
Abstract
A large body of evidence indicates that microRNAs play a critical role in tumor initiation and progression by negatively regulating oncogenes or tumor suppressor genes. Here, we report that the expression of miR-200a was notably downregulated in 45 renal cell carcinoma (RCC) samples. Restoration of miR-200a suppressed cell proliferation, migration, and invasion in two RCC cell lines. Furthermore, we used an epithelial-to-mesenchymal transition PCR array to explore the putative target genes of miR-200a. By performing quantitative real-time PCR, ELISA, and luciferase reporter assays, transforming growth factor beta2 (TGFB2) was validated as a direct target gene of miR-200a. Moreover, siRNA-mediated knockdown of TGFB2 partially phenocopied the effect of miR-200a overexpression. These results suggest that miR-200a suppresses RCC development via directly targeting TGFB2, indicating that miR-200a may present a novel target for diagnostic and therapeutic strategies in RCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma (RCC) originates from renal tubules. It contributes to about 3 % of adult malignancies and results in over 100,000 deaths per year [1]. It has been described as the most lethal malignancy of all the urological cancers. Although surgery is curative for the localized RCC, a remarkable proportion of patients will relapse or metastasize. Nearly 60 % of the RCC patients develop metastases, and the 5-year survival rate of metastatic RCC patients is approximately 10 % [2, 3]. Despite a series of new therapeutic drugs has been developed, it is still difficult to treat patients with metastatic RCC and the prognostic improvement cannot be accomplished. Therefore, new sensitive tumor markers and effective therapeutic methods are urgently needed to prolong a patient’s survival.
Emerging evidence suggests that microRNAs (miRNAs) are aberrantly expressed in human cancers, and they are involved in the cancer initiation, development, and metastasis [4]. MiRNAs are small (∼22 nucleotides) noncoding regulatory RNA molecules that function as negative gene regulators by repressing translation or cleaving RNA transcripts in a sequence-specific manner [5]. They can regulate a wide variety of developmental and physiological processes and function as oncogenes or tumor suppressors depending on their downstream target genes [6].
On the basis of recent studies describing miRNA signatures, dysregulation of several miRNAs has been reported in RCC [7–9]. In our previous study, we have identified numerous miRNAs which were abnormally expressed in RCC by digital gene expression sequencing, and miR-200a was one of the most downregulated miRNAs [10]. MiR-200a belongs to the miR-200 family, which plays an important role in the epithelial-to-mesenchymal transition (EMT) in human cancers [11]. Several studies show that miR-200a inhibited the metastatic potential and proliferation of cancer cells, indicating that miR-200a may function as a tumor suppressor in human cancers [12–15]. Furthermore, a previous study showed that miR-200a was downregulated in clinical RCC specimens compared with normal kidney tissues [16]. In this study, we performed quantitative real-time PCR in 45 pairs of RCC samples and the matched normal tissues to validate the aberrant expression of miR-200a in RCC. The tumor suppressive role of miR-200a in RCC was demonstrated by several functional experiments. To search for the putative target genes, an EMT PCR array was carried out and transforming growth factor beta2 (TGFB2) was subsequently validated as the direct target of miR-200a. Tumor suppressive miR-200a and its novel molecular target, TGFB2, may provide new insights into the potential mechanisms of RCC, and miR-200a may serve as a promising target for further therapeutic approaches.
Materials and methods
Ethics statement
The collection and use of the patient samples were approved by Institutional Review Board of Peking University Shenzhen Hospital. Written consent was obtained from all patients in the study.
Patient specimens
A total of 45 pairs of RCC and adjacent noncancerous tissue (36 clear cell RCC, 5 papillary RCC, and 4 chromophobe RCC) were obtained from the Sun Yat-sen University Cancer Center and The First Affiliated Hospital of Anhui Medical University. Histopathological diagnoses were performed according to the WHO classification system, and nuclear grade was scored according to the Fuhrman classification system [17]. The clinicopathological information of the patients was shown in previous study [18].
Cell culture
Two human RCC cell lines ACHN and 786-O were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were incubated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum and 1 % antibiotics (100 U/ml penicillin and 100 mg/ml streptomycin sulfates). Cells were maintained in humidified incubators (5 % CO2) at 37 °C.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from tissue samples or the RCC cells using the miRNeasy Mini Kit (QIAGEN, Valencia, CA, USA). All isolated RNA was quantified by NanoDrop® spectrophotometer. Only those RNA samples with 260/280 ratios of 1.8–2.0 were used for further investigation. cDNA was generated from 1 μg total RNA using the miScript II RT Kit (QIAGEN) and the RevertAid First Strand cDNA Synthesis Kit (Fermentas, Vilnius, Lithuania). The analysis of qRT-PCR was performed using the miScript SYBR® Green PCR Kit (QIAGEN Co. Ltd.) and the SYBR Premix Ex Taq™ (Takara Bio Inc.) on the Roche LightCycler 480 Real-Time PCR System. The expression of miR-200a was normalized to U6 small nuclear RNA (RNU6B). GAPDH was selected as the endogenous control for TGFB2. The following primers were used: miR-200a forward primer, 5′-TAA CAC TGT CTG GTA ACG ATG T-3′; RNU6B forward primer, 5′-ACG CAA ATT CGT GAA GCG TT-3′ (both of the reverse primers were provided by the miScript SYBR® Green PCR Kit); TGFB2 forward, 5′-TTA GAA ATG TGC AGG ATA ATT GCT-3′, reverse, 5′-CAG AAG TTG GCA TTG TAC CCT-3′; GAPDH forward, 5′-ATG GGG AAG GTG AAG GTC GG-3′, reverse, 5′-GAC GGT GCC ATG GAA TTT GC-3′. Amplification reactions were performed in a 20-μl final volume and were done for 40 cycles (15 s at 95 °C, 20 s at 55 °C, and 30 s at 70 °C). The mRNA and miRNA expression levels were determined using the delta–delta Ct method.
Mature miRNA and siRNA transfection
ACHN and 786-O cells were transfected with 50 nM miR-200a mimics or the negative control (RiboBio, Guangzhou, China) using Lipofectamine 2000 (Invitrogen, CA, USA) and Opti-MEM™ (Invitrogen). For knockdown of TGFB2, ACHN and 786-O cells were transfected with TGFB2 siRNA (si-TGFB2, GenePharma, Shanghai, China) or the negative control siRNA (si-NC) according to the manufacturer’s instructions.
Cell proliferation assay
Proliferation of ACHN and 786-O cells was measured by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT, Sigma Aldrich, St. Louis, MO, USA). Cells (approximately 6 × 103 cells) were seeded into a 96-well culture plate 24 h prior to transfection. The blank control was set up with a medium only. At 0, 24, 48, or 72 h post-transfection, 20 μl of MTT (5 mg/ml) was added to each well, and plates were incubated for 4 h at 37 °C. The precipitate was solubilized in 150 μl of dimethyl sulfoxide (DMSO), and absorbance was measured at a wavelength of 490 nm (with 630 nm as the reference wavelength) using an ELISA microplate reader (Bio-Rad, Hercules, CA, USA). Experiments were repeated at least three times.
Cell migration assay
Cell migration was examined by scratch assay according to the methods previously described [19]. Briefly, ACHN and 786-O cells were seeded to 6-well plates. At 24 h after transfection, an artificial homogenous wound was created onto the monolayer with a sterile 200-μl tip. After scratching, the cells were washed with phosphate buffer saline (PBS) medium three times and incubated at 37 °C. Images of cell migration were captured at time points of 0, 6, 12, 18, and 24 h by digital camera system. The software program MIAS-2000 was used to determine the migration distance. Experiments were run in three independent repeats in triplicate and analyzed in a double-blind fashion by at least two observers.
Cell invasion assay
Matrigel (1:4, 60 μl/well, BD Biosciences, Heidelberg, Germany) was added to Transwell chambers (8.0-μm pore size, Corning, Cambridge, MA) in a 24-well plate. At 24 h after transfection, ACHN and 786-O cells were harvested and resuspended in a serum-free DMEM medium. Aliquots (2 × 104 cells) of the prepared cell suspension were added into the upper chamber, and the lower chamber was filled with 1 ml DMEM supplemented with 10 % FBS. After 48 h incubation at 37 °C with 5 % CO2, non-invading cells in the upper chamber were removed with a cotton swab and washed three times with PBS. Then, the invasive cells were fixed in 100 % ethanol and stained with 0.1 % crystal violet. Invading cells were counted by taking photomicrographs in three fields. Experiments were performed in triplicate.
Epithelial-to-mesenchymal transition (EMT) PCR array
The RT2 Profiler PCR array for human EMT (QIAGEN/SABiosciences, which consists of 84 EMT-related genes, was used for the identification of the EMT-related genes. Total RNA isolated from the ACHN and 786-O cells which transfected with miR-200a or the negative control was used for screening by real-time PCR according to the manufacturer’s instructions.
Luciferase reporter assay
The 3′UTR of the TGFB2 was PCR amplified and inserted between the XhoI–NotI restriction sites in the 3′UTR of the hRluc gene in the psiCHECK™-2 luciferase vector (Promega, Madison, WI, USA). Primer sequences for the 3′UTR of TGFB2 mRNA (forward primer 5′-CCG CTC GAG GAT GAC GAC GAC AAC GAT GAT GC-3′ and reverse primer 5′-ATT TGC GGC CGC GGG GCT TTA CTC AAA AGT ACT GG-3′) were designed. In order to verify the binding sites of the miR-200a, we synthesized two short fragments of TGFB2 3′UTR which contains the potential binding sites. Then, we manually mutated the potential binding sites by exchanging the G and T, A and C. The sequences of the short fragments were shown in Fig. 4e. All the fragments were cloned into psiCHECKTM-2 luciferase vector, and all the constructs were verified by sequencing. Then, the luciferase reporter constructs, together with miR-200a mimics or the negative control, were transfected into ACHN cells. At 48 h after transfection, dual luciferase assay system (Promega, Madison, WI, USA) was used to detect the luciferase activity. Normalized data were calculated as the quotient of Renilla/firefly luciferase activities. The experiments were performed in duplicate and repeated at least three times.
ELISA
After 24 h, a serum-free medium was cultured, the conditioned medium of the transfected ACHN and 786-O cells was collected. Then, ELISA was performed to measure the concentration of TGFB2 in the cells culture supernatants using the human transforming growth factor beta 2 ELISA Kit (Yifeng, Shanghai, China) according to the manufacturer’s instructions.
Results
miR-200a is downregulated in RCC
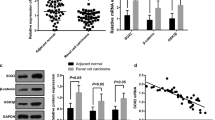
In order to investigate the expression profile of miR-200a in RCC, we performed qRT-PCR in 45 pairs of RCC specimens and their adjacent normal kidney tissues. As shown in Fig. 1a, the expression of miR-200a was downregulated in 38 samples (84.4 %), with an average of 3.42-fold downregulation (p < 0.01), suggesting that miR-200a may function as a tumor suppressor in RCC.

miR-200a was downregulated in RCC and inhibited RCC cell proliferation. a Quantitative RT-PCR of miR-200a expression relative to U6 expression in RCC tumor samples and adjacent normal kidney tissue of 45 renal cancer patients. The 2−ΔΔCT method was used to analyze the data, and the data were shown in log2 (cancer/normal). b, c miR-200a inhibited cell proliferation in ACHN cells and 786-O cells. Cell growth was measured by MTT assay at different time intervals. ANOVA was used for the comparison of cell growth curves
miR-200a inhibits cell proliferation
To investigate the functional significance of miR-200a in RCC, we performed gain-of-function studies using transient transfection with mature miR-200a mimics. The MTT assay demonstrated that the cell proliferation in miR-200a transfectants was significantly decreased by 19.3 % (24 h), 18.1 % (48 h), and 15.9 % (72 h) in ACHN cells (p < 0.05, Fig. 1b) and was decreased by 17.3 % (24 h), 17.4 % (48 h), and 13.6 % (72 h) in 786-O cells (p < 0.05, Fig. 1c). These data provided evidence showing that overexpression of miR-200a inhibited RCC cell proliferation.
miR-200a inhibits cell migration and invasion
To investigate the role of miR-200a in tumor metastasis, we performed cell migration and invasion assay in RCC cell lines ACHN and 786-O. As shown in Fig. 2a, b, cell migration was significantly decreased in miR-200a transfectants compared with negative control. The inhibition rates of migration were 25.4 % for ACHN cells (p < 0.01) and 41.5 % for 786-O cells (p < 0.01), respectively. We then performed cell invasion assays in ACHN and 786-O cells. The number of invading cells was significantly decreased by 46.9 % in ACHN (p < 0.01) and by 31.9 % in 786-O cells (p < 0.05) in the miR-200a transfectants compared with the negative control (Fig. 2c), suggesting that miR-200a inhibited RCC cell migration and invasion.

miR-200a inhibited cell migration and invasion. a miR-200a inhibited cell migration in ACHN cells. b miR-200a inhibited cell migration in 786-O cells. c miR-200a inhibited cell invasion in ACHN and 786-O cells. All experiments were performed three times, and a representative picture was shown. *p < 0.05, **p < 0.01
miR-200a regulates TGFB2 expression in RCC cells
The miR-200 family has been shown to inhibit the initiation step of metastasis, EMT, by maintaining the epithelial phenotype in cancer cells [11, 20]. Since miR-200a significantly decreased cellular migration and invasion in RCC cells, we searched for the downstream EMT-related genes that might be regulated by miR-200a using the EMT PCR array. Of the 84 genes tested, 18 genes and 14 genes were downregulated after miR-200a treatment (>3-fold) in ACHN and 786-O cells respectively, including ZEB1 and ZEB2 which were validated as the direct target genes of miR-200a in previous studies [21, 22]. Conversely, only two genes and three genes were upregulated in miR-200a transfectants in ACHN and 786-O cells, respectively (Fig. 3).

miR-200a transfections in ACHN and 786-O cells led to differential expressions of EMT-related genes. a Overexpression of miR-200a in ACHN cells led to differential expressions of genes represented on an EMT PCR array. b Overexpression of miR-200a in 786-O cells led to differential expressions of genes represented on an EMT PCR array. Gene expressions were represented in a scatter plot with a 3-fold cutoff displaying fold changes between miR-200a and negative control transfected cells. Genes represented as down- or upregulated in the plot were listed with fold-change. Heat maps of the EMT PCR array were shown in the lower panels: green square (downregulated), red square (upregulated), black square (not changed), gray square (undetermined)
Among the most downregulated genes, TGFB2 was found to have the putative binding site of miR-200a. Therefore, we performed qRT-PCR to verify the result of EMT array. As shown in Fig. 4a, the mRNA levels of TGFB2 were significantly downregulated in miR-200a transfectants compared with the negative control. Moreover, as detected by ELISA, the protein levels of TGFB2 in the conditioned media from miR-200a transfected cells were significantly decreased compared with the negative control (Fig. 4b). These data clearly demonstrate that miR-200a represses TGFB2 expression at both mRNA and protein levels in RCC cells.

TGFB2 was a direct target gene of miR-200a. a miR-200a downregulated TGFB2 mRNA. qRT-PCR was used to measure the expression level of TGFB2 mRNA. b miR-200a downregulated TGFB2 protein. The supernatants of ACHN and 786-O cells were collected, and ELISA was used to measure the concentration of TGFB2. c Putative duplex formation between miR-200a and the TGFB2 3′UTR. The binding site of TGFB2 3′UTR was highly conserved among several mammalian species. The seed-recognizing sites were marked in bold type and underlined. d The luciferase reporter constructs which contained 3′UTR of TGFB2, together with miR-200a mimics or the negative control were transfected into ACHN cells. A total of 48 h after transfection, luciferase activity was detected. Normalized data were calculated as the quotient of Renilla/firefly luciferase activities. e We synthesized a 23-nucleotide-long fragments of TGFB2 3′UTR which contained the wild-type (WT) potential binding sites. And then, the mutated (MU) fragments were generated by exchanging the G and T, A and C on the putative binding sites. Samples were assayed as in d. The experiments were performed in duplicate and repeated three times. *p < 0.05, **p < 0.01
TGFB2 is a direct target of miR-200a
As illustrated in the Fig. 4c, TGFB2 showed the putative target site of miR-200a at 95–116 bp of 3′UTR with an exact match in the seed region at 109–115 bp. The target site is located in conserved regions of the TGFB2 3′UTR among several species (Fig. 4c). To investigate whether TGFB2 is a direct target of miR-200a, we generated a reporter construct containing the 3′UTR of TGFB2 downstream of the luciferase gene. The luciferase reporter assay indicated that miR-200a significantly inhibited the luciferase activity (Fig. 4d). To further verify the site of interaction, we generated two luciferase reporter constructs containing the wild-type or mutated-type target sites of TGFB2 3′UTR. As shown in Fig. 4e, the luciferase activity was significantly suppressed by miR-200a in wild-type constructs, whereas the inhibitory effect of miR-200a was abolished in the mutated-type constructs. These results confirmed TGFB2 as a direct target of miR-200a and identified the binding site in the 3′UTR of TGFB2 mRNA.
TGFB2 knockdown inhibits cell proliferation
To analyze whether TGFB2 affects cell proliferation and invasion of RCC, TGFB2 siRNA (si-TGFB2) and control siRNA (si-NC) were transfected into ACHN and 786-O cells. The expression of TGFB2 mRNA was significantly downregulated in si-TGFB2 transfected cells (Fig. 5a). Furthermore, ELISA showed that the protein level of TGFB2 in the conditioned media from si-TGFB2 transfected cells was significantly decreased compared with the si-NC transfectants (Fig. 5b). To determine the effect of TGFB2 knockdown on cell proliferation, ACHN and 786-O cells were transfected with si-TGFB2 or si-NC. The MTT assay demonstrated that cell proliferation was decreased by 13.0 % (24 h), 24.4 % (48 h), and 17.8 % (72 h) in ACHN cells (p < 0.05, Fig. 5c) and was decreased by 18.8 % (24 h), 19.4 % (48 h), and 13.8 % (72 h) in 786-O cells (p < 0.05, Fig. 5d). The results demonstrated that knockdown of TGFB2 inhibited RCC cell proliferation.

TGFB2 knockdown inhibited cell proliferation. a si-TGFB2 downregulated TGFB2 mRNA. The expression level of TGFB2 mRNA was measured by qRT-PCR. b si-TGFB2 downregulated TGFB2 protein. The expression level of TGFB2 protein was measured by ELISA. c, d TGFB2 knockdown inhibited cell proliferation in ACHN and 786-O cells. ANOVA was used for the comparison of cell growth curves
TGFB2 knockdown inhibits cell migration and invasion
Finally, we performed cell migration and invasion assay in ACHN and 786-O after si-TGFB2 transfection. As shown in Fig. 6a, b, knockdown of TGFB2 significantly suppressed cell migration in ACHN and 786-O cells. The inhibition rates of migration were 47.3 % for ACHN cells (p < 0.05) and 37.6 % for 786-O cells (p < 0.05), respectively. Cell invasion assay showed that the number of invading cells was significantly decreased by 70.1 % in ACHN (p < 0.01) and by 74.8 % in 786-O cells (p < 0.01) in the si-TGFB2 transfectants compared with the negative control (Fig. 6c). Taken together, knockdown of TGFB2 dramatically decreased RCC cell migration and invasion.

TGFB2 knockdown inhibited cell migration and invasion. a TGFB2 knockdown inhibited cell migration in ACHN cells. b TGFB2 knockdown inhibited cell migration in 786-O cells. c TGFB2 knockdown inhibited cell invasion in ACHN and 786-O cells. All experiments were performed three times, and a representative picture was shown. *p < 0.05, **p < 0.01
Discussion
The miR-200 family contains five members (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) that are found in two clusters which are located on chromosomes 1 and 12 [11]. There is growing evidence that miR-200 plays a vital role in tumor suppression by inhibiting EMT process, the initiating step of metastasis [23, 24]. Downregulation of miR-200a has been reported in several human cancers, including nasopharyngeal carcinoma [12], meningiomas [25], and breast cancer [26]. Moreover, restoration of miR-200a inhibited cell proliferation, migration, and invasion of hepatocellular carcinoma [13], nasopharyngeal carcinoma [12], prostate cancer [14], and ovarian cancer [15]. Conversely, inhibition of miR-200a promoted cancer cell development [12], suggesting that miR-200a may act as a tumor suppressor in human cancer.
Our previous study has identified plenty of miRNAs which were abnormally expressed in RCC tissues by digital gene expression sequencing [10]. Among them, miR-200a was one of the most downregulated miRNAs. To further validate the sequencing results, we performed qRT-PCR to analyze the expression of miR-200a in other 45 samples of RCC patients. Our data showed that the expression of miR-200a was significantly downregulated in RCC samples compared with that in the adjacent normal tissues, which were consistent with a previous study. To further characterize the effect of miR-200a in RCC cell lines, a series of function assays was performed. Our results demonstrated that miR-200a inhibited cell proliferation, migration, and invasion, which provided compelling evidence to support the anticancer effect of miR-200a on RCC.
EMT is the process by which epithelial cells lose their cell polarity and adhesion and acquire migratory and invasive properties to become mesenchymal cells. EMT is a critical step in the initiation of metastasis for cancer progression and correlates positively with poor prognosis [27, 28]. Cancer cells in primary tumor lose cell–cell adhesion mediated by E-cadherin repression, get through the basement membrane with enhanced invasive properties, and enter the bloodstream through intravasation [29]. It has been well established that miR-200a is closely involved in the process of EMT [11, 21, 22]; we therefore performed an EMT PCR array to search the potential EMT-related pathway that is affected by miR-200a restoration in RCC cells. The EMT array contains 84 genes including morphogenesis, extracellular matrix and cell adhesion, cytoskeletal genes, and transcription factor genes that cause EMT. Our data showed that miR-200a regulates the expression of a variety of genes involved in EMT, suggesting that the downregulation of miR-200a in RCC may provide advantages for tumor metastasis by modulating a set of genes required for EMT.
Among the dysregulated EMT-related genes, we chose TGFB2 for further analysis as it has a putative binding site on the 3′UTR for miR-200a. TGFB is a protein secreted by most cells that controls cell proliferation, differentiation, motility, apoptosis, and immune regulation [30]. Dual functions of TGFB have been reported in human cancer. TGFB is an important cytostatic regulator in epithelial cells, acting as an antiproliferative factor in normal epithelial cells and at early stages of oncogenesis due to its ability to activate the transcription of cell-cycle inhibitors and/or apoptotic regulators [31–33]. However, the tumor-suppressive arm of the TGFB pathways is selectively inhibited as a tumor progresses. In the meanwhile, cancer cells can mutate parts of the TGFB signaling pathway and result in increased EMT, invasion, and metastasis [33, 34]. Moreover, TGFB can cause immunosuppression and angiogenesis through acting on the surrounding stromal cells, immune cells, endothelial, and smooth muscle cells, which makes the cancer more invasive [35]. Downregulation and the pro-oncogenic effect of TGFB2 have been reported in human cancer [36, 37]. However, little is known whether TGFB2 is involved in RCC. Our study clearly showed that miR-200a repressed the expression of TGFB2 in both mRNA and protein levels in RCC cell lines. Furthermore, luciferase reporter assay showed that miR-200a directly bind to the TGFB2 3′UTR and inhibits its luciferase activity. There results show that TGFB2 is a direct target of miR-200a in RCC, which is consistent with a previous study that miR-200a regulates the expression of TGFB2 in a rat kidney tubular epithelial cell [38]. To validate whether miR-200a has a tumor suppressive role through TGFB2, we knocked down TGFB2 in ACHN and 786-O cells by siRNA technique. TGFB2 mRNA and protein were significantly downregulated after si-TGFB2 transfection. Similar to the results of miR-200a, si-TGFB2 transfection suppressed the proliferation and invasion of RCC cell.
It has been well established that miR-200a inhibited the EMT of cancer cells through directly targeting E-cadherin transcriptional repressors (ZEB1 and ZEB2), and knockdown of miR-200a induced decreased level of E-cadherin and thus promoted EMT [39, 40]. By contrast, ZEB1 repressed the expression of miR-200 through directly binding to the E-box sites present upstream of miR-200 promoters [41]. ZEB1 silencing in mesenchymal cancer cells induced miR-200 expression [41, 42], indicating that ZEB and miR-200 were reciprocally linked in a feedback loop. Interestingly, sustained expression of ZEB requires the autocrine TGFB signaling, thus, contributing to the maintenance of the mesenchymal phenotype [43]. Moreover, continuous expression of TGFB led to reversible DNA methylation of the miR-200 promoters and subsequently resulted in miR-200 repression [43]. In theory, adding TGFb2 back to the culture medium will definitely rescue the effect of miR-200a. One possibility is that exogenous TGFb2 really rescued the downregulation of endogenous TGFb2 by miR-200a, while the other possibility is that TGFb2 itself could affect cell migration and invasion and reverse the effect of miR-200a. According to these results, we suppose that there also has a feedback loop between miR-200 and TGFB. The miR-200/ZEB/TGFB signaling network may play a crucial role in EMT and tumor metastasis.
In conclusion, miR-200a was significantly downregulated in clinical RCC specimens and functioned as a tumor suppressor in RCC by directly targeting TGFB2. Our study may lead to a better understanding of miR-200/ZEB/TGFB signaling network. Recently, preclinical studies have shown the potential application of anti-TGFB therapies on cancer targeted treatment [33]. Many of these strategies are currently in clinical trials. We suppose that miR-200a may also serve as a novel therapeutic strategy for patients with RCC.
References
Rini BI, Campbell SC, Escudier B. Renal cell carcinoma. Lancet. 2009;373(9669):1119–32.
Campbell SC, Flanigan RC, Clark JI. Nephrectomy in metastatic renal cell carcinoma. Curr Treat Options Oncol. 2003;4(5):363–72.
Sun M, Lughezzani G, Perrotte P, Karakiewicz PI. Treatment of metastatic renal cell carcinoma. Nat Rev Urol. 2010;7(6):327–38.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66.
Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129(7):1401–14.
Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer. 2006;6(4):259–69.
Nakada C, Matsuura K, Tsukamoto Y, Tanigawa M, Yoshimoto T, Narimatsu T, et al. Genome‐wide microRNA expression profiling in renal cell carcinoma: significant down‐regulation of miR‐141 and miR‐200c. J Pathol. 2008;216(4):418–27.
White N, Khella H, Grigull J, Adzovic S, Youssef Y, Honey R, et al. miRNA profiling in metastatic renal cell carcinoma reveals a tumour-suppressor effect for miR-215. Br J Cancer. 2011;105(11):1741–9.
Hidaka H, Seki N, Yoshino H, Yamasaki T, Yamada Y, Nohata N, et al. Tumor suppressive microRNA-1285 regulates novel molecular targets: aberrant expression and functional significance in renal cell carcinoma. Oncotarget. 2012;3(1):44.
Zhou L, Chen J, Li Z, Li X, Hu X, Huang Y, et al. Integrated profiling of microRNAs and mRNAs: microRNAs located on Xq27.3 associate with clear cell renal cell carcinoma. PLoS One. 2010;5(12):e15224.
Mongroo PS, Rustgi AK. The role of the miR-200 family in epithelial-mesenchymal transition. Cancer Biol Ther. 2010;10(3):219–22.
Xia H, Ng SS, Jiang S, Cheung WK, Sze J, Bian X-W, et al. miR-200a-mediated downregulation of ZEB2 and CTNNB1 differentially inhibits nasopharyngeal carcinoma cell growth, migration and invasion. Biochem Biophys Res Commun. 2010;391(1):535–41.
Feng J, Wang J, Chen M, Chen G, Wu Z, Ying L, et al. miR-200a suppresses cell growth and migration by targeting MACC1 and predicts prognosis in hepatocellular carcinoma. Oncol Rep. 2015;33(2):713–20.
Barron N, Keenan J, Gammell P, Martinez VG, Freeman A, Masters JR, et al. Biochemical relapse following radical prostatectomy and miR‐200a levels in prostate cancer. Prostate. 2012;72(11):1193–9.
Hu X, Macdonald DM, Huettner PC, Feng Z, El Naqa IM, Schwarz JK, et al. A miR-200 microRNA cluster as prognostic marker in advanced ovarian cancer. Gynecol Oncol. 2009;114(3):457–64.
Yoshino H, Enokida H, Itesako T, Tatarano S, Kinoshita T, Fuse M, et al. Epithelial-mesenchymal transition-related microRNA-200s regulate molecular targets and pathways in renal cell carcinoma. J Hum Genet. 2013;58(8):508–16.
Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol. 1982;6(7):655–64.
Lu R, Ji Z, Li X, Zhai Q, Zhao C, Jiang Z, et al. miR-145 functions as tumor suppressor and targets two oncogenes, ANGPT2 and NEDD9, in renal cell carcinoma. J Cancer Res Clin. 2014;140(3):387–97.
Zhai Q, Zhou L, Zhao C, Wan J, Yu Z, Guo X, et al. Identification of miR-508-3p and miR-509-3p that are associated with cell invasion and migration and involved in the apoptosis of renal cell carcinoma. Biochem Biophys Res Commun. 2012;419(4):621–6.
Korpal M, Kang Y. The emerging role of miR-200 family of microRNAs in epithelial-mesenchymal transition and cancer metastasis. RNA Biol. 2008;5(3):115–9.
Park S-M, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22(7):894–907.
Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283(22):14910–4.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10(5):593–601.
Becker LE, Takwi AAL, Lu Z, Li Y. The role of miR-200a in mammalian epithelial cell transformation. Carcinogenesis. 2014;36(1):2–12.
Saydam O, Shen Y, Würdinger T, Senol O, Boke E, James MF, et al. Downregulated microRNA-200a in meningiomas promotes tumor growth by reducing E-cadherin and activating the Wnt/β-catenin signaling pathway. Mol Cell Biol. 2009;29(21):5923–40.
Eades G, Yao Y, Yang M, Zhang Y, Chumsri S, Zhou Q. miR-200a regulates SIRT1 expression and epithelial to mesenchymal transition (EMT)-like transformation in mammary epithelial cells. J Biol Chem. 2011;286(29):25992–6002.
Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420.
Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, et al. Epithelial—mesenchymal and mesenchymal—epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213(2):374–83.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64.
Glasgow E, Mishra L. Transforming growth factor-β signaling and ubiquitinators in cancer. Endocr Relat Cancer. 2008;15(1):59–72.
Hill JJ, Tremblay T-L, Cantin C, O'Connor-McCourt M, Kelly JF, Lenferink A. Glycoproteomic analysis of two mouse mammary cell lines during transforming growth factor (TGF)-beta induced epithelial to mesenchymal transition. Proteome Sci. 2009;7(2):1–17.
Sánchez-Capelo A. Dual role for TGF-β1 in apoptosis. Cytokine Growth Factor Rev. 2005;16(1):15–34.
Smith AL, Robin TP, Ford HL. Molecular pathways: targeting the TGF-β pathway for cancer therapy. Clin Cancer Res. 2012;18(17):4514–21.
Padua D, Massagué J. Roles of TGFβ in metastasis. Cell Res. 2008;19(1):89–102.
Epstein FH, Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342(18):1350–8.
Semczuk A, Zakrzewski P, Forma E, Cygankiewicz A, Semczuk-Sikora A, Bryś M, et al. TGFβ-pathway is down-regulated in a uterine carcinosarcoma: a case study. Pathol Res Pract. 2013;209(11):740–4.
Beisner J, Buck MB, Fritz P, Dippon J, Schwab M, Brauch H, et al. A novel functional polymorphism in the transforming growth factor-β2 gene promoter and tumor progression in breast cancer. Cancer Res. 2006;66(15):7554–61.
Wang B, Koh P, Winbanks C, Coughlan MT, McClelland A, Watson A, et al. miR-200a prevents renal fibrogenesis through repression of TGF-β2 expression. Diabetes. 2011;60:280–7.
Xiong M, Jiang L, Zhou Y, Qiu W, Fang L, Tan R, et al. The miR-200 family regulates TGF-β1-induced renal tubular epithelial to mesenchymal transition through Smad pathway by targeting ZEB1 and ZEB2 expression. Am J Physiol Renal Physiol. 2012;302(3):F369–79.
Gregory PA, Bracken CP, Smith E, Bert AG, Wright JA, Roslan S, et al. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22(10):1686–98.
Bracken CP, Gregory PA, Kolesnikoff N, Bert AG, Wang J, Shannon MF, et al. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008;68(19):7846–54.
Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S, et al. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008;9(6):582–9.
Gregory PA. An autocrine TGF-β/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol Biol Cell. 2011;22:1686–98.
Acknowledgments
This project was supported by Science and Technology Planning Project of Shenzhen in China (no. JCYJ20140415162543037) and Health and Family Planning Scientific Research Project of Shenzhen in China (no. 201401049).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding authors
Additional information
Ruijing Lu and Ziliang Ji contributed equally to this work.
Rights and permissions
About this article

Cite this article
Lu, R., Ji, Z., Li, X. et al. Tumor suppressive microRNA-200a inhibits renal cell carcinoma development by directly targeting TGFB2 . Tumor Biol. 36, 6691–6700 (2015). https://doi.org/10.1007/s13277-015-3355-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3355-9