
Abstract
Majority of women with estrogen receptor (ER)-positive breast cancers initially respond to hormone therapies such as tamoxifen (TAM; antagonist of estrogen). However, many tumors eventually become resistant to TAM. Therefore, understanding the various cellular components involved in causing resistance to TAM is of paramount importance in designing novel entities for efficacious hormone therapy. Previously, we found that suppression of BIK gene expression induced TAM resistance in MCF-7 breast cancer cells. In order to understand the response of these cells to TAM and its association with resistance, a microarray analysis of gene expression was performed in the BIK-suppressed MCF-7 cells and compared it to the TAM-only-treated cells (controls). Several genes participating in various cellular pathways were identified. Molecules identified in the drug resistance pathway were 14-3-3z or YWHAZ, WEE1, PRKACA, NADK, and HSP90AA 1. Further, genes involved in cell cycle control, apoptosis, and cell proliferation were also found differentially expressed in these cells. Transcriptional and translational analysis of key molecules such as STAT2, AKT 3, and 14-3-3z revealed similar changes at the messenger RNA (mRNA) as well as at the protein level. Importantly, there was no cytotoxic effect of TAM on BIK-suppressed MCF-7 cells. Further, these cells were not arrested at the G0-G1 phase of the cell cycle although 30 % of BIK-suppressed cells were arrested at the G2 phase of the cycle on TAM treatment. Furthermore, we found a relevant interaction between 14-3-3z and WEE1, suggesting that the cytotoxic effect of TAM was prevented in BIK-suppressed cells because this interaction leads to transitory arrest in the G2 phase leading to the repair of damaged DNA and allowing the cells to proliferate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Antiestrogens such as tamoxifen (TAM) are widely used in the clinic to treat estrogen receptor (ER)-positive breast tumors. About 70 % of all patients with breast cancer overexpress nuclear estrogen receptor alpha (ER-α) [1, 2], rendering it an excellent candidate for endocrine therapy. TAM is an ER antagonist that competitively inhibits the interaction of estrogen with ER, thus repressing ER activity and is commonly administered as a first-line adjuvant treatment for an ER-α-positive breast tumor [3, 4].
To develop efficacious treatment for TAM-resistant breast cancers, it is important to first understand the complex signal transduction pathways [5]. Studies have already revealed several mechanisms of TAM resistance, including increased TAM metabolism [6], loss or alteration of ER-α and ER-β expression [7–9], estrogen gap needed between hypersensitivity, and [8] altered expression of co-regulators [9], altered signaling pathway mediated by tyrosine kinases, inhibition of apoptosis regulated by the Bcl-2 protein family, cell cycle, autophagy, and microRNA (miRNA) [10, 11]; however, global analysis of changes in gene expression and signaling pathways associated with the development of TAM resistance are not completely understood. Previously, we observed that BIK is associated with TAM resistance because suppression of BIK using small interfering RNA interference (siRNAi) induced TAM resistance in MCF-7 cells, while knockdown of endogenous BIK decreased TAM-induced apoptosis in MCF-7 cells, due to inhibition of the intrinsic pathway and transition in mitochondrial permeability.
These data would be useful for future studies in understanding the mechanism of regulation of TAM resistance in breast cancers [11]. However, it is necessary to determine whether other mechanisms of resistance exit in which BIK might be involved especially in coordination [12, 13].
To further characterize the mechanisms of TAM resistance induced by silenced to BIK expression, we conducted a microarray analysis in BIK-suppressed MCF-7 cells treated with TAM and found changes in the expression of certain genes that could promote resistance to TAM. These genes are associated with signaling pathways involved in the processes of apoptosis, cell cycle control, and proliferation.
Methods
Cell cultures
MCF-7 human breast cancer cells (American Type Culture Collection, ATCC, USA) were maintained in Dulbecco’s modified Eagle’s medium F:12 (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10 % fetal bovine serum (FBS) containing penicillin (100 U/mL) and streptomycin (100 μg/mL) (Life Technologies, Inc. BLR, Grand Island, NY, USA). The cells were grown in 75-cm2 tissue culture flasks in 5 % CO2 at 37 °C. Before each experiment, cells were seeded in 3.5-cm-diameter tissue culture plates.
Suppression of BIK expression with siRNAi
Cells were plated in antibiotic-free DMEM-F12 at a density of 2.5 × 105 cells; when 50 % confluence was reached, the cells were transfected with Oligofectamine™ reagent (Invitrogen) and 100 nmol/L BIK RNAi (oligoduplex 5′-AAGACCCCUCUCCAGAGACAU-3′, 5′-AAAUGUCUCUGGAGAGGGGUC-3′) (Labs and Integrated DNA Technologies [IDT]) or control scrambled sequence (Silencer Negative Control #3 RNA (siRNA); (Ambion)), composed of a 19-bp scrambled sequence without significant homology to any known gene sequences from mouse, rat, or human. Briefly, 10-μL Oligofectamine™ was diluted 7.5-fold in reduced serum medium (Opti-MEM®) and incubated at room temperature for 10 min. In parallel, a separate tube containing 5 μL of 50 μmol/L siRNA was diluted in 425 μL of Opti-MEM®. Diluted Oligofectamine™ (75 μL) was added to the diluted siRNA and the complex was incubated for 20 min at room temperature. Cells were washed with 2 mL of Opti-MEM™. siRNA + Oligofectamine™ complex (500 μL) was gently added to the dish. The final concentration of siRNA was 100 mol/L. After 6 h, 1.25 mL of 3× serum-free medium was added to the dish without removing the transfection mix. The medium was exchanged for serum-containing medium after 6 h, and the cells were further cultured for 48 h. The experiments were repeated 2–3 times.
Hybridization and analysis of microarray data
Ten micrograms of total RNA was used for complementary DNA (cDNA) synthesis and labeling with Super Script II kit Invitrogen, using in array dUTP-Cy3 incorporation for MCF-7 TAM (control) and dUTP-Cy5 MCF-7 siRNA BIK with TAM; incorporation efficiency was analyzed by measuring absorbance at 555 nm for Cy3 and 655 nm for Cy5. Fluorophore-labeled cDNA were hybridized to the oligonucleotides collection 50-mer Human 10K from MWG Biotech Oligo Bio Sets (Germany). Images of the microarrays were acquired and quantified in the Scan Array 4000 using the Quant Array software from Packard BioChips (USA). Analysis of the images and their data was performed using Array-Pro Analyzer software from Media Cybernetics (USA).
Microarray data analysis was performed using the free software program GenArise that was developed in the Computing Unit of the Cellular Physiology Institute of UNAM (http://www.ifc.unam.mx/genarise/). GenArise conducted the following transformations: background correction, normalization, intensity filter, replicate analysis, and selection of differentially expressed genes. The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE60802 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE60802).
GenArise was used to identify the genes that were differentially expressed by calculating an intensity-dependent Z score. Genes with a Z score ≥2 were considered with altered expression (Table 1 Z score).
One hundred forty-nine genes were selected (82 down and 67 upregulated) as central genes in our study considering genes that show the biggest number of interactions in HIPPIE database (7 as minimal interaction number) and its participation in cancer. Hierarchical clustering of the data was performed using Cluster program with the Spearman rank correlation analysis and visualized using Java TreeView.
To determine gene interactions and molecular pathways potentially altered in MCF-7 cells transfected with siRNA to BIK treated with TAM, we used KEGG database (Kyoto Encyclopedia of Genes and Genomes) (http://www.genome.jp/kegg/pathway.html). The cutoff values for inclusion in these analyses were Z score of 2.0 and seven interactions as minimal (HIPPIE database).
Cell cycle analysis by flow cytometry (FACS)
Cells were harvested directly from culture plates. After centrifugation at 1400 rpm, at 4 °C for 5 min, the pellet was washed with phosphate-buffered saline/bovine serum albumin (PBS/BSA) 1 % and resuspended in 500 mL PBS. Cells were fixed by adding 1.5 mL 100 % cold ethanol and left for at least 2 h at −20 °C. Then, cells were washed with 4 mL of 1 % PBS/BSA. After centrifugation at 1400 rpm and 4 °C by 5 min, the pellet was resuspended in buffer: 500 μL PBS, RNase A 1 mg/mL, propidium iodide 10 μg/mL, and incubated for 30 min at 37 °C in the dark. Cell cycle profile was analyzed with FACS Calibur™ flow cytometry (Becton-Dickinson) using CellQuest™ software (Becton-Dickinson).
Cytotoxic effect of TAM
Cells (3 × 103 per well) were seeded in minimal essential medium (MEM, 180 μL) onto 96-well plates. After 4 h, hydroxytamoxifen (Sigma-Aldrich) dissolved in PBS solution (20 μL) was added at final concentrations of 60 μM and incubated at 37 °C with 5 % CO2. In previous experiments, it was determined that the half maximal inhibitory concentration (IC50) for TAM was 6.0 μM. After culturing for 24 and 48 h, MTT solution (2 mg/mL, 20 μL) was added to each well and incubated for 4 h to induce cellular conversion of the tetrazolium salt into a formazan product. The supernatant was then removed and dimethyl sulfoxide (DMSO) (200 μL) was added to dissolve the formazan, which was detected by spectrophotometry at 570 nm, and this provided a relative estimate of the cell population.
Western blot analysis
Batches of MCF-7 cells (non-treated, scrambled, siRNA BIK, and siRNA BIK with TAM) were cultured overnight at room temperature in 3.5-cm-diameter tissue culture plates at a density of 1 × 105 cells/plate. Cells were transferred into 100 μL of lysis buffer (RIPA-Tris buffer (mM: EGTA 2; NaCl 316; Na2MoO4 20; NaF 50; Tris-HCl 20; Na3VO4 100; PMSF 100 and EDTA 100; 0.1 % of leupeptin and aprotinin, SDS 0.2 %, and Triton-X100 2 %) and maintained under constant shaking for 2 h at 4 °C. Subsequently, the sample was centrifuged for 5 min at 20,800 rpm and the supernatant (30 μg of protein) was denatured in Laemmli sample buffer [14], resolved through 12 % sodium dodecyl sulfate (SDS) polyacrylamide gels, and electroblotted onto polyvinylidene difluoride (PVDF) membranes. Blots were stained with Ponceau S to confirm that protein loading was identical in all lanes. Membranes were soaked in PBS to remove the Ponceau S and incubated for 90 min in Tris-buffered saline (TBS) containing 5 % dried skimmed milk and 0.1 % Tween 20 to block non-specific protein binding sites. Subsequently, the membranes were incubated for 14 h at 4 °C with the primary antibody 1:1000: STAT2; AKT3 from Santa Cruz Biotechnology (Santa Cruz, CA, USA). BIK 1:100 and 14-3-3z 1:500 from Abcam were diluted in TBS-Tween 20 0.1 % including 5 % dried skimmed milk. Then, these were washed and incubated with peroxidase-conjugated secondary antibodies 1:10,000. Protein was detected using an ECL Western blot detection kit (Millipore). The blots were subjected to densitometry analysis and data were analyzed using GraphPad Prism 5 software (GraphPad Software, San Diego, CA, USA). Western blots were repeated three times.
14-3-3z interactome
Protein data mining was performed with the HIPPIE (http://cbdm.mdc-berlin.de/tools/hippie/information.php) database, which integrates information from the HPRD, BioGRID, IntAct, MINT, Rual05, Lim06, Bell09, Stelzl05, DIP, BIND, Colland04, Lehner04, Albers05, MIPS, Venkatesan09, Kaltenbach07, and Nakayama02 databases [15]. Analysis was achieved with the open source bioinformatics platform Cytoscape [16] and BisoGenet Plugin, utilizing the proteins identified as bait nodes and adding edges as follows: Organism > Homo sapiens, protein identifiers only; Data Settings > protein-protein interactions, all data sources and all experimental methods, and method > by adding edges connecting input nodes and as Output > Proteins.
Data analysis
Data were presented as mean ± standard deviation (SD) (n = 3). Statistical comparisons were performed by one-way analysis of variance (ANOVA) with Tukey test. Significant differences were indicated as p < 0.05 or 0.01.
Results
BIK suppression
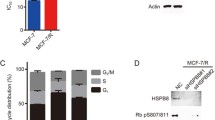
In order to evaluate whether BIK expression was blocked using siRNA, we compared the levels of messenger RNA (mRNA) and protein in the various MCF-7 cell groups: non-treated, scrambled sequence, siRNAiBIK, and TAM and siRNAiBIK/TAM. Transient transfection of BIK siRNA reduced BIK mRNA expression levels by about 55 ± 0.106 % in MCF-7 cells compared with the controls (non-treated and scrambled), while in the TAM group, BIK mRNA levels increased 2-fold with respect to control groups. In the siRNAiBIK group, TAM induced a slight increase in BIK, reaching values similar to those of the non-treated group, that is, 2.4-fold lower than those of the TAM-treated group. These results were confirmed by Western blot analysis (Fig. 1a, b).

Changes monitored at the transcriptional and at the translational levels in BIK-suppressed MCF-7 cells. a Silencing of the BIK gene in MCF-7 cells was achieved by transiently transfecting small interfering RNA and scrambled sequence was use as control. The cells were cultured in the presence and absence of 6 μM TAM for 24 h, followed by the evaluation of mRNA levels. b Western blot analysis of the same samples was conducted using β-actin as control. One representative of at least three independent experiments with similar results is shown. The groups were analyzed by one-way analysis of variance (ANOVA) with Tukey test. *p < 0.05 and ***p < 0.001 vs. non-treated
Transcriptional profiles induced by TAM in siRNAiBIK
In order to have a comprehensive analysis of changes in transcripts, we performed hierarchical clustering analysis of differentially expressed genes when siRNAiBIK-treated cells were exposed to TAM 6 μM, in relation with TAM-treated MCF-7 cells (Fig. 2 a), with the expectation that some of these genes can explain why TAM does not exert a cytotoxic effect on BIK-suppressed MCF-7 cells (TAM resistance).

Transcriptional profiles induced by TAM in siRNAiBIK cells. a Heat map of mRNA statistically significant between MCF-7 cells treated with TAM at a concentration of 6.0 μM. MCF-7 cells transfected with small interfering RNA interference (siRNAi) BIK exposed to TAM 6.0 μM. b Validation at protein level of the expression of AKT 3, STAT2, and 14-3-3z in BIK-suppressed MCF-7 cells. Cell lysates were prepared and subjected to Western blot analyses for STAT2, AKT3, and 14-3-3z. One representative of at least three independent experiments with similar results is shown. c, d, e Band intensities of STAT2, AKT3, and 14-3-3z were quantified by densitometric analysis. The groups were analyzed by one-way analysis of variance (ANOVA) with Tukey test. *p < 0.05; **p < 0.01 vs. non-treated sample representative figures of three independent experiments carried out in triplicate are shown
Of a total of 1755 genes upregulated and 943 genes downregulated after TAM treatment of BIK-transfected cells, 149 genes were selected (82 down- and 67 upregulated) as central genes in our study, considering genes that exhibited the largest number of interactions in the HIPPIE database (seven as minimal interaction number) and their participation in cancer (Tables 1 and 2). To identify genes with significant changes, we selected genes with a 2-fold differential expression. Some of the TAM-induced overexpressed genes in BIK-suppressed cells have been associated with resistance to chemotherapeutic agents, including X-linked inhibitor of apoptosis (XIAP) [17]; WEE1 G2 checkpoint kinase (WEE1) [18]; heat shock protein 90 kDa alpha (cytosolic) class A member 1 (HSP90AA1) [19]; protein kinase, cAMP dependent, catalytic alpha (PRKACA) [20]; Cbl proto-oncogene B, E3 ubiquitin protein ligase (CBLB) [21]; breast cancer 2, early onset (BRCA2): RAB GTPase activating protein 1-like (RABGAP1L); Unc-51-like autophagy activating kinase 2 (ULK2) [22]; checkpoint with forkhead and ring finger domains, E3 ubiquitin protein ligase (CHFR) [23]; RPTOR-independent companion of mammalian target of rapamycin (mTOR), complex 2 (RICTOR) [24]; and tyrosine 3 monooxygenase/tryptophan 5-monooxygenase activation protein, zeta polypeptide (YWHAZ) [25, 26], also known as 14-3-3z. Another group of overexpressed genes in this treatment has been associated with increased cancer cell motility and invasiveness and includes breast carcinoma amplified sequence 2 (BCAS2) [27]; syndecan-2 (SDC2) [28]; plasminogen (PLG) [29]; and Rac/Cdc42 guanine nucleotide exchange factor (GEF) 6 (ARHGEF6) (Table 1). However, other genes associated with breast cancer invasiveness were downregulated, including 3-phosphoinositide-dependent protein kinase-1 (PDPK1) [30] and v-Akt murine thymoma viral oncogene homolog 3 (AKT3) (Table 2).
Analysis of signaling pathways involved in the response to TAM in BIK-suppressed MCF-7 cells
The main aim of our study was to translate the list of differentially expressed genes into a better understanding of the underlying biological phenomena. To this end, we selected genes that exhibited a change of at least 2.0-fold. In the analysis carried out with KEGG, we found some differentially expressed genes that were associated with cell cycle regulation, mitogen-activated protein kinases (MAPK), insulin, Janus activated kinase signal transducers and activators of transcription (JAK-STAT), adherent junctions, Erb2, WNT, mTOR, vascular endothelial growth factor (VEGF), phosphatidylinositol, apoptosis, and autophagy signaling pathways (Table 3).
Derived from the analysis based on the KEGG database, we selected central genes, such as 14-3-3z, AKT3, and STAT2, because of their participation in several regulatory pathways, such as MAPK, JAK-STAT, and insulin signaling pathways, or due to their role in cell cycle control (Table 3). Importantly, we observed that the YWHAZ [26] gene (also known as 14-3-3z) was one of the differentially expressed genes that was most significantly altered.
Differential levels of some regulatory proteins in TAM-treated BIK-suppressed MCF-7 cells
To complement the microarray data, we evaluated STAT2, 14-3-3z, and AKT3 at the protein level because all of these have been associated with cancer progression [31–34]. Western blot analysis was performed comparing the following three conditions: control cells, control treated with TAM 6.0 μM, and siRNAiBIK/TAM 6.0 μM. Enhanced levels of STAT2 and 14-3-3z were found in MCF-7 cells transfected with BIK exposed to TAM in comparison with control groups and the TAM group (Fig. 2 e).
On the other hand, in siRNAiBIK/TAM cells, 0.68-fold decreased expression of AKT3 was found on comparison with the control group and the TAM group. These data validate that differential changes in messenger RNA (mRNA) as well as protein levels of STAT2, 14-3-3z, and AKT3 followed a similar trend in response to TAM (Fig. 2b–e).
Suppression of BIK induced G2 arrest in response to TAM
In order to correlate how changes at the expression level and in regulatory pathways affect the cellular processes, cell cycle was evaluated by flow cytometry with propidium-iodide stain. About 52 ± 2.98 % TAM-treated MCF-7 cells remained in the G0 phase, and the number of cells in the S phase was reduced in relation with those of the control group not treated with TAM, indicating that TAM induced G0 arrest and growth inhibition in MCF-7 cells, while in BIK-transfected cells exposed to tamoxifen, we found an increased proportion of cells in G2-M phase (38 ± 4.01 %), whereas the percentage of cells in G2-M was similar in the control group and in the group of MCF-7 treated with TAM. Additionally, the proportion of cells in G0 in BIK-suppressed cells treated with TAM did not change significantly in relation to the control group (Fig. 3a). These results suggest that suppression of BIK plays a role in regulating the cell cycle in response to TAM; in this case, cells are not arrested in G0, which is probably related with the prevention of apoptosis; however, cells are arrested in G2, which can be associated with the function of 14-3-3z as a checkpoint kinase regulator. Next, we evaluated by MTT assay whether changes observed in the cell cycle were reflected in differences in cell population growth (Fig. 3b). In TAM-exposed control cells, cell number decreased by about 50 % with respect to control cells at 24, 48, and 72 h of treatment. Further, a significant reduction in cell viability was observed, while the siRNAiBIK/TAM group exhibited cell population growth in the presence of TAM, although at a lower proportion than the control group, suggesting that these cells are resistant to the cytotoxic effect of TAM (Fig. 3b).

Tamoxifen (TAM)-induced arrest in G2 and increased cell population in BIK-suppressed MCF-7 cells. a Cell cycle distribution of control, small interfering RNA interference (siRNAi) BIK, and siRNAiBIK/TAM cells was analyzed by FACS after propidium-iodide staining, and data were represented graphically. b Total cell population obtained by MTT assay after 24-h exposure to TAM. The groups were analyzed by one-way analysis of variance (ANOVA) followed by Tukey test.*p < 0.05; **p < 0.01; ***p < 0.001 vs. non-treated. Representative figures of three independent experiments carried out in triplicate are shown
Potential role of 14-3-3z in regulating the cell cycle in BIK-transfected cells on treatment with tamoxifen
To broaden our perspective of other molecules that could regulate the cell cycle, we performed an interactome analysis employing the HIPPIE program. We found that 14-3-3z interacts with 690 proteins; of these proteins, 55 interacting proteins are involved in cell cycle regulation (Fig. 4). In BIK-transfected cells exposed to TAM, we detected significant interaction of 14-3-3z with the checkpoint kinase WEE1, known to be involved in the control of the G2 phase of the cell cycle. Further, its mRNA levels also increased on TAM treatment of BIK-suppressed MCF-7 cells (Table 1). The signaling pathways participating in causing resistance to tamoxifen in BIK a suppressed MCF-7 cell are depicted in Fig. 5.

14-3-3z interactome. According to the Human Integrated Protein-Protein Interaction (HIPPIE) database, the interactome revealed that 14-3-3z interacts with 690 proteins. Of these proteins, 55 interacting proteins are involved in cell cycle regulation. WEE1 had high expression in the analysis carried out, which suggests that it protects breast cancer cells against DNA damage and cell death when BIK is suppressed in MCF-7 cells treated with TAM

Signaling pathways involved in small interfering RNA interference (siRNAi)/BIK cells exposed to tamoxifen (TAM). Red boxes: upregulated genes; green boxes: downregulated genes; and blue boxes: biological mechanism. (┴) inhibitory signals; (↓) activating signals. Signaling pathways that participate in siRNAiBIK cells exposed to TAM could be regulated by key proteins, such as 14-3-3z, which is known to be involved in various physiological processes such as intracellular signaling (e.g., RAF, MEKK, PI-3 kinase, IRS-1), cell cycling (e.g., Cdc25, WEE1), and apoptosis, which could promote cell survival by sequestering and inactivating several proapoptotic proteins, including BAD and FOXO3a, after their phosphorylation by survival-inducing kinases such as AKT
Discussion
Breast cancer is the most frequently detected neoplasm in women worldwide and is heterogeneous in nature when analyzed at the clinical, histological, as well as at the molecular levels. TAM, an antagonist of estrogen, has been used in the clinic to treat breast cancers; however, resistance to TAM has been the bottleneck in achieving successful outcomes. Although some key molecules and mechanisms related to TAM resistance have been identified, other molecules are emerging for understanding the mechanisms of TAM resistance. BIK has been associated with tumor reversion in several cell lines, and it has been proposed as a target for inducing apoptosis in cancer, including breast cancer. In transformed mice fibroblasts, the capacity of v-Src to prevent apoptosis is based on inhibition of the mitochondrial pathway of apoptosis through the increase of the degradation rate of BIK. This effect is dependent on the activation of the RAS-RAF-MEK1/2-ERK1/2 pathway. In this case, phosphorylation of the Thr124 of BIK leads to ubiquitination of the Lys33 of BIK and its degradation by the proteasome, which in turn leads to tumor progression [12, 35].
Previously, we found that BIK gene suppression in MCF-7 breast cancer cells induced TAM resistance because these cells escape from the intrinsic apoptosis pathway induced by TAM [11]. To understand the response of these cells to TAM, and why these cells escape from the toxic effects of TAM, a microarray analysis of gene expression during TAM treatment of MCF-7 suppressed for BIK was conducted and compared with TAM-treated MCF-7 cells. It was found that TAM induced changes in the mRNA levels of several genes, including enhanced expression of STAT2 and 14-3-3z and reduced expression of AKT3 in BIK-suppressed cells, but not in non-suppressed cells. Levels of the proteins encoded by these genes were modified in a similar way by TAM, and global analysis of differential gene expression in TAM-treated BIK-suppressed cells indicated an important role for MAPK, insulin, JAK-STAT, 14-3-3z, and WEE1 leading to the prevention of the cytotoxic effect of TAM.
Further, these pathways were grouped considering a higher number of genes involved in them and their p value. The following describe each signaling pathway.
Apart from various signaling pathways, the MAPK signaling pathway has been reported to be mediating response to TAM [36]. Our analysis of KEGG signaling pathways in siRNAiBIK cells exposed to TAM demonstrate some patterns of gene expression indicating the involvement of this pathway, including high expression of KRAS and CDc42/Rac. Several oncogenic mutations have been described in the KRAS gene, which result in its constitutive activation and in autonomous, non-regulated proliferation of the transformed cells, as well as their resistance to apoptosis [37]. High expression of the RAS gene and activation of the RAS/RAF-1/MAPK pathway could lead to overexpression of HER2 [ErbB2/epidermal growth factor receptor (EGFR) 2/Neu], which is associated with poor prognosis, especially in TAM-treated patients. Moreover, 14-3-3z proteins form a complex comprising many signaling molecules, including RAF-1 kinase. In the in vitro studies, RAF-1 must be complexed with 14-3-3z for efficient recruitment and its activation by oncogenic Ras [38]. On the other hand, Rac/CDc42 guanine nucleotide exchange factor or GEF6 activates CDc42/Rac that, together with P120GAP, appears to regulate the cytoskeletal dynamics of resistant cells, leading to a more motile and aggressive phenotype.
Another important molecule in regulating response to TAM is AKT3, which participates in the insulin signaling pathway. Although activation of the mTOR-AKT pathway has been associated with TAM resistance in MCF-7 cells, it is surprising that recently, some hormone resistance sublines have been derived from MCF-7, which have reduced mTOR signaling and reduced phosphorylation/activation of AKT [39], indicating that under some conditions, such as BIK suppression, the reduced AKT activity or expression could be associated with TAM resistance.
The protein AKT3 has been related with cancer progression, and high expression of this protein has been found in ER-negative cells and associated with a more aggressive phenotype [40]. It has been suggested that inhibition of AKT causes autophagy in human endothelial cells, and this process is increased in AKT3-deficient mice, due to the increase in phagosome formation in an AKT1/mTOR-independent pathway [41, 42] It has been considered that autophagy (also referred to as macroautophagy) is critical for the development of antiestrogen resistance. This pro-survival role for autophagy in antiestrogen-treated breast cancer cells is consistent with one function of the autophagosomes, which is to recycle metabolites from degraded cellular constituents to support a basal level of cell maintenance under starvation and oxidative stress (OS), thus rendering it essential for cellular viability [42]. Our observation of reduced expression of AKT3 in siRNAiBIK/TAM could be associated with increased autophagy.
Analysis of expression of molecules in the JAK/STAT signaling pathway revealed the overexpression of STAT2 in BIK-suppressed MCF-7 cells when treated with TAM. The JAK/STAT pathway is known to mediate the action of proinflammatory cytokines and growth factors, which favors cancer progression [31]; particularly STAT2 has been recently associated with the growth of MCF-7 cell population [52], although its role in breast cancer progression remains to be well studied. Our data suggest that TAM could promote MCF-7 cell survival through the activation of STAT2 when the gene BIK is suppressed [52]. Also STAT2 can promote a proinflammatory environment as reported of its action in skin and colorectal cancers [31]. In these cancers, it has been proposed that the overexpression of STAT2 induces the production of proinflammatory cytokines like IL6, creating an environment that promote the activation of proteins associated with cancer development, including the STAT3 oncogenic pathway [31].
Pathways leading to the abrogation of apoptosis and cell cycle arrest also play a central role in regulating tumorigenesis and tumor growth in different types of cancer including breast cancer. In siRNAiBIK cells exposed to TAM, we found some overexpressed genes that could be associated in the prevention of the toxic effect of TAM, since they have been associated with cell cycle regulation and resistance to chemotherapy, such as 14-3-3z and WEE1 [18, 43–45] (Table 3).
14-3-3 proteins modified the activity and location of several proteins by its binding when they are phosphorylated on serine-threonine residues, participating in multiple cell functions. Low expression of 14-3-3z inhibits proliferation and sensitizes cells to stimuli that induce apoptosis; therefore, increased expression of 14-3-3z could result in TAM resistance through its action in pathways regulating cell cycle, proliferation, and cancer progression. Our observation of increased expression of 14-3-3z in BIK-transfected cells treated with TAM could play a role in mediating TAM resistance of these cells. Overexpression of 14-3-3z in BIK-suppressed cells treated with TAM could have an inhibitory or preventive action on apoptosis [20–22], to promote cell survival and proliferation or to promote the reparative response to DNA damage. Previous studies have reported that 14-3-3 proteins bind to members of the Bcl-2 family, including BAD and BAX, thereby inhibiting the proapoptotic activity of BAX and permeabilization of mitochondrial membrane [21–23]. 14-3-3 overexpression also blocks apoptosis by inhibiting promoters of cell death, like apoptosis signal-regulating kinase 1 (ASK1) [23] and forkhead box O3 (FOXO 3) [24].
14-3-3z may play an important role in signaling pathway in breast cancer. Also, a high 14-3-3z expression could positively regulate growth factor receptors and protein kinase pathways (e.g., EGFR, HER2, MAPK, and AKT), stimulating the activity of pathways that promote survival, antiapoptotic signaling, and conferring endocrine resistance in breast cancer cells. Within this context, 14-3-3z has been considered a target for manipulating multiple cancer cell pathways [46, 47].
Furthermore, by means of in silico prediction of other molecules regulated by 14-3-3z implicated in cell cycle progression in siRNAiBIK/TAM cells, a theoretical interactome was carried out. From this analysis, we found that the interaction of 14-3-3z with proteins such as FOX 1 and WEE1 leads to arrest in G2 phase (Fig. 4). Consistent with this conclusion, the array analysis demonstrated that WEE1 was highly expressed in siRNAiBIK/TAM cells. WEE1 is known to regulate the repair of DNA damage. Previous studies have found that WEE1, together with CHK1 (another cell cycle regulator), is overexpressed in cisplatin-resistant cells [48]. Importantly, the therapeutic inhibition of WEE1 in combination with chemotherapy is currently in preclinical assays for the treatment of cancer [57].
Moreover, WEE1 has been found to be highly expressed in various cancer types and could participate in their transformation, as well as in the acquisition of resistance to DNA-damaging agents [49, 50]. In our studies, the overexpression of WEE1 associated with TAM resistance could protect breast cancer cells against DNA damage and cell death [51, 52].
In conclusion, our data demonstrated that suppression of BIK in ER-positive MCF-7 cells prevents the cytotoxic effect of TAM and favors a more aggressive phenotype, due to the molecular change of different pathways involved in different cell processes such as apoptosis, cell cycle, proliferation, invasion, and cell migration (Fig. 5). Knowledge of these mechanisms will permit the development of therapeutic strategies to improve treatment efficiency and to prevent or overcome drug resistance in cancer.
References
Gherardi E, Rota E, Calandra S, Genova R, Tamborino A. Relationship among the concentrations of serum lipoproteins and changes in their chemical composition in patients with untreated nephrotic syndrome. Eur J Clin Invest. 1977;7(6):563–70.
Vargo-Gogola T, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7(9):659–72. doi:10.1038/nrc2193.
Jordan VC, Murphy CS. Endocrine pharmacology of antiestrogens as antitumor agents. Endocr Rev. 1990;11(4):578–610.
Sheen YY, Ruh TS, Mangel WF, Katzenellenbogen BS. Antiestrogenic potency and binding characteristics of the triphenylethylene H1285 in MCF-7 human breast cancer cells. Cancer Res. 1985;45(9):4192–9.
Gee JM, Harper ME, Hutcheson IR, Madden TA, Barrow D, Knowlden JM, et al. The antiepidermal growth factor receptor agent gefitinib (ZD1839/Iressa) improves antihormone response and prevents development of resistance in breast cancer in vitro. Endocrinology. 2003;144(11):5105–17. doi:10.1210/en.2003-0705en.2003-0705.
Hoskins JM, Carey LA, McLeod HL. CYP2D6 and tamoxifen: DNA matters in breast cancer. Nat Rev Cancer. 2009;9(8):576–86. doi:10.1038/nrc2683nrc2683.
Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, et al. Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005;23(11):2469–76. doi:10.1200/JCO.2005.01.172.
Shi L, Dong B, Li Z, Lu Y, Ouyang T, Li J, et al. Expression of ER-{alpha}36, a novel variant of estrogen receptor {alpha}, and resistance to tamoxifen treatment in breast cancer. J Clin Oncol. 2009;27(21):3423–9. doi:10.1200/JCO.2008.17.2254JCO.2008.17.2254.
Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, et al. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. 2008;26(22):3727–34. doi:10.1200/JCO.2007.14.296826/22/3727.
Viedma-Rodriguez R, Baiza-Gutman L, Salamanca-Gomez F, Diaz-Zaragoza M, Martinez-Hernandez G, Ruiz Esparza-Garrido R, et al. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review). Oncol Rep. 2014. doi:10.3892/or.2014.3190.
Viedma-Rodriguez R, Baiza-Gutman LA, Garcia-Carranca A, Moreno-Fierros L, Salamanca-Gomez F, Arenas-Aranda D. Suppression of the death gene BIK is a critical factor for resistance to tamoxifen in MCF-7 breast cancer cells. Int J Oncol. 2013;43(6):177786. doi:10.3892/ijo.2013.2127.
Lopez J, Hesling C, Prudent J, Popgeorgiev N, Gadet R, Mikaelian I, et al. Src tyrosine kinase inhibits apoptosis through the Erk1/2-dependent degradation of the death accelerator Bik. Cell Death Differ. 2012;19(9):1459–69. doi:10.1038/cdd.2012.21cdd201221.
Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res. 2007;67(8):3734–40. doi:10.1158/0008-5472.CAN-06-4594.
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–5.
Schaefer MH, Fontaine JF, Vinayagam A, Porras P, Wanker EE, Andrade-Navarro MA. HIPPIE: integrating protein interaction networks with experiment based quality scores. PLoS One. 2012;7(2):e31826. doi:10.1371/journal.pone.0031826PONE-D-11-20814.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. doi:10.1101/gr.123930313/11/2498.
Rajput S, Kumar BN, Sarkar S, Das S, Azab B, Santhekadur PK, et al. Targeted apoptotic effects of thymoquinone and tamoxifen on XIAP mediated Akt regulation in breast cancer. PLoS One. 2013;8(4):e61342. doi:10.1371/journal.pone.0061342PONE-D-12-38938.
Pouliot LM, Chen YC, Bai J, Guha R, Martin SE, Gottesman MM, et al. Cisplatin sensitivity mediated by WEE1 and CHK1 is mediated by miR-155 and the miR-15 family. Cancer Res. 2012;72(22):5945–55. doi:10.1158/0008-5472.CAN-12-14000008-5472.CAN-12-1400.
Chu SH, Liu YW, Zhang L, Liu B, Li L, Shi JZ. Regulation of survival and chemoresistance by HSP90AA1 in ovarian cancer SKOV3 cells. Mol Biol Rep. 2013;40(1):1–6. doi:10.1007/s11033-012-1930-3.
Moody SE, Schinzel AC, Singh S, Izzo F, Strickland MR, Luo L, et al. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene. 2014. doi:10.1038/onc.2014.153onc2014153.
Carraway 3rd KL. E3 ubiquitin ligases in ErbB receptor quantity control. Semin Cell Dev Biol. 2010;21(9):936–43. doi:10.1016/j.semcdb.2010.09.006S1084-9521(10)00153-9.
Liu B, Wen X, Cheng Y. Survival or death: disequilibrating the oncogenic and tumor suppressive autophagy in cancer. Cell Death Dis. 2013;4:e892. doi:10.1038/cddis.2013.422cddis2013422.
Privette LM, Petty EM. CHFR: a novel mitotic checkpoint protein and regulator of tumorigenesis. Transl Oncol. 2008;1(2):57–64.
Jordan NJ, Dutkowski CM, Barrow D, Mottram HJ, Hutcheson IR, Nicholson RI, et al. Impact of dual mTORC1/2 mTOR kinase inhibitor AZD8055 on acquired endocrine resistance in breast cancer in vitro. Breast Cancer Res. 2014;16(1):R12. doi:10.1186/bcr3604bcr3604.
Bergamaschi A, Christensen BL, Katzenellenbogen BS. Reversal of endocrine resistance in breast cancer: interrelationships among 14-3-3zeta, FOXM1, and a gene signature associated with mitosis. Breast Cancer Res. 2011;13(3):R70. doi:10.1186/bcr2913bcr2913.
Frasor J, Chang EC, Komm B, Lin CY, Vega VB, Liu ET, et al. Gene expression preferentially regulated by tamoxifen in breast cancer cells and correlations with clinical outcome. Cancer Res. 2006;66(14):7334–40. doi:10.1158/0008-5472.CAN-05-4269.
Zeitz MJ, Ay F, Heidmann JD, Lerner PL, Noble WS, Steelman BN, et al. Genomic interaction profiles in breast cancer reveal altered chromatin architecture. PLoS One. 2013;8(9):e73974. doi:10.1371/journal.pone.0073974PONE-D-13-13446.
Sun M, Gomes S, Chen P, Frankenberger CA, Sankarasharma D, Chung CH, et al. RKIP and HMGA2 regulate breast tumor survival and metastasis through lysyl oxidase and syndecan-2. Oncogene. 2014;33(27):3528–37. doi:10.1038/onc.2013.328onc2013328.
Dutta S, Bandyopadhyay C, Bottero V, Veettil MV, Wilson L, Pins MR, et al. Angiogenin interacts with the plasminogen activation system at the cell surface of breast cancer cells to regulate plasmin formation and cell migration. Mol Oncol. 2014;8(3):483–507. doi:10.1016/j.molonc.2013.12.017S1574-7891(13)00189-0.
Nagashima K, Shumway SD, Sathyanarayanan S, Chen AH, Dolinski B, Xu Y, et al. Genetic and pharmacological inhibition of PDK1 in cancer cells: characterization of a selective allosteric kinase inhibitor. J Biol Chem. 2011;286(8):6433–48. doi:10.1074/jbc.M110.156463M110.156463.
Gamero AM, Young MR, Mentor-Marcel R, Bobe G, Scarzello AJ, Wise J, et al. STAT2 contributes to promotion of colorectal and skin carcinogenesis. Cancer Prev Res (Phila). 2010;3(4):495–504. doi:10.1158/1940-6207.CAPR-09-01051940-6207.CAPR-09-0105.
Chin YR, Yoshida T, Marusyk A, Beck AH, Polyak K, Toker A. Targeting Akt3 signaling in triple-negative breast cancer. Cancer Res. 2014;74(3):964–73. doi:10.1158/0008-5472.CAN-13-21750008-5472.CAN-13-2175.
Rehman SK, Li SH, Wyszomierski SL, Wang Q, Li P, Sahin O, et al. 14-3-3zeta orchestrates mammary tumor onset and progression via miR-221-mediated cell proliferation. Cancer Res. 2014;74(1):363–73. doi:10.1158/0008-5472.CAN-13-20160008-5472.CAN-13-2016.
Neal CL, Yao J, Yang W, Zhou X, Nguyen NT, Lu J, et al. 14-3-3zeta overexpression defines high risk for breast cancer recurrence and promotes cancer cell survival. Cancer Res. 2009;69(8):3425–32. doi:10.1158/0008-5472.CAN-08-27650008-5472.CAN-08-2765.
Ballesta A, Lopez J, Popgeorgiev N, Gonzalo P, Doumic M, Gillet G. Data-driven modeling of SRC control on the mitochondrial pathway of apoptosis: implication for anticancer therapy optimization. PLoS Comput Biol. 2013;9(4):e1003011. doi:10.1371/journal.pcbi.1003011PCOMPBIOL-D-12-01842.
McGlynn LM, Kirkegaard T, Edwards J, Tovey S, Cameron D, Twelves C, et al. Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin Cancer Res. 2009;15(4):1487–95. doi:10.1158/1078-0432.CCR-07-496715/4/1487.
Sanchez-Munoz A, Gallego E, de Luque V, Perez-Rivas LG, Vicioso L, Ribelles N, et al. Lack of evidence for KRAS oncogenic mutations in triple-negative breast cancer. BMC Cancer. 2010;10:136. doi:10.1186/1471-2407-10-1361471-2407-10-136.
Roy S, McPherson RA, Apolloni A, Yan J, Lane A, Clyde-Smith J, et al. 14-3-3 facilitates Ras-dependent Raf-1 activation in vitro and in vivo. Mol Cell Biol. 1998;18(7):3947–55.
Leung EY, Kim JE, Askarian-Amiri M, Joseph WR, McKeage MJ, Baguley BC. Hormone resistance in Two MCF-7 breast cancer cell lines is associated with reduced mTOR signaling, decreased glycolysis, and increased sensitivity to cytotoxic drugs. Front Oncol. 2014;4:221. doi:10.3389/fonc.2014.00221.
Faridi J, Wang L, Endemann G, Roth RA. Expression of constitutively active AKT3 in MCF-7 breast cancer cells reverses the estrogen and tamoxifen responsivity of these cells in vivo. Clin Cancer Res. 2003;9(8):2933–9.
Corum DG, Tsichlis PN, Muise-Helmericks RC. AKT3 controls mitochondrial biogenesis and autophagy via regulation of the major nuclear export protein CRM-1. FASEB J. 2014;28(1):395–407. doi:10.1096/fj.13-235382fj.13-235382.
Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5(3):400–3.
Hamilton DH, Huang B, Fernando RI, Tsang KY, Palena C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer Res. 2014;74(9):2510–9. doi:10.1158/0008-5472.CAN-13-18940008-5472.CAN-13-1894.
Danes CG, Wyszomierski SL, Lu J, Neal CL, Yang W, Yu D. 14-3-3 zeta down-regulates p53 in mammary epithelial cells and confers luminal filling. Cancer Res. 2008;68(6):1760–7. doi:10.1158/0008-5472.CAN-07-317768/6/1760.
Matta A, Siu KW, Ralhan R. 14-3-3 zeta as novel molecular target for cancer therapy. Expert Opin Ther Targets. 2012;16(5):515–23. doi:10.1517/14728222.2012.668185.
van Hemert MJ, Steensma HY, van Heusden GP. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. Bioessays. 2001;23(10):936–46. doi:10.1002/bies.1134.
Tzivion G, Gupta VS, Kaplun L, Balan V. 14-3-3 proteins as potential oncogenes. Semin Cancer Biol. 2006;16(3):203–13. doi:10.1016/j.semcancer.2006.03.004.
Coleman TR, Dunphy WG. Cdc2 regulatory factors. Curr Opin Cell Biol. 1994;6(6):877–82.
Lal S, Burkhart RA, Beeharry N, Bhattacharjee V, Londin ER, Cozzitorto JA, et al. HuR posttranscriptionally regulates WEE1: implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014;74(4):1128–40. doi:10.1158/0008-5472.CAN-13-191574/4/1128.
Ghiasi N, Habibagahi M, Rosli R, Ghaderi A, Yusoff K, Hosseini A, et al. Tumour suppressive effects of WEE1 gene silencing in breast cancer cells. Asian Pac J Cancer Prev. 2013;14(11):6605–11.
Magnussen GI, Holm R, Emilsen E, Rosnes AK, Slipicevic A, Florenes VA. High expression of Wee1 is associated with poor disease-free survival in malignant melanoma: potential for targeted therapy. PLoS One. 2012;7(6):e38254. doi:10.1371/journal.pone.0038254PONE-D-12-03007.
Mir SE, De Witt Hamer PC, Krawczyk PM, Balaj L, Claes A, Niers JM, et al. In silico analysis of kinase expression identifies WEE1 as a gatekeeper against mitotic catastrophe in glioblastoma. Cancer Cell. 2010;18(3):244–57. doi:10.1016/j.ccr.2010.08.011S1535-6108(10)00308-9.
Acknowledgments
We thank Jorge Ramírez-Salcedo and collaborators for their technical assistance in the microarray analysis. This study was performed in partial fulfillment of the requirements for the Ph.D. degree in Biomedical Sciences of RV-R, who was the recipient of doctoral fellowship provided by CONACyT-México (grant no. 207148), at the Universidad Nacional Autónoma de México. This study was supported by grants Salud-2007-785-063 from CONACyT-México and PAPIIT, DGAPA, UNAM, project IN223014.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Additional information
Diego Arenas-Aranda passed away last 2014 (1958–2014).
Rights and permissions
About this article

Cite this article
Viedma-Rodríguez, R., Ruiz Esparza-Garrido, R., Baiza-Gutman, L.A. et al. Involvement of multiple cellular pathways in regulating resistance to tamoxifen in BIK-suppressed MCF-7 cells. Tumor Biol. 36, 6991–7005 (2015). https://doi.org/10.1007/s13277-015-3374-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3374-6