J Nephrol. 2005;25:365–72.
Vielhauer V, Berning E, Eis V, Kretzler M, Segerer S, Strutz F, et al. CCR1 blockade reduces interstitial inflammation and fibrosis in mice with glomerulosclerosis and nephrotic syndrome. Kidney Int. 2004;66:2264–78.
Ajuebor MN, Hogaboam CM, Le T, Proudfoot AE, Swain MG. CCL3/MIP-1alpha is pro-inflammatory in murine T cell-mediated hepatitis by recruiting CCR1-expressing CD4(+) T cells to the liver. Eur J Immunol. 2004;34:2907–18.
Yang X, Lu P, Fujii C, Nakamoto Y, Gao JL, Kaneko S, et al. Essential contribution of a chemokine, CCL3, and its receptor, CCR1, to hepatocellular carcinoma progression. Int J Cancer J Int Cancer. 2006;118:1869–76.
Reichel CA, Khandoga A, Anders HJ, Schlondorff D, Luckow B, Krombach F. Chemokine receptors Ccr1, Ccr2, and Ccr5 mediate neutrophil migration to postischemic tissue. J Leukoc Biol. 2006;79:114–22.
Bonville CA, Lau VK, DeLeon JM, Gao JL, Easton AJ, Rosenberg HF, et al. Functional antagonism of chemokine receptor CCR1 reduces mortality in acute pneumovirus infection in vivo. J Virol. 2004;78:7984–9.
Ness TL, Carpenter KJ, Ewing JL, Gerard CJ, Hogaboam CM, Kunkel SL. CCR1 and CC chemokine ligand 5 interactions exacerbate innate immune responses during sepsis. J Immunol. 2004;173:6938–48.
Yun JJ, Whiting D, Fischbein MP, Banerji A, Irie Y, Stein D, et al. Combined blockade of the chemokine receptors CCR1 and CCR5 attenuates chronic rejection. Circulation. 2004;109:932–7.
Gao W, Topham PS, King JA, Smiley ST, Csizmadia V, Lu B, et al. Targeting of the chemokine receptor CCR1 suppresses development of acute and chronic cardiac allograft rejection. J Clin Investig. 2000;105:35–44.
Yoshimura T, Oppenheim JJ. Chemokine-like receptor 1 (CMKLR1) and chemokine (C-C motif) receptor-like 2 (CCRL2); two multifunctional receptors with unusual properties. Exp Cell Res. 2011;317:674–84.
Otero K, Vecchi A, Hirsch E, Kearley J, Vermi W, Del Prete A, et al. Nonredundant role of CCRL2 in lung dendritic cell trafficking. Blood. 2010;116:2942–9.
Migeotte I, Franssen JD, Goriely S, Willems F, Parmentier M. Distribution and regulation of expression of the putative human chemokine receptor HCR in leukocyte populations. Eur J Immunol. 2002;32:494–501.
Georges RB, Adwan H, Hamdi H, Hielscher T, Linnemann U, Berger MR. The insulin-like growth factor binding proteins 3 and 7 are associated with colorectal cancer and liver metastasis. Cancer Biol Ther. 2011;12:69–79.
Georges R, Bergmann F, Hamdi H, Zepp M, Eyol E, Hielscher T, et al. Sequential biphasic changes in claudin1 and claudin4 expression are correlated to colorectal cancer progression and liver metastasis. J Cell Mol Med. 2012;16:260–72.
Team RDC. R: a language and environment for statistical computing. Vienna, Austria R Foundation for Statistical Computing. 2011.
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6.
Ghadjar P, Coupland SE, Na IK, Noutsias M, Letsch A, Stroux A, et al. Chemokine receptor CCR6 expression level and liver metastases in colorectal cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2006;24:1910–6.
Ghadjar P, Rubie C, Aebersold DM, Keilholz U. The chemokine CCL20 and its receptor CCR6 in human malignancy with focus on colorectal cancer. Int J Cancer J Int Cancer. 2009;125:741–5.
Rubie C, Frick VO, Pfeil S, Wagner M, Kollmar O, Kopp B, et al. Correlation of IL-8 with induction, progression and metastatic potential of colorectal cancer. World J Gastroenterol WJG. 2007;13:4996–5002.
Rubie C, Frick VO, Wagner M, Schuld J, Graber S, Brittner B, et al. ELR+ CXC chemokine expression in benign and malignant colorectal conditions. BMC Cancer. 2008;8:178.
Rubie C, Frick VO, Ghadjar P, Wagner M, Justinger C, Faust SK, et al. Cxc receptor-4 mRNA silencing abrogates cxcl12-induced migration of colorectal cancer cells. J Transl Med. 2011;9:22.
Brand S, Olszak T, Beigel F, Diebold J, Otte JM, Eichhorst ST, et al. Cell differentiation dependent expressed CCR6 mediates ERK-1/2, SAPK/JNK, and Akt signaling resulting in proliferation and migration of colorectal cancer cells. J Cell Biochem. 2006;97:709–23.
Juremalm M, Nilsson G. Chemokine receptor expression by mast cells. Chem Immunol Allergy. 2005;87:130–44.
Schober A, Zernecke A. Chemokines in vascular remodeling. Thromb Haemost. 2007;97:730–7.
Williams IR. Chemokine receptors and leukocyte trafficking in the mucosal immune system. Immunol Res. 2004;29:283–92.
Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate. 2006;66:124–34.
Moller C, Stromberg T, Juremalm M, Nilsson K, Nilsson G. Expression and function of chemokine receptors in human multiple myeloma. Leukemia. 2003;17:203–10.
Wu X, Fan J, Wang X, Zhou J, Qiu S, Yu Y, et al. Downregulation of CCR1 inhibits human hepatocellular carcinoma cell invasion. Biochem Biophys Res Commun. 2007;355:866–71.
Shimada T, Matsumoto M, Tatsumi Y, Kanamaru A, Akira S. A novel lipopolysaccharide inducible C-C chemokine receptor related gene in murine macrophages. FEBS Lett. 1998;425:490–4.
Brouwer N, Zuurman MW, Wei T, Ransohoff RM, Boddeke HW, Biber K. Induction of glial L-CCR mRNA expression in spinal cord and brain in experimental autoimmune encephalomyelitis. Glia. 2004;46:84–94.
Zuurman MW, Heeroma J, Brouwer N, Boddeke HW, Biber K. LPS-induced expression of a novel chemokine receptor (L-CCR) in mouse glial cells in vitro and in vivo. Glia. 2003;41:327–36.
Zabel BA, Nakae S, Zuniga L, Kim JY, Ohyama T, Alt C, et al. Mast cell-expressed orphan receptor CCRL2 binds chemerin and is required for optimal induction of IgE-mediated passive cutaneous anaphylaxis. J Exp Med. 2008;205:2207–20.
Oostendorp J, Hylkema MN, Luinge M, Geerlings M, Meurs H, Timens W, et al. Localization and enhanced mRNA expression of the orphan chemokine receptor L-CCR in the lung in a murine model of ovalbumin-induced airway inflammation. J Histochem Cytochem Off J Histochem Soc. 2004;52:401–10.
Kitamura T, Kometani K, Hashida H, Matsunaga A, Miyoshi H, Hosogi H, et al. SMAD4-deficient intestinal tumors recruit CCR1+ myeloid cells that promote invasion. Nat Genet. 2007;39:467–75.
Hirai H, Fujishita T, Kurimoto K, Miyachi H, Kitano S, Inamoto S, et al. CCR1-mediated accumulation of myeloid cells in the liver microenvironment promoting mouse colon cancer metastasis. Clin Exp Metastasis. 2014;31:977–89.
Reufsteck C, Lifshitz-Shovali R, Zepp M, Bauerle T, Kubler D, Golomb G, et al. Silencing of skeletal metastasis-associated genes impairs migration of breast cancer cells and reduces osteolytic bone lesions. Clin Exp Metastasis. 2012;29:441–56.
Kovacheva M, Zepp M, Berger SM, Berger MR. Sustained conditional knockdown reveals intracellular bone sialoprotein as essential for breast cancer skeletal metastasis. Oncotarget. 2014;5:5510–22.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Israa G. Akram and Rania Georges contributed equally to this work.
Rights and permissions
About this article

Cite this article
Akram, I.G., Georges, R., Hielscher, T. et al. The chemokines CCR1 and CCRL2 have a role in colorectal cancer liver metastasis. Tumor Biol. 37, 2461–2471 (2016). https://doi.org/10.1007/s13277-015-4089-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4089-4