
Abstract
TP53 gene defects represent a strong adverse prognostic factor for patient survival and treatment resistance in chronic lymphocytic leukemia (CLL). Although various methods for TP53 mutation analysis have been reported, none of them allow the identification of all occurring sequence variants, and the most suitable methodology is still being discussed. The aim of this study was to determine the limitations of commonly used methods for TP53 mutation examination in CLL and propose an optimal approach for their detection. We examined 182 CLL patients enriched for high-risk cases using denaturing high-performance liquid chromatography (DHPLC), functional analysis of separated alleles in yeast (FASAY), and the AmpliChip p53 Research Test in parallel. The presence of T53 gene mutations was also evaluated using ultra-deep next generation sequencing (NGS) in 69 patients. In total, 79 TP53 mutations in 57 (31 %) patients were found; among them, missense substitutions predominated (68 % of detected mutations). Comparing the efficacy of the methods used, DHPLC and FASAY both combined with direct Sanger sequencing achieved the best results, identifying 95 % and 93 % of TP53-mutated patients. Nevertheless, we showed that in CLL patients carrying low-proportion TP53 mutation, the more sensitive approach, e.g., ultra-deep NGS, might be more appropriate. TP53 gene analysis using DHPLC or FASAY is a suitable approach for mutation detection. Ultra-deep NGS has the potential to overcome shortcomings of methods currently used, allows the detection of minor proportion mutations, and represents thus a promising methodology for near future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chronic lymphocytic leukemia (CLL), the most frequently diagnosed adult leukemia in Western countries, is characterized by considerable biological and clinical heterogeneity. CLL prognosis is based mainly on clinical staging, detection of recurrent cytogenetic aberrations [del (13) (q14), +12, del (11) (q22), del (17) (p13.1)] and immunoglobulin heavy-chain variable region gene (IGHV) mutation status determination [1–4]. Moreover, mutations in the tumor suppressor gene TP53 have been associated with substantially shortened overall survival, short time to treatment, and resistance to fludarabine-based therapies [5–10], even in cases with low proportion of TP53-mutated subclones [11]. In addition, the cancer cells carrying TP53 mutations might be selected during CLL course [12, 13]; therefore, a TP53 mutational status examination is recommended before each therapy [14].
TP53 gene encodes the key transcriptional factor acting in response to genotoxic stress. Defects in p53 pathway impair correct DNA repair and apoptosis and result in increased genomic instability and abnormal cell proliferation. In various cancer types, p53 protein is most often inactivated due to mutation in the TP53 gene accompanied by deletion of the other allele (locus 17p.13.1). In CLL, TP53 defects have been observed in ∼5–10 % patients at diagnosis, with an increased frequency in progressive and chemo-refractory disease. Moreover, the negative clinical impact of sole TP53 mutations [in the absence of del (17) (p13.1)] has been shown in CLL [9, 10]; these sole TP53 mutations cannot be recognized by routinely used cytogenetic examination of 17p locus.
TP53 mutation analysis is becoming routine in research institutes and medical centers examining CLL patients; nevertheless, the methods applied and detection conditions vary considerably, and therefore, somewhat inconsistent results might be achieved [5–8, 14, 15]. To reduce the interlaboratory variability, the European Research Initiative on CLL (ERIC) has recently published criteria for TP53 analysis together with recommendation of the most suitable methodologies for TP53 mutation detection. These include (i) direct Sanger sequencing, (ii) denaturing high-performance liquid chromatography (DHPLC) or single-strand conformation analysis, (iii) functional analysis of separated alleles in yeast (FASAY), (iv) chip-based arrays, and (v) next generation sequencing (NGS) [14].
Since each of these methodologies shows some limitations and the optimal approach is still a matter of intensive discussion in CLL community, we present here a follow-up report to the ERIC recommendations [14] comparing the detection efficacy of DHPLC, FASAY, and the AmpliChip p53 Research Test. All three methods were employed in parallel to examine a cohort of 182 CLL patients; additionally, in 69 patients, the results were complemented by ultra-deep NGS.
Materials and methods
Patients’ cohort
The cohort examined included 182 CLL patients monitored and treated at the Department of Internal Medicine-Hematology and Oncology, University Hospital Brno in agreement with National Cancer Institute-sponsored Working Group guidelines [16, 17]. In order to collect a sufficient number of clinically relevant mutations, the patients with unfavorable prognosis including advanced disease stage, unmutated IGHV gene status, and/or del (17p) were preferentially selected (Table 1). The peripheral blood samples and buccal swabs were obtained between the years 2004–2014 under written informed consent according to the Declaration of Helsinki and University Hospital Brno Ethics Committee regulations.
TP53 mutation analysis using a combination of detection methods
Mutations in the TP53 gene were examined using FASAY, DHPLC, and the AmpliChip p53 Research Test in 182 CLL patients in parallel. Since both FASAY and DHPLC represent highly sensitive but only prescreening detection methods [10, 14, 18], mutations in the samples manifesting TP53 variations in the analyses were confirmed by conventional Sanger sequencing. Moreover, in 69 patients examined (32 TP53-wild-type and 37 TP53-mutated), the results were also verified using ultra-deep NGS. The nucleic acid samples were isolated from peripheral blood mononuclear cells (Histopaque®-1077; Sigma Aldrich) and/or separated CD19+ cells of the patients analyzed (Ficoll-Paque PLUS; GE Healthcare, complemented with RosetteSep™ kits; Stemcell™ Technologies). In the two cases harboring TP53 mutations without functional impact, as assessed by FASAY and the International Agency for Research on Cancer (IARC) TP53 database [19], the corresponding buccal swabs were sequenced to verify the somatic origin of the mutations.
Yeast functional analysis
The experimental setting of FASAY with appropriate modifications for CLL patients including details about the optimization process was previously described [10, 18]. Briefly, the cDNA amplified using proof-reading Pfu DNA polymerase (exons 4–10; Agilent Technologies, Inc.) was transformed into ADE2− LEU2− modified yeast strain together with an open reading frame expression vector containing ADE2 gene under the control of p53-responsive promoter and selectable LEU marker. In a medium deficient for adenine, transcripts coding for transcriptionally inactive p53 give rise to easily distinguishable growth-restricted red colonies in contrast to white ones harboring wild-type p53. The background of FASAY was determined at 10 % of red colonies covering the alterations caused by sample processing, low input RNA quality, or PCR amplification errors [18]. The presence of TP53 mutations was confirmed using direct Sanger sequencing of corresponding DNA isolated from the red colonies (Big Dye chemistry; Applied Biosystems).
DHPLC and DNA sequencing
Mutational screening using DHPLC (Varian Inc.) encompassed exons 4–9 and bordering intron TP53 gene sequences. DNA samples were amplified according to the IARC recommendations [19] using proof-reading Optimase Polymerase (Transgenomic) to minimize artificial mismatches. Each sample was prepared in duplicate; one aliquot was mixed with 25 % of TP53-wild-type DNA to efficiently recognize fully selected mutations. After renaturation, the samples harboring sequence variations were distinguished from wild-type PCR products based on the different column-retention time upon partially denaturing conditions (available upon request).
Among 728 PCR products tested using DHPLC, 154 TP53 wild-type amplicons and all 172 TP53-aberrant amplicons were sequenced on an ABI PRISM® 3700 Genetic Analyzer (Applied Biosystems) and compared to the reference sequences [GenBank: NG_017013.2; NC_000017: c7531642-7512445]. The functional effect of the TP53 variants was verified using the IARC TP53 database [19] to distinguish polymorphisms and functional variants from deleterious mutations. The estimated threshold for direct Sanger sequencing was ∼10 % of mutated DNA (Mutation Surveyor DNA Variant Analysis Software; Softgenetics®).
AmpliChip p53 Research Test
TP53 gene analysis using the AmpliChip p53 Research Test, a microarray-based assay, was performed according to the manufacturer̕s instructions in cooperation with Roche Molecular Systems Inc. Pleasanton, CA. Briefly, exons 2–11 of the TP53 gene (including splicing sites) were amplified in two master mix reactions. The generated PCR products were fragmented with DNase I, end-labeled using TdT enzyme, and then loaded onto the Amplichip p53 microarray containing oligonucleotide probes corresponding to the wild-type and mutant TP53 sequences. Hybridization, staining, and washing procedures were performed using a GeneChip® Fluidics Station 450 (Affymetrix); the microarrays were scanned on an Affymetrix GeneChip® Scanner 3000 7G. Data were analyzed, and results were evaluated by Roche Molecular Systems Inc. Pleasanton, CA.
The AmpliChip p53 Research Test was designed to detect single base pair substitutions and single nucleotide deletions in the whole coding region and splice-sites of the TP53 gene [8, 20]. The declared threshold for the TP53-specific resequencing microarray was ∼25 % of mutated DNA.
Ultra-deep NGS
Ultra-deep next generation sequencing of TP53 exons 4–10 and corresponding splicing sites was performed on a MiSeq platform (Illumina). The experimental design and reaction conditions followed the manufacturer recommendations. Briefly, DNA samples were amplified using proof-reading Q5® High-Fidelity DNA Polymerase (New England Biolabs; primers’ sequences in Online Resource, Table 1S). Each PCR product was purified separately with Agencourt® AMPure® XP (Beckman Coulter) and quantified using a Qubit® dsDNA HS Assay Kit (Life Technologies). The purified amplicons were mixed at equimolar ratios according to the number of molecules and diluted to a final amount of 1 ng. The indexed paired-end library was prepared with a Nextera XT DNA Sample Preparation Kit (Illumina) and sequenced using a MiSeq Reagent Kit v2 (300 cycles; Illumina). The achieved median per base coverage was 27,538 reads (range 2096–88,976).
To call the sequence variants, an in-house bioinformatics pipeline was established [21]. Sequencing reads were preprocessed and aligned to the reference sequence [GenBank: GRCh37.p9] using CLC Genomic Workbench version 6.0.4 (CLC Bio). Variant calling was performed using the deepSNV R-package [21] with a statistical approach applying the shearwater algorithm to compute Bayes classifiers based on a betabinomial model [22, 23]. From the reproducibility test, we disclosed that we were able to reliably distinguish point mismatches and ≥2 nucleotide insertions/deletions (indels) at the level of 0.2 % of variant reads, and 1-nucleotide deletions at the level of 1 % of variant reads as these may be artificially introduced during the sequencing and alignment process [21].
Cytogenetic analysis using I-FISH and SNP-based arrays
The del (17) (p13.1) was examined together with other common cytogenetic aberrations [i.e., del (13) (q14), +12 and del (11) (q22.3)] in 179/182 patients investigated using interphase fluorescence in situ hybridization (I-FISH) with locus-specific probes (Abbott Molecular Inc.) [10]. In 15 of the TP53-mutated patients, TP53 gene abnormalities or copy-neutral loss of heterozygozity (cn-LOH) were analyzed using Cytogenetics Whole-Genome 2.7 M Array (n = 9; Affymetrix) and CytoScan® High Density Array (n = 6; Affymetrix). The cytogenetic array analyses were performed according to the manufacturer̕s instructions.
Results
Frequency and localization of TP53 mutations
The presence of TP53 mutations was examined in all patients (n = 182) using three methods in parallel: FASAY and DHPLC both complemented with direct Sanger sequencing and the AmpliChip p53 Research Test. Moreover, ultra-deep NGS was performed in 32 TP53 wild-type and 37 TP53-mutated patients. Only TP53 mutations identified using at least two different methodologies were considered as true variants.
In total, 79 TP53 mutations in 31 % (57/182) of patients were detected; multiple TP53 mutations were found in 17 of them (2–3 mutations per patient; Online Resource, Table 2S). The silent mutations and intron variants (with the exception of splice-site mutations) were not further considered since their prognostic impact is mostly unknown and it is generally supposed to be minimal. The observed high TP53 mutation frequency attributes to the unfavorable profile of analyzed cohort (Table 1).
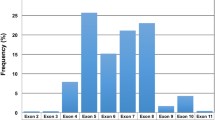
Among all TP53 mutations detected, missense substitutions predominated (n = 54; 68 %), followed by frameshift mutations (n = 12; 15 %), splice-site mutations (n = 6; 8 %), in-frame deletions (n = 4; 5 %), and nonsense mutations (n = 3; 4 %). The occurrence of the mutations identified was limited to exons 4–10 (between amino acids 109–346; Fig. 1). In exons 2, 3, and 11, which were analyzed using the AmpliChip p53 Research Test only, no TP53 mutation was detected confirming that they are rare in these loci [8, 24].

Localization and frequency of 79 identified TP53 mutations according to the codon distribution including splice-sites. In case of deletions and insertions, only the first affected codon was considered. The region examined using all three detection methods spanned codons 35–331. The most frequently mutated loci are shown
The most frequently mutated regions included the well-known codons 234 (n = 3), 248 (n = 3), 249 (n = 3), 273 (n = 4), 277 (n = 4) [19, 24], and unexpectedly also the splice site c.673-2 nt (n = 4) leading to the aberrant splicing of exon 7. Similar to the TP53 mutation pattern in other cancers [19], 95 % of mutations (n = 75) occurred within the p53 DNA-binding domain (codons 101–300 including splice-sites). The remaining 4 mutations were found outside this region, namely in exons 9 (n = 3) and 10 (n = 1) (Figs. 1 and 2). Interestingly, we observed a high proportion of non-missense TP53 mutations in exon 6 (6/12 mutations detected in this region); in contrast, in exon 7, only missense substitutions were found (n = 15) (Fig. 2).

Localization and frequency of 79 detected TP53 mutations according to the exon-intron distribution. In case of introns, only splice-site mutations were considered. The region examined using all detection methods spanned exons 4–9
Association between TP53 mutations and del (17) (p13.1)
Using I-FISH, del (17) (p13.1) was observed in 18 % (32/179) of patients examined; in one patient (no. 287), the del (17) (p13.1) was selected later during the disease course in addition to the already present TP53 mutation. The del (17) (p13.1) significantly correlated with the presence of TP53 mutations (P < 0.0001); 81 % (26/32) of patients with del (17) (p13.1) harbored TP53 mutations, and reciprocally, in 46 % (26/56) of patients with TP53 mutations, the del (17) (p13.1) was found. Overall, the concurrent presence of TP53 mutations and del (17) (p13.1) was observed in 42 % (26/62) of patients with TP53 defects (Fig. 3).

Characterization of CLL patients according to the presence and type of TP53 defects. Concurrent data on the 17p13.1 locus deletion (examined by I-FISH) and TP53 mutational status (investigated using FASAY, DHPLC, and the AmpliChip p53 Research Test) were available in 179 of 182 analyzed patients. The presence of cn-LOH assessed by SNP-based arrays and direct sequencing and the occurrence of additional low-level TP53 mutations detected using ultra-deep NGS only are not shown
Within the group harboring TP53 mutation (s) but not del (17) (p13.1) (n = 30), two or more different TP53 mutations were found in 33 % (10/30) of patients. Using FASAY, which is based on subcloning, we were able to confirm that in all cases with two TP53 mutations, they were present on separate alleles. However, unless single-cell analysis is used, it is not possible to decide whether the mutations are present in different subclones or if both TP53 alleles in one cell are affected. In the remaining 20 patients without del (17) (p13.1), a single TP53 mutation was observed (Fig. 3).
To investigate the occurrence of cn-LOH or additional defects in the TP53 gene, the SNP-based arrays were performed in available samples from 15/30 patients with sole TP53 mutation (s). Using this approach, the deletion of exon 11 in the TP53 gene was detected in one patient (patient no. 373). The cn-LOH was observed in 6/30 TP53-mutated patients without del (17) (p13.1) and corresponded to the DNA sequencing results (mutation proportion >50 % of DNA determined using Mutation Surveyor DNA Variant Analysis Software; Online Resource, Table 2S).
Screening of TP53 mutations using FASAY
Mutation analysis using FASAY detected 78 % (62/79) of TP53 mutations in 93 % (53/57) of TP53-mutated patients. The causal TP53 mutations leading to transcriptional p53-inactivation were identified by DNA sequencing from FASAY-generated red colonies. We particularly noticed a decreased ability of FASAY to detect truncating TP53 mutations presumably causing nonsense-mediated mRNA decay (nonsense, frameshift, and splice-site mutations); only 9/21 of these mutations were found (Table 2). However, 10/12 undetected truncating mutations accompanied other TP53 mutation (s) identified by FASAY; therefore, the patients carrying these mutations were recognized as TP53-mutated (Online Resource, Table 2S). Moreover, owing to the high detection efficacy of FASAY followed by cloned DNA sequencing (background ∼10 % of red colonies), all 4 in-frame deletions and 49/54 missense substitutions were detected (Table 2). Among five unidentified missense substitutions, two of them (p.Q317K, p.R283C) were TP53 variants with preserved transcriptional activity (patient no. 194, 868; Online Resource, Table 2S). In line with its functional read-out, these mutations were present in white FASAY colonies and were not therefore recognized in the original analysis. In these cases, the paired nontumor DNA was analyzed using Sanger sequencing, and the germinal origin of the mutations was proven. The other three undetected missense substitutions were present in a small proportion of cancer cells (<10 % of DNA) and accompanied dominant TP53 mutation (s) recognized by FASAY (patient no. 480, 653, 414; Online Resource, Table 2S).
Analysis of TP53 mutations using DHPLC and DNA sequencing
Mutational screening based on DHPLC combined with direct Sanger sequencing detected 87 % (69/79) of TP53 mutations in 95 % (54/57) of TP53-mutated patients; in 95 DHPLC-abnormal PCR products, annotated TP53 polymorphisms were found (p.P36P; p.R72P; p.R213R). DHPLC supplemented with Sanger sequencing identified all frameshift mutations, in-frame deletions, and nonsense mutations (n = 19; Table 2). On the other hand, this approach not detected 9/54 missense substitutions and 1/6 splice-site mutations most likely due to the lower detection efficacy of conventional Sanger sequencing utilized as a confirmatory method (∼10 % of mutated DNA; Online Resource, Table 2S). None of the 154 amplicons determined as TP53 wild-type in DHPLC analysis carried TP53 variations according to Sanger sequencing.
Detection of TP53 mutations using the AmpliChip p53 Research Test
The AmpliChip p53 Research Test was developed as a specific array for TP53 mutation detection and was performed in collaboration with Roche Molecular Systems Inc. In total, 71 % (56/79) of TP53 mutations in 81 % (46/57) of TP53-mutated patients were detected using this array (Table 2). Considering these results in the context of the detection limits declared (threshold 25 % of mutated DNA, recognition of single base pair substitutions and deletions), the AmpliChip p53 Research Test correctly identified 91 % (31/34) of the mutations in 91 % (29/32) of TP53-mutated patients. The TP53 mutations undetected included single nucleotide deletions (n = 2; patient no. 91, 399) and one missense substitution (n = 1; patient no. 6007). Interestingly, among the remaining 45 TP53 mutations that were present under the declared limit of the AmpliChip p53 Research Test detection, this approach recognized 25 of them (19 missense substitutions, 3 splice-site mutations, 2 multiple nucleotide deletions, and 1 duplication; Online Resource, Table 2S). The spectrum and frequency of TP53 polymorphisms identified using this method were exactly the same as in the DHPLC analysis combined with DNA sequencing (see above).
Confirming the TP53 mutations’ presence using ultra-deep NGS
To verify the results obtained using FASAY, DHPLC, and/or the AmpliChip p53 Research Test, ultra-deep NGS on the Illumina MiSeq platform was performed in 69 patients. In total, 58 TP53 mutations found by the three tested methods in 37 patients were examined, and the presence of all these mutations was confirmed using ultra-deep NGS. Moreover, in 24 of these patients, additional low-level TP53 mutations occurring below 10 % of DNA were detected (Online Resource, Table 2S) [21].
In the 32 TP53 wild-type patients analyzed, the ultra-deep NGS revealed very low level TP53 mutations occurring under the detection limit of all tested methods in 8 of them (mutation proportion 0.2–3.8 %). In 5 of these patients, an expansion of the particular TP53 mutations was noticed using FASAY in the available follow-up sample (data not shown) [21].
Minor proportion TP53 mutations
Among 79 TP53 mutations detected, 16 low-level mutations present in <10 % of DNA were observed in 26 % (15/57) of TP53-mutated patients using a combination of FASAY, DHPLC, and the AmpliChip p53 Research Test. As assessed by ultra-deep NGS and FISH analyses, 2 of these patients carried the low-level TP53 mutation as a single abnormality; in 13 patients, the minor proportion TP53 mutations were accompanied by other TP53 defects [TP53 mutation (s) and/or del (17p)]. The low-level TP53 mutations identified included 12 missense substitutions, 1 frameshift mutation, 2 splice-site mutations, and 1 in-frame deletion. The presence of all these mutations was independently confirmed by ultra-deep NGS (Online Resource, Table 2S).
Comparing the efficiency of the methods applied with respect to the minor proportion mutations’ detection, FASAY achieved the best results, identifying 63 % (10/16) of the low-level mutations, and importantly, all 15 of the patients examined were recognized as TP53-mutated (Table 3). DHPLC combined with Sanger sequencing showed a similar detection efficacy to the AmpliChip p53 Research Test in these cases (recognition of 50 and 50 % minor proportion TP53 mutations and identification 12 and 10 patients as TP53-mutated, respectively; Table 3).
Discussion
The independent poor prognostic impact of TP53 defects (gene deletion and/or mutations) on disease course and patient prognosis has been repeatedly proven in CLL [5–8, 25, 26]. The proper assessment of TP53 status is especially crucial in CLL therapy management as patients with TP53 defects should be considered for allogeneic stem cell transplantation or enrollment to clinical trials testing new perspective drugs [2–4]. Regarding this, the detection limit of a particular methodology is important as the clinical impact of small TP53-mutated subclones has very recently been proven in CLL [11, 21]. Besides standardized investigation of TP53 allele deletion at 17p locus using interphase FISH, various approaches with different detection efficacy have been applied to examine TP53 mutations [5–8, 14, 15]. Since an optimal methodology has been intensively discussed in the CLL community during the last few years because of TP53 defects’ heterogeneity [14], we report here a single-center study reflecting on the limitations of the major methods recently used in TP53 mutation analysis.
To explore the disparity in TP53 mutation detection, we examined a cohort of 182 CLL patients enriched for unfavorable cases using FASAY, DHPLC, and the AmpliChip p53 Research Test in parallel. In total, 79 TP53 mutations were identified and verified in 57 (31 %) patients. The localization and spectrum of the detected mutations were comparable to other CLL cohorts, including, e.g., 2 nt deletion in codon 209 [19, 24]. Of note, we observed quite a high frequency of TP53 mutations in the splice-site c.673-2 nt (n = 4). Mutations in this position lead to the aberrant splicing of TP53 exon 7 and have been sporadically observed in solid tumors, e.g., in lung or urinary tract cancers [19, 27].
TP53 gene analysis generally focuses on exons 4–10 and the corresponding splice-sites, where up to 95 % of mutations is supposed to be detected [19, 24]. In CLL, the presence of TP53 mutations in exons 2, 3, and 11 has been shown to be extremely rare, and their examination is not recommended in routine practice [8, 14]. No mutation in this region was also detected in our patients using the AmpliChip p53 Research Test.
In accordance with the reported results [7, 9, 10, 24], the presence of TP53 mutations was significantly associated with del (17) (p13.1) in our cohort (P < 0.0001). However, in 30 TP53-defected patients, the sole TP53 mutation (s) occurred without del(17) (p13.1). In addition to the dominant TP53 mutations detected, minor proportion TP53 mutations (present in <10 % of DNA) were observed in 16 TP53-mutated patients using FASAY, DHPLC, and the AmpliChip p53 Research Test. Nevertheless, due to the low mutation load, the detection of minor proportion TP53 mutations might be problematic using conventional methods, and a more sensitive approach, e.g., next generation sequencing should be applied in these cases [11, 21].
In addition to somatic mutations leading to p53 dysfunction, two germinal missense TP53 substitutions with a preserved p53 transcriptional-activation function [19] were identified in two patients using DHPLC and the AmpliChip p53 Research Test. Q317K, the first of the TP53 mutations identified, has been sporadically observed in some solid tumors [19]. The second, R283C, has already been detected in a CLL patient, who had acquired the mutation after allogeneic stem cell transplantation from the sibling and the mutation was proven to be present in the donor DNA without any effect on donor health [28]. The impact of these mutations and other functional TP53 variants including germinal mutations on CLL prognosis is highly improbable, and their reporting without the appropriate description might be misleading.
TP53 mutational analysis using FASAY, DHPLC, and the AmpliChip p53 Research Test revealed 93 %, 95 %, and 81 % of TP53-mutated patients, respectively. Despite the high detection efficacy, some mutations may escape identification using any of these methods [14] due to the TP53 mutations’ heterogeneity. Direct Sanger sequencing is considered to be the gold standard for TP53 mutation analysis [14]. Nevertheless, the low sensitivity of this approach, generally reaching ∼20 % of mutated DNA and a relatively high direct cost per sample, has resulted in a combination of Sanger sequencing with prescreening methods. Among them, DHPLC and FASAY are frequently used for TP53 testing in CLL as they are able to detect even subclonal TP53 mutations [8, 10, 14].
Considering the limits of the methods examined, FASAY detected fewer truncating TP53 mutations leading to nonsense-mediated mRNA decay than DHPLC and the AmpliChip p53 Research Test. On the other hand, only FASAY identified all 15 patients carrying minor proportion TP53 mutations. FASAY can also recognize patients with multiple low-level TP53 mutations in the absence of any dominant TP53 mutation as the overall percentage of red colonies equals the sum of all present mutations [29]. In addition, only deleterious TP53 mutations leading to the transcriptional p53-inactivation and therefore a presumed negative impact on disease prognosis are identified using FASAY [10, 18].
DHPLC complemented with DNA sequencing showed the best efficacy from the three methods tested for non-missense TP53 mutations detection. However, utilizing direct Sanger sequencing as a confirmatory method in DHPLC analysis somewhat reduces the sensitivity of this approach [8]. Of note, using prescreening methods such as DHPLC and FASAY, the detection limit reached strongly depends on the input sample quality and the precise experimental setting optimization [8, 10, 18], which may be relatively laborious. Since false positive or false negative results caused by inadequate sample preparation and processing might be produced by these methods, it is recommended to perform external data validation in cooperation with centers experienced in TP53 mutational analysis [14].
In contrast, it is not necessary to optimize any ready-to-use method for TP53 analysis such as the AmpliChip p53 Research Test, which has recently been tested especially in leukemia and breast cancer patients [8, 15, 20]. However, this approach fails to recognize insertions and multiple nucleotide deletions as the array is not designed for their detection [15].
In view of the reported limitations in each method tested [14], utilizing NGS seems to be a promising approach for TP53 mutation detection. In our study, the ultra-deep NGS confirmed the presence of all TP53 mutations assessed; of note, this methodology enabled the detection of additional low-level TP53 mutations in CLL cells, which might be the subject of further clonal selection [11–13, 21, 30, 31]. However, using NGS, determining the noise level is important as errors might be induced during PCR amplification of the samples [11, 21]. Despite the obvious benefits such as sensitivity and time efficiency, the expensive laboratory equipment and high direct costs together with the necessity of a background in bioinformatics still represent the major limitations of wider NGS utilization in TP53 mutation analysis [14].
Since TP53 gene analysis itself cannot resolve all CLL treatment-refractory cases [32], a lot of functional tests have been developed to study ATM-p53 DNA-damage-response pathway impairment [33]. Among them, the most clinically applicable assays include the following: (i) p53-target genes’ expression analysis (e.g., CDKN1A, MIR34A) using real-time polymerase chain reaction [32, 34]; (ii) p53-regulated apoptotic gene expression examination (e.g., BAX, PUMA, CD95) using reverse transcription-multiplex ligation-dependent probe amplification [35]; (iii) p53-p21 protein level detection using Western blot or fluorescence activated cell sorting [34–36]. However, in the case of low sample purity or when a small proportion of TP53-mutated clones or TP53 truncating mutations are present, the sensitivity of the functional tests have been reported to be considerably reduced [35, 36]; therefore, their routine utilization is rather questionable [14, 35].
We conclude that DHPLC or FASAY followed by mutation presence confirmation and identification using direct Sanger sequencing represent suitable methods for TP53 mutation analysis, since both approaches detected TP53 mutations in more than 90 % of TP53-mutated patients. Owing to the adverse prognostic impact of TP53 mutations on CLL prognosis, it is strongly recommended to check the functionality of mutations and the frequency of their occurrence using the IARC TP53 database [19] or The TP53 mutant Web site [27]. In the near future, next generation sequencing including commercially available assays is likely to become a standard approach for TP53 mutation analysis as its costs are supposed to decrease and appropriate statistical tools are being developed and widely tested.
References
Butler T, Gribben JG. Biologic and clinical significance of molecular profiling in chronic lymphocytic leukemia. Blood Rev. 2010;24(3):135–41.
Pettitt AR, Jackson R, Carruthers S, Dodd J, Dodd S, Oates M, et al. Alemtuzumab in combination with methylprednisolone is a highly effective induction regimen for patients with chronic lymphocytic leukemia and deletion of TP53: final results of the national cancer research institute CLL206 trial. J Clin Oncol. 2012;30(14):1647–55.
Moreno C, Montserrat E. New prognostic markers in chronic lymphocytic leukemia. Blood Rev. 2008;22(4):211–9.
Hillmen P. Using the biology of chronic lymphocytic leukemia to choose treatment. Hematology Am Soc Hematol Educ Program. 2011;2011:104–9.
Gonzalez D, Martinez P, Wade R, Hockley S, Oscier D, Matutes E, et al. Mutational status of the TP53 gene as a predictor of response and survival in patients with chronic lymphocytic leukemia: results from the LRF CLL4 trial. J Clin Oncol. 2011;29(16):2223–9.
Zenz T, Eichhorst B, Busch R, Denzel T, Häbe S, Winkler D, et al. TP53 mutation and survival in chronic lymphocytic leukemia. J Clin Oncol. 2010;28(29):4473–9.
Rossi D, Cerri M, Deambrogi C, Sozzi E, Cresta S, Rasi S, et al. The prognostic value of TP53 mutations in chronic lymphocytic leukemia is independent of Del17p13: implications for overall survival and chemorefractoriness. Clin Cancer Res. 2009;15(3):995–1004.
Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, et al. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia. 2009;23(1):117–24.
Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, Benner A, et al. Monoallelic TP53 inactivation is associated with poor prognosis in chronic lymphocytic leukemia: results from a detailed genetic characterization with long-term follow-up. Blood. 2008;112(8):3322–9.
Malcikova J, Smardova J, Rocnova L, Tichy B, Kuglik P, Vranova V, et al. Monoallelic and biallelic inactivation of TP53 gene in chronic lymphocytic leukemia: selection, impact on survival, and response to DNA damage. Blood. 2009;114(26):5307–14.
Rossi D, Khiabanian H, Spina V, Ciardullo C, Bruscaggin A, Famà R, et al. Clinical impact of small TP53 mutated subclones in chronic lymphocytic leukemia. Blood. 2014;123(14):2139–47.
Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell. 2013;152(4):714–26.
Schuh A, Becq J, Humphray S, Alexa A, Burns A, Clifford R, et al. Monitoring chronic lymphocytic leukemia progression by whole genome sequencing reveals heterogeneous clonal evolution patterns. Blood. 2012;120(20):4191–6.
Pospisilova S, Gonzalez D, Malcikova J, Trbusek M, Rossi D, Kater AP, et al. ERIC recommendations on TP53 mutation analysis in chronic lymphocytic leukemia. Leukemia. 2012;26(7):1458–61.
Chiaretti S, Tavolaro S, Marinelli M, Messina M, Del Giudice I, Mauro FR, et al. Evaluation of TP53 mutations with the AmpliChip p53 research test in chronic lymphocytic leukemia: correlation with clinical outcome and gene expression profiling. Genes Chromosomes Cancer. 2011;50(4):263–74.
Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ, O’Brien S, et al. National Cancer Institute-sponsored Working Group guidelines for chronic lymphocytic leukemia: revised guidelines for diagnosis and treatment. Blood. 1996;87(12):4990–7.
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Döhner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the international workshop on chronic lymphocytic leukemia updating the national cancer institute-working group 1996 guidelines. Blood. 2008;111(12):5446–56.
Smardová J, Pavlová S, Koukalová H. Determination of optimal conditions for analysis of p53 status in leukemic cells using functional analysis of separated alleles in yeast. Pathol Oncol Res. 2002;8(4):245–51.
Petitjean A, Mathe E, Kato S, Ishioka C, Tavtigian SV, Hainaut P, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum Mutat. 2007;28(6):622–9.
Baker L, Quinlan PR, Patten N, Ashfield A, Birse-Stewart-Bell LJ, McCowan C, et al. p53 mutation, deprivation and poor prognosis in primary breast cancer. Br J Cancer. 2010;102(4):719–26.
Malcikova J, Stano-Kozubik K, Tichy B, Kantorova B, Pavlova S, Tom N, et al. Detailed analysis of therapy-driven clonal evolution of TP53 mutations in chronic lymphocytic leukemia. Leukemia. 2014.
Gerstung M, Beisel C, Rechsteiner M, Wild P, Schraml P, Moch H, et al. Reliable detection of subclonal single-nucleotide variants in tumour cell populations. Nat Commun. 2012;3:811.
Gerstung M, Papaemmanuil E, Campbell PJ. Subclonal variant calling with multiple samples and prior knowledge. Bioinformatics. 2014;30(9):1198–204.
Zenz T, Vollmer D, Trbusek M, Smardova J, Benner A, Soussi T, et al. TP53 mutation profile in chronic lymphocytic leukemia: evidence for a disease specific profile from a comprehensive analysis of 268 mutations. Leukemia. 2010;24(12):2072–9.
Döhner H, Fischer K, Bentz M, Hansen K, Benner A, Cabot G, et al. p53 gene deletion predicts for poor survival and non-response to therapy with purine analogs in chronic B-cell leukemias. Blood. 1995;85(6):1580–9.
Trbusek M, Smardova J, Malcikova J, Sebejova L, Dobes P, Svitakova M, et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol. 2011;29(19):2703–8.
Soussi T, Hamroun D, Hjortsberg L, Rubio-Nevado JM, Fournier JL, Béroud C. MUT-TP53 2.0: a novel versatile matrix for statistical analysis of TP53 mutations in human cancer. Hum Mutat. 2010;31(9):1020–5.
Pekova S, Mazal O, Cmejla R, Hardekopf DW, Plachy R, Zejskova L, et al. A comprehensive study of TP53 mutations in chronic lymphocytic leukemia: Analysis of 1287 diagnostic and 1148 follow-up CLL samples. Leuk Res. 2011;35(7):889–98.
Malcikova J, Smardova J, Pekova S, Cejkova S, Kotaskova J, Tichy B, et al. Identification of somatic hypermutations in the TP53 gene in B-cell chronic lymphocytic leukemia. Mol Immunol. 2008;45(5):1525–9.
Jethwa A, Hüllein J, Stolz T, Blume C, Sellner L, Jauch A, et al. Targeted resequencing for analysis of clonal composition of recurrent gene mutations in chronic lymphocytic leukaemia. Br J Haematol. 2013;163(4):496–500.
Ouillette P, Saiya-Cork K, Seymour E, Li C, Shedden K, Malek SN. Clonal evolution, genomic drivers, and effects of therapy in chronic lymphocytic leukemia. Clin Cancer Res. 2013;19(11):2893–904.
Zenz T, Häbe S, Denzel T, Mohr J, Winkler D, Bühler A, et al. Detailed analysis of p53 pathway defects in fludarabine-refractory chronic lymphocytic leukemia (CLL): dissecting the contribution of 17p deletion, TP53 mutation, p53-p21 dysfunction, and miR34a in a prospective clinical trial. Blood. 2009;114(13):2589–97.
Te Raa GD, Malčiková J, Mraz M, Trbusek M, Le Garff-Tavernier M, Merle-Béral H, et al. Assessment of TP53 functionality in chronic lymphocytic leukaemia by different assays; an ERIC-wide approach. Br J Haematol. 2014;167(4):565–9.
Pozzo F, Dal Bo M, Peragine N, Bomben R, Zucchetto A, Rossi F, et al. Detection of TP53 dysfunction in chronic lymphocytic leukemia by an in vitro functional assay based on TP53 activation by the non-genotoxic drug Nutlin-3: a proposal for clinical application. J Hematol Oncol. 2013;6:83.
Mohr J, Helfrich H, Fuge M, Eldering E, Bühler A, Winkler D, et al. DNA damage-induced transcriptional program in CLL: biological and diagnostic implications for functional p53 testing. Blood. 2011;117(5):1622–32.
Lin K, Adamson J, Johnson GG, Carter A, Oates M, Wade R, et al. Functional analysis of the ATM-p53-p21 pathway in the LRF CLL4 trial: blockade at the level of p21 is associated with short response duration. Clin Cancer Res. 2012;18(15):4191–200.
Acknowledgments
The authors would like to thank ERIC steering board for support, Hana Skuhrova-Francova, Lenka Jurackova and Jitka Kabathova for help with experimental analysis and Matthew Smith for language editing. This study was supported by VaVPI project MSMT CR CZ.1.05/1.1.00/02.0068 of Central European Institute of Technology (CEITEC) and projects of the Internal Grant Agency, Ministry of Health, Czech Republic IGA MZ CR NT13493-4/2012 and NT13519-4/2012.
Conflict of interest
The authors declare that they have no conflict of interests. Sim Truong and Nancy Patten are employed by Roche Molecular Systems, Inc.
Author information
Authors and Affiliations
Corresponding author
Additional information
Barbara Kantorova and Jitka Malcikova contributed equally to this study
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOCX 50 kb)
Rights and permissions
About this article

Cite this article
Kantorova, B., Malcikova, J., Smardova, J. et al. TP53 mutation analysis in chronic lymphocytic leukemia: comparison of different detection methods. Tumor Biol. 36, 3371–3380 (2015). https://doi.org/10.1007/s13277-014-2971-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2971-0