
Abstract
Japonicone A, which is a natural product isolated from the aerial part of Inula japonica Thunb., has a wide range of clinical applications, including anti-inflammation and anti-oxidation. This study investigated the effects of japonicone A on the growth of non-small cell lung cancer (NSCLC) cell lines. The results showed that japonicone A significantly inhibited the growth of NSCLC cell lines in a dose- and time-dependent manner. This product also blocked cell cycle progression at S phase and induced mitochondrial-related apoptosis by upregulating Bax, cleaved caspase-9, cleaved caspase-3, and cleaved poly(ADP-ribose) polymerase (PARP) protein levels and by downregulating Bcl-2, cyclin D1, CDC25A, and CDK2 protein levels. In vivo, japonicone A suppressed tumor growth via the same mechanism as that observed in vitro. In conclusion, our study is the first to report that japonicone A has an inhibitory effect on the growth of NSCLC cells, indicating that japonicone A administration is a potential therapeutic approach for future NSCLC treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lung cancer remains to be the leading cause of cancer-related deaths worldwide, accounting for 1.38 million annual deaths and representing 18.2 % of total deaths from cancer [1, 2]. Approximately 80 % of lung cancers are non-small cell lung cancer (NSCLC), most of which are subdivided into squamous cell carcinoma and adenocarcinoma [3]. Although surgery is the most effective curative treatment for early-stage NSCLC, up to 75 % of patients will present with advanced disease at diagnosis [4, 5]. Despite developments in diagnostic and therapeutic techniques, the prognosis of NSCLC patients has improved only minimally, with a 5-year survival rate of less than 15 % [6]. Therefore, identifying new therapeutic agents against NSCLC is critical for improving the health and survival of patients with NSCLC.
Traditional herbal medicines containing various biologically active natural compounds are believed to have therapeutic efficacy with minimal adverse effects [7]. Japonicone A is a natural product isolated from the aerial part of Inula japonica Thunb., which is a traditional medicine used to treat digestive disorders, bronchitis, inflammation, and bacterial and viral infections (including hepatitis) [8]. In recent studies, japonicone A was shown to directly bind to TNF-α, selectively inhibit its binding to tumor necrosis factor receptor 1 (TNFR1), effectively block TNFR1-mediated signaling in TNF-α-stimulated cells, and antagonize its proinflammatory activities in vivo without impairing host antiviral immunity [9]. Further studies have demonstrated that japonicone A displayed potent anticancer effects on Burkitt lymphoma cells in vitro and in vivo through targeting the NF-κB signaling cascade [7]. However, the effects of japonicone A and its mechanism of action on human NSCLC cells have never been elucidated. In the present study, we investigated the anti-neoplastic activity of japonicone A in the NSCLC cell lines H441 and H520 in vivo and in vitro and explored the possible molecular mechanisms underlying this action, providing experimental evidence for the potential application of japonicone A as a new natural anti-tumor medicine for NSCLC.
Methods
Reagents
Japonicone A, with purity greater than 97 %, was isolated from I. japonica Thunb. in the Natural Products Laboratory at the Second Military Medical University (Shanghai, People’s Republic of China). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies, Inc. All antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All cell culture supplies were obtained from Invitrogen Gibco Co.
Cell culture
The NSCLC cell lines H441 and H520 and a human bronchial epithelial cell (HBEC) line were purchased from the Shanghai Institutes for Biological Sciences in China. All the cells were maintained in a RPMI 1640 medium with 10 % (v/v) calf bovine serum and routinely cultivated in a humidified incubator at 37 °C and 5 % CO2.
Cell viability assay
Cell viability was determined with CCK-8 as instructed by the manufacturer. Briefly, cancer cells were seeded in 96-well plates and treated for either 48 h with japonicone A at serial concentrations or various times (0, 24, 48, or 72 h). After treatment, CCK-8 solution (10 μl) was added to each well, followed by 3 h of incubation. The absorbance was recorded at an optical density of 450 nm using a microplate reader (BioTek) to calculate the percentages of cell survival. Fifty percent inhibitive concentration (IC50) values were determined by plotting a linear regression curve.
Colony formation assay
H441 and H520 cell lines treated with different concentrations of japonicone A were counted and seeded in 12-well plates (in triplicate) at 100 cells per well. The medium was replaced with a fresh culture medium every 3 days. Colonies were counted only if they contained more than 50 cells; the number of colonies was counted from the 6th day after seeding. Then, the cells were stained using crystal violet. The rate of colony formation was calculated using the following equation: colony formation rate = (number of colonies / number of seeded cells) × 100 %.
Cell cycle and apoptosis analysis
Propidium iodide (PI) staining was used to analyze DNA content and cell cycle distribution. After being exposed to different concentrations of japonicone A for 24 h, the cells were harvested, fixed in 70 % ethanol, centrifuged (3000 rpm, 5 min), incubated with RNase (100 m g/ml) at 37 °C for 30 min, and stained with PI (50 mg/ml in phosphate-buffered saline (PBS)). The cellular DNA content and cell cycle distribution were analyzed by flow cytometry (BD FACSCalibur, USA).
Apoptosis analyses were conducted using an annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit (BioVision). Cells (5 × 105) were exposed to different concentrations of japonicone A for 24 h. Then, the cells were collected by centrifugation and resuspended in 500 μl of 1× binding buffer. Annexin V-FITC (5 μl) and PI (5 μl) were added to the cells. After being incubated at room temperature for 5 min in the dark, the cells were analyzed by fluorescence-activated cell sorting (FACS) by flow cytometry (BD FACSCalibur, USA). Cells that stained positive for early apoptosis markers (annexin V-FITC stained only) and for late apoptosis markers (annexin V-FITC and PI stained) were combined for analysis.
Mitochondrial membrane potential (ΔΨm) assay
After being treated with different concentrations of japonicone A for 48 h, the cells were collected and washed twice with cold PBS. Then, the cells were incubated with rhodamine 123 (Sigma-Aldrich) for 30 min in the dark. Subsequently, the cells were washed twice with cold PBS and analyzed by flow cytometry.
Detection of morphological apoptosis by Hoechst 33342 staining
NSCLC cell lines were treated for 48 h with different concentrations of japonicone A. Then, the cells were washed with PBS and fixed in methanol/acetic acid (3:1) for 15 min at room temperature. The fixed cells were washed with PBS and stained with 5 μg/ml Hoechst 33342 for 10 min. The morphological changes in the nuclei of cells, which were visible after Hoechst 33342 staining, were observed using a fluorescence microscope (Leica, Germany).
Western blot analysis
The cells were treated with different concentrations of japonicone A for 48 h, and then, the adherent and floating cells were harvested, washed twice with cold PBS, and lysed in RIPA buffer (Beyotime Institute of Biotechnology, Beijing, China) and protease inhibitor (Roche Applied Science, Indianapolis, IN, USA) at 4 °C for 5 min. After centrifugation at 14,000×g for 5 min, the protein concentration of the cell extracts was determined using the bicinchoninic acid (BCA) assay kit (Beyotime) according to the manufacturer’s instructions. Equal amounts of protein lysates (40 μg/lane) from each sample were separated by 10 % SDS-PAGE and then electrophoretically transferred to nitrocellulose membranes (Millipore, Bedford, MA, USA). Each membrane was blocked with 5 % skim milk and then incubated with the indicated primary antibodies against Bcl-2, Bax, cleaved caspase-3, cleaved caspase-9, cleaved poly ADP-ribose polymerase (PARP), CDC25A, cyclin D1, CDK2, and β-actin (1:1000) at 4 °C overnight. After washing with TBST buffer, the membrane was incubated with the secondary antibodies (HRP-conjugated goat anti-rabbit IgG, 1:5000; Abcam, Cambridge, UK) for 1 h at room temperature and the bands were visualized using a Gel Doc 2000 system (Bio-Rad, USA).
In vivo efficacy of japonicone A
BALB/c homozygous (nu/nu) nude mice (6–8 weeks old, 18–20 g body weight), bred in-house, were maintained in a specific pathogen-free environment. Exponentially growing H520 cells (2.5 × 106) were suspended in 100 μl PBS and subcutaneously injected into the left axilla of recipient mice. On day 5, tumor-bearing mice were randomly divided into three groups (one control group and two treatment groups), with six animals in each group. Japonicone A was administered to mice in the treatment groups by intraperitoneal (i.p.) injection every day at 25 and 50 mg/kg. Control animals received i.p. injections of Dulbecco's Modified Eagle Medium (DMEM). On day 27, all mice were euthanized and the tumors were dissected and weighed. Animal procedures were conducted in accordance with the institutional guidelines of Shanghai Jiao Tong University (Shanghai, China).
Immunohistochemistry
Immunohistochemical staining was performed using the streptavidin-peroxidase method. Briefly, the tissue sections were dehydrated with ethanol, washed three times with PBS (pH 7.4), and boiled for 8 min in a pressure cooker to retrieve the antigen. Then, endogenous peroxidase activities were inactivated in 3 % hydrogen peroxide for 10 min at room temperature. The sections were further blocked with 3 % normal goat serum for 10 min. After the serum was discarded, the sections were incubated with cleaved caspase-3 monoclonal mouse anti-human antibodies (1:200 dilution; Abcam, MA, USA) in a humid chamber at 4 °C overnight. The following day, the sections were further incubated with biotinylated IgG, followed by an avidin-biotin-peroxidase complex (Dingguo, Shanghai). Immunoreactions were visualized using diaminobenzidine tetrahydrochloride and counterstained with hematoxylin.
Statistical analysis
The results of each experiment are shown as the mean ± SD where applicable. Statistically significant differences in each assay were determined using Statistical Package for the Social Sciences (SPSS) version 18.0 software. The differences in each group were tested for significance using ANOVA. P < 0.05 was considered significant.
Results and discussion
Japonicone A inhibits cell proliferation and colony formation in H441 and H520 cells
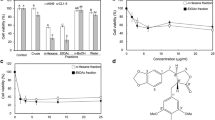
Deregulated cell proliferation is a hallmark of cancer [10]. To test the effects of japonicone A administration on NSCLC cell proliferation and independent growth, we investigated the proliferative activities and independent growth of H441 and H520 cell lines using CCK-8 and colony formation assays. The results demonstrated that japonicone A could significantly diminish the proliferative activities of both cancer cell lines in a dose- and time-dependent manner compared with the control group (Fig. 1b, c). The IC50 value of japonicone A for H441 and H520 cell viability was approximately 2 mM after 48 h. However, japonicone A did not show marked inhibition of HBECs, which demonstrated its lower cytotoxicity in these cells (Fig. 1b). In Fig. 1c, we found that the effects at 48 h were more obvious than those observed at 24 h and more stable than the groups at 72 h. Therefore, the groups at 48 h were chosen for further experiments to detect changes in molecular events. In addition, the colony formation rate of H441 and H520 cells in japonicone A-treated groups was also markedly lower than that of the control group (**P < 0.01, ***P < 0.001) (Fig. 1d–f).

Japonicone A inhibits cell proliferation and colony formation in H441 and H520 cells. a The chemical structure of japonicone A. b, c H441 and H520 cells were treated with various concentrations of japonicone A for 24, 48, and 72 h. Cell viability was assessed using the CCK-8 assay. d–f Japonicone A suppressed colony formation of H441 and H520 cells. Cells were treated with japonicone A and then cultured in a fresh medium for 14 days to form colonies. The values represent the mean ± SD of three independent experiments. **P < 0.01; ***P < 0.001
Japonicone A induces apoptosis in H441 and H520 cells
The induction of apoptotic activity is a good basis for anticancer therapies and a valuable guide to predict tumor response after anticancer treatment is administered [11]. Apoptotic cells with nuclear condensation and fragmentation can be visualized by Hoechst 33258 and DAPI staining. To determine the effects of japonicone A on NSCLC cell apoptosis, Hoechst 33258 staining and flow cytometric analysis were performed. After exposure to four concentrations of japonicone A (0, 1, 2, and 4 μM) for 48 h, NSCLC cell apoptosis was demonstrated by Hoechst 33258 staining, which revealed increases in cell membrane permeability and nuclear condensation (Fig. 2a). The numbers of apoptotic nuclei containing condensed chromatin increased significantly as the japonicone A concentration increased (Fig. 2b). Furthermore, the apoptosis index of NSCLC cells in the japonicone A-treated groups was markedly higher than that in the control group (*P < 0.05, **P < 0.01) (Fig. 2a–d).

Japonicone A induces apoptosis in NSCLC cells. a, b Changes in apoptotic nuclear morphology were observed by Hoechst 33342 staining and visualized by fluorescent microscopy. c, d H441 and H520 cells were analyzed by flow cytometry with annexin V-FITC/PI staining after japonicone A treatment. Annexin V vs. PI plots from the gated cells showed the populations corresponding to viable (annexin V−/PI−), necrotic (annexin V−/PI+), early (annexin V+/PI−), and late (annexin V+/PI+) apoptotic cells. The data represent the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01, compared with the control
As assessed by flow cytometry and as shown in Fig. 2c, d, japonicone A treatment reduced the number of surviving cells and increased the number of both early and late apoptotic cells in a dose-dependent manner. The early apoptotic population in the untreated cells was 2.5 ± 0.5 and 1.0 ± 0.51 % in H411 and H520. With the increasing of drug concentrations, it increased from 2.0 ± 0.3 to 15.0 ± 1.02 % in H441 and from 5.0 ± 0.31 to 15.1 ± 0.99 % in H520. These results from different apoptosis assays revealed significant features of apoptosis, which strongly suggested that the japonicone A-mediated inhibition of NSCLC cell growth closely correlated with the observed enhancement of apoptosis.
Japonicone A causes mitochondrial depolarization and activates the mitochondrial-related apoptotic pathway in H441 and H520 cells
The decline in mitochondrial membrane potential is a characteristic of apoptosis, which is reflected by changes in ΔΨm [12, 13]. In this study, ΔΨm in japonicone A-treated NSCLC cells was examined using rhodamine 123, which can be used to estimate integrity and changes in membrane potential. A dose-dependent decrease in the mitochondrial membrane potential was found after incubating the cells with japonicone A for 48 h (Fig. 3a–c). These findings suggest that japonicone A treatment could reduce the mitochondrial membrane potential and induce the mitochondrial dysfunction in NSCLC cells.

Japonicone A disrupts mitochondrial integrity in NSCLC cells. a, b Flow cytometric analysis of ΔΨm. H441 and H520 cells were treated with japonicone A, followed by rhodamine 123 staining. Cells with high ΔΨm are marked survival, and those with low ΔΨm are marked apoptosis. The percentages of cells with low ΔΨm (apoptosis) are shown. c Western blot analysis of apoptosis-related proteins in both cell lines. β-Actin was used as a loading control. The data represent the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01, compared with the control
Two major pathways are known to be involved with the initiation of apoptosis: the mitochondria-mediated intrinsic pathway and the death receptor-induced extrinsic pathway [14]. Our results showed that japonicone A treatment decreased the ΔΨm in all NSCLC cell lines, which indicates that japonicone A-induced apoptosis in NSCLC cells occurs via the mitochondria-related pathway. The disruption of the ΔΨm is recognized as an early stage of apoptosis, where the release of cytochrome c from mitochondria signals the initiation of apoptosis [15, 16]. The mitochondria-dependent pathway is regulated by Bcl-2 family proteins, which are involved in apoptosis involving the pro-apoptotic proteins Bax [17, 18]. In the present study, the results from Western blot analysis indicated that japonicone A promoted an increase in Bax/Bcl-2 levels (Fig. 3d), suggesting that the upregulation of Bax/Bcl-2 could be an important molecular mechanism by which japonicone A induced apoptosis in NSCLC cell lines.
The release of cytochrome c from mitochondria to the cytosol is an important step in the apoptotic pathway [19]. This release leads to the caspase-9-dependent activation of caspase-3 and of a cleaved form of PARP. In our experiments, cleaved caspase-3 activity was upregulated along with an increase in PARP cleavage (Fig. 3d) [20, 21]. These results indicate that japonicone A-induced apoptosis in NSCLC cells occurs via the mitochondria-caspase-dependent pathway.
Japonicone A induces G0/G1 arrest in H441 and H520 cells
Cell cycle arrest is closely linked to apoptosis [22, 23]. The deregulation of cell cycle progression is a hallmark of tumor growth [24, 25]. To determine whether japonicone A inhibited cell cycle progression, cell cycle distribution and related regulators were studied. For all NSCLC cells, japonicone A induced cell cycle arrest in the S phase in a concentration-dependent manner (30.25 ± 2.14, 40.19 ± 3.97, and 65.16 ± 4.28 % vs. 25.12 ± 3.23 % in the control group in H441 cells, P < 0.05; 29.80 ± 3.22, 40.53 ± 3.80, and 55.28 ± 3.61 % vs. 24.93 ± 3.11 % in the control group in H520 cells, P < 0.05; Fig. 4a–c). Notably, progression through the cell cycle depends on the activation of cyclin-dependent kinases (CDKs) and their regulatory subunits, the cyclins. In the present study, japonicone A treatment resulted in a time-dependent decrease in the protein expression of CDK2, CDC25A, and cyclin D1 (Fig. 4d, e). These results showed that S phase arrest also accounts for the antiproliferative effect of japonicone A in both cell lines possibly by disturbing the expression of cell cycle regulators.

Japonicone A induces cell cycle arrest at S phase and regulates the expression of cell cycle-related proteins in NSCLC cells. a, b H441 and H520 cells were treated with japonicone A (0, 1, 2, and 4 μmol/l) for 48 h. The cell cycle phases of the treated cells were evaluated by flow cytometry. The data are expressed as the mean ± SD (n = 3). c The expression levels of CDC25A, cyclin D1, and CDK2 were measured by Western blot analysis, and β-actin was used as a loading control. The results are representative of the three independent experiments
Japonicone A inhibits tumor xenograft growth
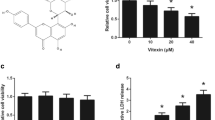
To further investigate the effect of japonicone A on tumor growth in vivo, we intraperitoneally injected DMEM or two different doses of japonicone A into nude mice with subcutaneous H520 tumor xenografts. Our results showed that the growth of tumors in the two treatment groups was significantly inhibited in a dose-dependent manner compared with tumors in the control group (Fig. 5a–c). Furthermore, Western blot and immunohistochemistry (IHC) showed a significant increase of active caspase-3 expression in tumors in the japonicone A-treated mice, which was consistent with the results in vitro (Fig. 5d).

Japonicone A represses tumor growth in a xenograft nude mice model by causing apoptotic cell death. Tumor xenografts were established by subcutaneous inoculation of H520 cells into the left flank of nude mice. a, b Then, the mice were administered 0.1 ml of vehicle (PBS) or japonicone A (25 and 50 mg/kg) intraperitoneally every day for up to 4 weeks. Tumor volumes were measured. c Tumors were excised from the animals and weighed. d Western blot and IHC analysis (×200 magnification) illustrated that active caspase-3 expression was observed in the japonicone A-treated tumors compared to the vehicle control groups. The data represent the mean ± SD of the three independent experiments. *P < 0.05; **P < 0.01, compared with the control
Conclusions
In summary, the current study presents the first evidence for the role of japonicone A in inducing cell cycle arrest and in promoting apoptosis in NSCLC cells and revealed that japonicone A exerts its functions by regulating the mitochondria-caspase-dependent pathway. From these anticancer properties, we expect that japonicone A may provide new hope for an effective therapeutic approach for NSCLC.
References
Kumar MS, Armenteros-Monterroso E, East P, Chakravorty P, Matthews N, Winslow MM, et al. HMGA2 functions as a competing endogenous RNA to promote lung cancer progression. Nature. 2014;505:212–7.
Al-Shahrabani F, Vallbohmer D, Angenendt S, Knoefel WT. Surgical strategies in the therapy of non-small cell lung cancer. World J of Clin Oncol. 2014;5:595–603.
Xiong F, Jiang M, Huang Z, Chen M, Chen K, Zhou J, et al. A novel herbal formula induces cell cycle arrest and apoptosis in association with suppressing the PI3K/AKT pathway in human lung cancer A549 cells. Integr Cancer Ther. 2014;13:152–60.
Smith SL, Palma D, Parhar T, Alexander CS, Wai ES. Inoperable early stage non-small cell lung cancer: comorbidity, patterns of care and survival. Lung Cancer. 2011;72:39–44.
Paz-Ares LG, Altug S, Vaury AT, Jaime JC, Russo F, Visseren-Grul C. Treatment rationale and study design for a phase III, double-blind, placebo-controlled study of maintenance pemetrexed plus best supportive care versus best supportive care immediately following induction treatment with pemetrexed plus cisplatin for advanced nonsquamous non-small cell lung cancer. BMC Cancer. 2010;10:85.
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90.
Li X, Yang X, Liu Y, Gong N, Yao W, Chen P, et al. Japonicone A suppresses growth of Burkitt lymphoma cells through its effect on NF-kappaB. Clin Cancer Res. 2013;19:2917–28.
Qin JJ, Jin HZ, Fu JJ, Hu XJ, Wang Y, Yan SK, et al. Japonicones A-D, bioactive dimeric sesquiterpenes from Inula japonica Thunb. Bioorg Med Chem Lett. 2009;19:710–3.
Hu Z, Qin J, Zhang H, Wang D, Hua Y, Ding J, et al. Japonicone A antagonizes the activity of TNF-alpha by directly targeting this cytokine and selectively disrupting its interaction with TNF receptor-1. Biochem Pharmacol. 2012;84:1482–91.
Luscan A, Shackleford G, Masliah-Planchon J, Laurendeau I, Ortonne N, Varin J, et al. The activation of the WNT signaling pathway is a hallmark in neurofibromatosis type 1 tumorigenesis. Clin Cancer Res. 2014;20:358–71.
Li HH, Su JH, Chiu CC, Lin JJ, Yang ZY, Hwang WI, et al. Proteomic investigation of the sinulariolide-treated melanoma cells A375: effects on the cell apoptosis through mitochondrial-related pathway and activation of caspase cascade. Mar Drugs. 2013;11:2625–42.
Chiou HL, Hsieh YS, Hsieh MR, Chen TY. HCV E2 may induce apoptosis of Huh-7 cells via a mitochondrial-related caspase pathway. Biochem Biophys Res Commun. 2006;345:453–8.
Xiang SS, Wang XA, Li HF, Shu YJ, Bao RF, Zhang F, et al. Schisandrin B induces apoptosis and cell cycle arrest of gallbladder cancer cells. Molecules. 2014;19:13235–50.
Liu JY, Liu Z, Wang DM, Li MM, Wang SX, Wang R, et al. Induction of apoptosis in K562 cells by dicyclohexylammonium salt of hyperforin through a mitochondrial-related pathway. Chem Biol Interact. 2011;190:91–101.
Wang XA, Xiang SS, Li HF, Wu XS, Li ML, Shu YJ, et al. Cordycepin induces S phase arrest and apoptosis in human gallbladder cancer cells. Molecules. 2014;19:11350–65.
Xue C, Pasolli HA, Piscopo I, Gros DJ, Liu C, Chen Y, et al. Mitochondrial structure alteration in human prostate cancer cells upon initial interaction with a chemopreventive agent phenethyl isothiocyanate. Cancer Cell Int. 2014;14:30.
Choi BH, Kim W, Wang QC, Kim DC, Tan SN, Yong JW, et al. Kinetin riboside preferentially induces apoptosis by modulating Bcl-2 family proteins and caspase-3 in cancer cells. Cancer Lett. 2008;261:37–45.
Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–80.
Vladimirov YA, Proskurnina EV, Alekseev AV. Molecular mechanisms of apoptosis. Structure of cytochrome c-cardiolipin complex. Biochemistry. 2013;78:1086–97.
Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43.
Tsai JR, Chong IW, Chen YH, Hwang JJ, Yin WH, Chen HL, et al. Magnolol induces apoptosis via caspase-independent pathways in non-small cell lung cancer cells. Arch Pharm Res. 2014;37:548–57.
Xu XY, Xia P, Yu M, Nie XC, Yang X, Xing YN, et al. The roles of REIC gene and its encoding product in gastric carcinoma. Cell Cycle. 2012;11:1414–31.
Li X, Cheung KF, Ma X, Tian L, Zhao J, Go MY, et al. Epigenetic inactivation of paired box gene 5, a novel tumor suppressor gene, through direct upregulation of p53 is associated with prognosis in gastric cancer patients. Oncogene. 2012;31:3419–30.
Li M, Lu J, Zhang F, Li H, Zhang B, Wu X, et al. Yes-associated protein 1 (YAP1) promotes human gallbladder tumor growth via activation of the AXL/MAPK pathway. Cancer Lett. 2014;355:201–9.
Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–7.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81402403), Shanghai Rising-Star Program (No. 15QA1403100), and the Natural Science Foundation of Shanxi Province (Nos. 20110313013-3 and 2014021037-3).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding authors
Additional information
Maolan Li and Jianqiang Li jointly directed this work.
Yan Du and Jiannan Gong contributed equally to this work.
Rights and permissions
About this article

Cite this article
Du, Y., Gong, J., Tian, X. et al. Japonicone A inhibits the growth of non-small cell lung cancer cells via mitochondria-mediated pathways. Tumor Biol. 36, 7473–7482 (2015). https://doi.org/10.1007/s13277-015-3439-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3439-6