
Abstract
Glycolysis has been shown to be required for the cell growth and proliferation in several cancer cells. However, prostate cancer cells were accused of using more fatty acid than glucose to meet their bioenergetic demands. The present study was designed to evaluate the involvement of hexokinase and CPT-1 in the cell growth and proliferation of human prostate cancer cell lines, PC3, and LNCaP-FGC-10. Hexokinase and CPT-1 activities were examined in the presence of different concentrations of their inhibitors, lonidamine and etomoxir, to find the concentration of maximum inhibition ([I max]). To assess cell viability and proliferation, dimethylthiazol (MTT) assay was carried out using [I max] for 24, 48, and 72 h on PC3 and LNCaP cells. Apoptosis was determined using annexin-V, caspase-3 activity assay, Hoechst 33258 staining, and evaluation of mitochondrial membrane potential (MMP). Moreover, ATP levels were measured following lonidamine and etomoxir exposure. In addition, to define the impact of exogenous fatty acid on the cell growth and proliferation, CPT-1 activity was evaluated in the presence of palmitate (50 μM). Hexokinase and CPT-1 activities were significantly inhibited by lonidamine [600 μM] and etomoxir [100 μM] in both cell lines. Treatment of the cells with lonidamine [600 μM] resulted in a significant ATP reduction, cell viability and apoptosis, caspase-3 activity elevation, MMP reduction, and appearance of apoptosis-related morphological changes in the cells. In contrast, etomoxir [100 μM] just decreased ATP levels in both cell lines without significant cell death and apoptosis. Compared with glucose (2 g/L), palmitate intensified CPT-1 activity in both cell lines, especially in LNCaP cells. In addition, activity of CPT-1 was higher in LNCaP than PC3 cells. Our results suggest that prostate cancer cells may metabolize glucose as a source of bioenergetic pathways. ATP could also be produced by long-chain fatty acid oxidation. In addition, these data might suggest that LNCaP is more compatible with palmitate.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
According to Warburg phenomenon, cancer cells consume elevated amounts of glucose through aerobic glycolysis even in the presence of adequate oxygen [1]. This enhanced glucose uptake and utilization constitutes some advantages for tumor growth, such as survival in condition of fluctuating oxygen tension [2] and generation of lactic acid [3], which favor tumor invasion [4]. In addition, metabolizing glucose through the pentose phosphate pathway generates NADPH which, as an antioxidant, confers protection against unfriendly microenvironment and chemotherapeutic agents [5]. The most important advantage of Warburg phenomenon is that some of the intermediates of glycolysis are shunted toward anabolic reactions, e.g., DHAP for triacylglycerol and phospholipid synthesis or pyruvate for malate and alanine synthesis [5].
However, regarding glucose utilization by prostate cancer cells, there are some contradictions as follow:
2-Deoxyglucose (hexokinase inhibitor) induces apoptosis in LNCaP cells [6], and inhibition of glucose uptake sensitizes PPC-1, but not DU-145, to FAS-induced cell death [7]. In contrast, it was reported that LNCaP cells grow and proliferate at control rates even in the presence of 0.05 g/l glucose, but DU-145 cells are more sensitive to glucose deprivation, and their growth decreased significantly even in the medium containing 0.5 g/l glucose [8].
As rapid cell proliferation requires increased energy demand, if glucose consumption is not increased proportionally, alternative metabolic source, especially fatty acid oxidation, is needed to provide bioenergy for this abnormal proliferation. According to some reports, fatty acid oxidation may be the dominant bioenergetic pathway in prostate cancer [9], and in LNCaP and PC3, uptake of palmitate was at least tenfold higher than that of glucose [10]. Elevated activity of some enzymes of β-oxidation pathway, such as α-methylacyl-CoA racemase [11–15], and upregulation of peroxisomal D-bifunctional protein (involved in branched-chain fatty acid β-oxidation) [16] provide more evidences.
Therefore, the present study was designed to evaluate the effect of two critical enzymes in glycolysis and β-oxidation, i.e., hexokinase (HK) and carnitine–palmitoyl CoA transferase 1 (CPT-1), on the androgen-dependent and androgen-independent prostate cancer cell lines, LNCaP and PC3.
So we used chemical inhibitors of the HK and CPT-1 enzymes, lonidamine and etomoxir.
Lonidamine (1-[2, 4-dichlorobenzyl]-1Hindazole-3-carboxylic acid) is a small molecule that inhibits glycolysis and the inhibition of HK by lonidamine is well established [17]. Lonidamine stimulates aerobic glycolysis in normal differentiated cells but suppresses that of neoplastic cells [18]. This selective action of lonidamine on the tumor glycolysis pathway may be modulated through its inhibitory effect on mitochondrial-bound HK (HK type II) [19], which is present in considerable amounts on the outer membrane of tumor mitochondria but not in those of normal differentiated cells [20]. To inhibit CPT-1 enzyme, the cells were treated with etomoxir, 2-[6-(4-chlorophenoxy)hexyl]oxirane-2-carboxylate. etomoxir is absorbed in the intestine and transported, similar to fatty acids, to the tissues. Once converted to its CoA ester in the cells, it acts as a strong and irreversible inhibitor of mitochondrial CPT-1 [21], through which the transport of long-chain acyl-CoA into mitochondria is inhibited [22, 23].
Materials and methods
Materials
All materials used in the experiment were of high quality, appropriate for cell culture research and purchased as follow: RPMI-1640 (Sigma-Aldrich), fetal bovine serum (FBS) (Gibco). Cell culture plastic ware (Orange scientific), etomoxir (Sigma-Aldrich), lonidamine (Sigma-Aldrich), palmitic acid (Sigma-Aldrich), fatty acid-free BSA (Sigma-Aldrich), protease inhibitor cocktail (Roche), glucose 6 phosphate dehydrogenase (Sigma-Aldrich), NADP(Sigma), l-carnitine (Sigma), palmitoyl CoA (Sigma-Aldrich), DTBN (Sigma), dimethylthiazol (MTT) (Merck), Hoechst 33258 (Sigma–Aldrich), annexin-V-FITC apoptosis detection kit (Biovision), ApoSensor Cell Viability Assay Kit (Biovision), Oxoplate (gift from Precision Sensing GmbH), caspase-3 fluorometric assay kit (Biovision), JC-1 Mitochondrial Membrane Potential Assay Kit (Cayman chemical).
Cell culture and treatments
LNCaP-FGC-10 (LNCaP) and PC3 cell lines were obtained from the National Cell bank affiliated to Pasteur Institute (Tehran, Iran). LNCaP and PC3 cell lines were cultured in RPMI-1640 (Gibco) supplemented with 10 % fetal bovine serum (FBS, Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco), at 5 % CO2 and 95 % humidity at 37 °C. Where indicated, the cells were grown in glucose-free RPMI-1640, supplemented with BSA-conjugated palmitate.
Preparation of bovine serum albumin (BSA)-conjugated palmitate
Palmitate was used to assess the effect of exogenous fatty acid. BSA-conjugated palmitate was prepared by the method of Listenberger LL [24]. Briefly, 2 mM solution of palmitic acid in 0.01 M NaOH was incubated at 70 °C for 30 min. The resultant palmitate soap was conjugated with 0.34 mM fatty acid-free BSA solution while stirring at 37 °C for 1 h.
Total hexokinase activity
To determine the maximum inhibitory concentration ([I max]) of lonidamine, total hexokinase activity was measured using a glucose 6-phosphate dehydrogenase (G6PD)-coupled assay as described previously [25] with minor modifications. LNCaP and PC3 cells were cultured in RPMI-1640, containing 2 g/L glucose. After treatment of the cells with lonidamine at concentrations 300, 450, 600, 750, and 850 μM for 1 h, trypsinization, and centrifugation, the pellets were sonicated in ice-cold homogenization buffer (protease inhibitor, 45 mM Tris–HCl, 50 mM KH2PO4, 10 mM glucose, 11.1 mM monothioglycerol, 0.5 mM EDTA, 0.2 % (v/v) Triton X-100, pH 8.2). The final assay mixture consisted of 10 μl freshly lysed cell supernatant (obtained through centrifugation at 12,000×g, 10 min), 1 unit/ml G6PD, 0.5 mg/ml NADP, 6.7 mM ATP, 7 mM MgCl2, 4 mM glucose, 2.5 mM KH2PO4, 1 mM NaH2PO4, 11.1 mM monothioglycerol, 0.01 % (v/v) Triton X-100, 25 μM EDTA, and 45 mM Tris–HCl, pH 8.5. Hexokinase activity was determined by following the conversion of NADP to NADPH spectrophotometrically at 340 nm and defined as millimolars of NADPH per mg protein at 37 °C.
CPT-1 activity
To determine the [I max] of etomoxir on CPT-1, LNCaP and PC3 cells were treated with etomoxir 0, 25, 50, 100, 200, and 400 μM in the presence of glucose (2 g/L) or palmitate (50 μM) for 1 h. CPT-1activity in the cell homogenates were assayed spectrophotometrically by following the release of CoA-SH from palmitoyl-CoA using the general thiol reagent DTNB (5,5′-dithio-bis-(2-nitrobenzoic acid)) as described by Karlic et al. [26]. Briefly, cell homogenates were prepared in buffer (0.25 M sucrose, 1 mM EDTA, and protease inhibitors) followed by sonication and centrifugation at 12,000×g, 10 min. Reaction mixtures containing DTNB buffer and cell lysate were incubated at room temperature for 30 min, and the resulting absorbance at 405 nm was defined as background. To start reaction, substrates, i.e., palmitoyl-CoA (final concentration 100 μM) and l-carnitine solution (final concentration 1 mM) were added to the reaction mixture. Reaction mixtures were incubated for 30 min at 37 °C, and the absorbance was measured at 405 nm. The difference between absorbance with and without substrates measured CPT-1 activity which was defined as millimolars CoA-SH released per milligram protein.
MTT assay
The MTT assay was optimized for the LNCaP and PC3 cell lines in our experiments. Briefly, LNCaP and PC3 cells were incubated with etomoxir (in the presence of glucose or palmitate) or lonidamine at [I max] for 24, 48, and 72 h, and then 0.5 mg/ml of MTT was added to the culture medium for 4 h. DMSO was added, after gentle shaking and incubation for 15 min at 37 °C; absorbance was read at 570 nm using the microplate spectrophotometer system (TECAN™ sunrise model, Austria). Results were presented as percentage of the control value.
Intracellular ATP level
LNCaP and PC3 Cells were exposed to lonidamine or etomoxir at [I max] for 0, 1, 2, and 4 h. Following cell lysis by nuclear releasing buffer and subsequent adding ATP monitoring enzyme (both supplied by ApoSensor Cell Viability Assay Kit), luminescent intensity from each well was monitored on Bio-Tek Synergy HT Microplate Reader in the luminescence mode. Normalized to the protein concentration in each sample, ATP levels were calculated as a percentage of control.
Oxygen consumption rate
Oxygen consumption rate (OCR) was measured using oxoplate, according to the manufacture protocol as following:
The plate contains two different dyes; fluorescence intensity of the indicator dye depends on the oxygen content of the sample while the fluorescence intensity of the reference dye is independent of the oxygen content. Oxygen partial pressure can be calculated using intensity of the indicator/intensity of the reference ratio (IR) and two-point calibration; oxygen-free water (1 g/L of sodium sulfite, Calibrator 0) and air-saturated water (vigorously shook water for 2 min, calibrator 100), as follow:
Briefly, 50 μl of culture medium of control and etomoxir-treated cells (for 0, 1, 2 and 4 h) were transferred to the oxoplate and then fluorescent emissions were read at 650 nm (indicator) and 590 nm (reference) following excitation at 540 nm.
Annexin-V staining for determining cell death mechanism
To determine the mechanism of cell death following treatment of LNCaP and PC3 with lonidamine and etomoxir (in the presence of glucose or palmitate), 50,000 cells/well were seeded and treated with lonidamine and etomoxir at [I max]. Then the cells were harvested after 48 h through trypsinization, washed in PBS, and centrifuged at 200×g for 5 min. The pellet was then stained for 15 min at room temperature with annexin-V-FITC and PI (supplied by Annexin-V-FITC apoptosis detection kit) and examined using FACS Calibur flow cytometer (USA). Analyses were performed by the software supplied with the instrument.
Caspase-3 activity assay
Contribution of caspases-3 in the lonidamine or etomoxir-induced apoptosis was investigated using Caspase-3 Fluorometric Assay Kit according to the manufacturer’s instruction. Briefly, LNCaP and PC3 cells were incubated for 12, 24, 36, 48, and 72 h with lonidamine or etomoxir (in the presence of glucose or palmitate) at [I max]. After incubation of the cells with cell lysis buffer on ice for 30 min, 50 μl of cell lysate, 50 μl of 2× reaction buffer containing 10 mM DTT and DEVD-AFC substrate (50 μM final concentration) were mixed and incubated at 37 °C for 2 h. Samples were read in Bio-Tek Synergy HT Microplate Reader equipped with a 360/40 nm excitation and 528/20 nm emission filter. Fold-increase in caspases-3 activity/mg protein was determined by comparing the results with the level of the untreated control.
Morphological aspects of apoptosis
Nuclear morphology was assessed as described previously by Salami et al. [27]. Briefly, LNCaP and PC3 cells were plated in eight-well chamber slides and allowed to adhere. Then the cells were treated with lonidamine or etomoxir at [I max] for 48 h. The cells were fixed with methanol–acetic acid 3:1 (v/v) for 10 min, after which staining was carried out with Hoechst 33258 (10 μg/mL) at 37 °C in dark (15 min). Slides were then washed in distilled water and examined by fluorescence microscope (Micros, Austria). Apoptotic cells were defined on the basis of chromatin condensation and fragmentation, cytoplasm shrinkage and plasma membrane blabbing.
Mitochondrial potential measurements
JC-1 dye (Cayman’s JC-1 Mitochondrial Membrane Potential Assay Kit) was used to examine mitochondrial membrane potential (ΔΨm). In the cells with intact mitochondria, JC-1 accumulates in the mitochondria in proportion to ΔΨm as red fluorescent aggregates while, in depolarized mitochondria, red fluorescence fades and green fluorescence of JC-1 monomers predominates [28].
LNCaP and PC3 cells were plated and allowed to adhere. Then the cells were treated with lonidamine and etomoxir at [I max] for 12, 24, and 36 h. After incubation for 15 min at 37 °C in the presence of JC-1 dye and washing twice with assay buffer, fluorescence of JC-1 was quantified by the use of Bio-Tek Synergy HT Microplate Reader equipped with excitation and emission pairs 560/595 nm for red and 485/535 nm for green fluorescence.
ΔΨm was expressed as a ratio of red to green fluorescence. Lower ratio correlates with more positive ΔΨm [29]. Therefore, these ratios were used as an indicator of mitochondria health.
Statistical analysis
Non-parametric unpaired t test and ANOVA with Dunnett’s test were used respectively for comparison of two and more groups, using Graphpad Prism software 5. P < 0.05 was considered significant. All data are expressed as mean ± SD.
Results
Effect of lonidamine on total hexokinase activity
LNCaP and PC3 cell lines were treated with lonidamine at concentrations of 0, 300, 450, 600, 750, and 850 μM for 1 h, and then HK activity was determined. As shown in Fig. 1a, HK activity decreased in a dose-dependent manner in LNCaP (P = 0.0020) and PC3 (P = 0.0007). The first significant inhibitory effect was seen at 600 μM for LNCaP (46.19 %) and PC3 (48.15 %). Since there were no significant differences between 850 μM inhibition and that of 600 μM, this concentration of lonidamine was considered [I max] for next experiments.

Effect of various concentrations of lonidamine and etomoxir on the activity of hexokinase and CPT-1, respectively, a lonidamine significantly reduced hexokinase activity in a dose-dependent manner. LNCaP and PC3 were cultured in RPMI-1640 containing 2 g/L glucose and then treated with different concentrations of lonidamine for 1 h. LNCaP (b) and PC3 (c) were treated with various concentrations of etomoxir for 1 h, and then CPT-1 activity was determined. Etomoxir resulted in the dose-dependent decrease in CPT-1 activity. Results were presented as mean and P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were considered significant
Effect of etomoxir on CPT-1 activity in the presence of glucose (2 g/L)
To determine CPT-1 activity in response to etomoxir, LNCaP and PC3 cells were exposed to etomoxir at concentrations of 25, 50,100, 200, and 400 μM for 1 h, and then CPT-1 activity was determined. Exposure of LNCaP and PC3 to etomoxir resulted in a significant and dose-dependent decrease of CPT-1 activity in LNCaP (P = 0.0009) and PC3 (P = 0.0015) cell lines. As it is shown in Fig. 2, the highest decrease in CPT-1 activity occurred at concentration of 100 μM, 57 and 63.5 % for LNCaP and PC3 cells respectively (Fig. 1b, c). Therefore, this concentration of etomoxir was considered [I max] for the next experiments.

Inhibition of cell viability upon exposure of LNCaP and PC3 cell lines to lonidamine and etomoxir. Lonidamine (600 μM) inhibited the viability of LNCaP and PC3 cells (a). LNCaP (b) and PC3 (c) were cultured in medium containing glucose (2 g/L) or palmitate (50 μM) and then treated with etomoxir [100 μM] for 24, 48, and 72 h. Results (mean ± SD) were calculated as percent of corresponding control values. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) are considered significant
Effect of etomoxir on CPT-1 activity in the presence of palmitate (50 μM)
To validate the effect of exogenous fatty acid (palmitate), CPT-1 activity was determined after treating LNCaP and PC3 cells with etomoxir at 0, 25, 50, 100, 200, and 400 μM in the presence of palmitate (50 μM) for 1 h. A fall in the activity of CPT-1 in response to etomoxir in the presence of palmitate was dose-dependent (P = 0.0014 for PC3 and 0.001 for LNCaP cells), and 100 μM concentration of etomoxir lowered CPT-1 activity up to 51 and 60 % for PC3 and LNCaP, respectively. Therefore, this dose of etomoxir was used as [I max] for next experiments. As it is evident in Fig. 1b and c, in the presence of palmitate, activity of CPT-1 was significantly higher compared with its activity in the presence of glucose (unpaired t test P = 0.037 and 0.0218 for PC3 and LNCaP). In addition, CPT-1 was approximately 37 % more active in the presence of palmitate in LNCaP cells than PC3 cells (non-parametric unpaired t test P = 0.0453). These data show that palmitate induces CPT-1 activity especially in LNCaP cells.
Lonidamine decreased viability of prostate cancer cells
To evaluate the effect of hexokinase inhibition on the cell viability, PC3 and LNCaP cell lines were treated with lonidamine [600 μM] for 24, 48, and 72 h, and the decline of cell viability was measured by MTT assay. MTT assay showed a time-dependent decrease in the viability of both cell lines. As it is observed in Fig. 2a, lonidamine induced a significant reduction (P < 0.001, one-way ANOVA) in PC3 cells viability (69.5 ± 10.6 % for 24 h to 19.8 ± 3.5 % for 72 h vs. control). Growth inhibitory effects of lonidamine on LNCaP cells were also time-dependent and significant (P < 0.001, one-way ANOVA) so that viability of LNCaP cells were decreased from 85.5 ± 5.45 % at 24 h to 31.3 ± 2.9 % at 72 h vs. control.
Inhibitory effect of lonidamine on cell viability was more pronounced on PC3 cells in comparison to LNCaP cells, and this effect was significant for 72 h treatment (P = 0.013, non-parametric unpaired t test).
These preparatory results suggesting that hexokinase activity is necessary for the prostate cancer cells viability have been confirmed using flow cytometery analysis (Annexin-V-FITC).
Viability assay in response to etomoxir treatment
Non-significant cytotoxic effect of etomoxir on LNCaP and PC3 cells
LNCaP and PC3 cells were exposed to etomoxir [100 μM] for 24, 48, and 72 h to determine its effect on the cell viability using MTT assay. By increasing duration of exposure to etomoxir, viability was decreased in both cell lines in the presence of glucose or palmitate. However, etomoxir was unable to exert significant decline in cell viability of LNCaP and PC3 cells in the presence of glucose or palmitate (Fig. 2b, c). Therefore, CPT-1 activity and maybe palmitate are not vital for prostate cancer cells.
Intracellular ATP levels
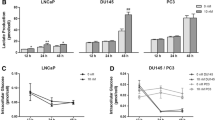
Having established that treating LNCaP and PC3 cells with lonidamine and etomoxir resulted in decreased activity of hexokinase and CPT-1, respectively, we then investigated whether they depend on glucose or palmitate for ATP supply. As shown in Fig. 3a, ATP levels of LNCaP and PC3 cells exposed to lonidamine [600 μM] declined significantly (P < 0.0001 for both cell lines) to approximately 40 % of the control. However, decrease in ATP levels were not significant for 2 and 4 h compared with 1 h exposure.

Alteration of ATP level and oxygen consumption rate following hexokinase and CPT-1 inhibition. Results are expressed as relative content of ATP as compared with control. Lonidamine [600 μM] caused a significant ATP level depletion in LNCaP and PC3 cells (a). LNCaP (b) and PC3 (c) cells were administered etomoxir [100 μM] exhibited reductions in the ATP levels. d (LNCaP) and e (PC3) represent oxygen consumption rates from control and treated cell lines with etomoxir [100 μM]
In response to CPT-1 inhibition by etomoxir, the ATP levels fell significantly in the presence of glucose (2 g/L) (P = 0.0294 and 0.0079 for PC3 and LNCaP) and palmitate (P = 0.0192 and 0.0022 for PC3 and LNCaP). Compared with 1 h treatment with etomoxir, decrease in ATP levels were not significant during 2 and 4 h exposure (Fig. 3b, c).
These data raised the possibility that hexokinase inhibition may have direct impact on the cellular energy metabolism.
Oxygen consumption rate
To measure mitochondrial fatty acid oxidation by LNCaP and PC3 cells, in response to CPT-1 inhibition, OCR was determined after treatment of the cells with etomoxir [100 μM] for 0, 1, 2, and 4 h.
In the presence of palmitate, treating with etomoxir resulted in a significant time-dependent decrease in the OCR for LNCaP (39–48 %, P = 0.001) and PC3 (21–32 %, P = 0.011) cells (Fig. 3d, e). However, reduction of OCR following treatment with etomoxir, in the presence of glucose was not significant for both cell lines (Fig. 5). These data reflect that CPT-1 inhibition decreases mitochondrial β-oxidation and hence O2 utilization.
Unlike etomoxir, lonidamine induces apoptosis in prostate cancer cells
To more directly examine the effect of lonidamine and etomoxir on cell viability and also determining underlying mechanism of cell death, PC3 and LNCaP cells were exposed to the lonidamine [600 μM] or etomoxir [100 μM] for 48 h and analyzed using annexin-V and PI double staining. Following exposure to lonidamine, PC3 cells undergone a significant increase in early (non-parametric unpaired t test P = 0.0002) and late (non-parametric unpaired t test P = 0.0001) apoptosis (Fig. 4a). Lonidamine also induced a significant increase in the percentage of both early (non-parametric unpaired t test P = 0.0003) and late (non-parametric unpaired t test P = 0.0004) apoptosis in LNCaP cells (Fig. 4a). In contrast, the apoptotic changes in response to etomoxir were not significant in both cell lines (Fig. 4b, c). Therefore, inhibition of hexokinase activity has resulted in the apoptotic cell death which did not occur by CPT-1 inhibition. Moreover, the overall cell death frequency was in accordance with MTT assay.

To determine the mechanism of cell death following lonidamine and etomoxir, flow cytometry analysis using annexin-V-FITC was done. Induction of apoptosis by lonidamine [600 μM] in LNCaP and PC3 cell lines was significant (a), while etomoxir [100 μM] did not induce significant apoptosis in LNCaP (b) and PC3 (c) cell lines. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were considered significant. Statistical analysis was performed by non-parametric unpaired t test. Each bar represents mean ± SD, n = 3
Caspase-3 participated in the apoptosis induction of lonidamine on LNCaP and PC3 cells
To examine the contribution of caspases in the lonidamine induced apoptosis, the activity of caspase-3 was investigated. These data demonstrated that the activity of caspase-3 increased markedly in a time-dependent manner in PC3 (4.98 times more than the control) and LNCaP cells (5.45 times more than the control) after treatment with lonidamine (600 μM) (P < 0.0001 ANOVA for both cells). Caspase-3 activity reached the peak value at 48 h (Fig. 5a). The date suggest that, in these cell lines, lonidamine-induced apoptosis is caspase-3-dependent. On the other hand, treatment of LNCaP and PC3 cells with etomoxir (100 μM) induced caspase-3 activity both in the presence of glucose and palmitate, but not significantly (Fig. 5b, c).

Evaluation of caspase-3 activity in response to the treatment of LNCaP and PC3 cell lines with lonidamine and etomoxir. Activity of caspase-3 in PC3 and LNCaP cell lines increased significantly following treatment with lonidamine [600 μM] (a); in contrast, treatment of LNCaP (b) and PC3 (c) cells with etomoxir [100 μM] had not resulted in a significant increase of caspase-3 activity. Each bar represents mean ± SD (n = 3). P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were considered significant
Role of mitochondrial membrane potential (MMP) in lonidamine and etomoxir induced cell apoptosis
One of the early markers of the apoptosis process is depletion of mitochondria membrane potential (ΔΨm). Therefore, alteration in ΔΨm was evaluated after treatment of LNCaP and PC3 cells with lonidamine [600 μM] for 24, 36, and 48 h. The results revealed a progressive and significant decrease of the red/green ratio of jc-1 fluorescence intensity for PC3 (P = 0.0068) and LNCaP (P = 0.0228) cell lines. As indicated in Fig. 6a, the lowest ratio was seen following 36 (LNCaP) or 48 h (PC3) treatment. Therefore, apoptosis following lonidamine treatment was derived from mitochondrial alterations.

Mitochondrial membrane potential as estimated by JC-1 after treatment of LNCaP and PC3 cell lines for 24, 36, and 48 h with lonidamine and etomoxir. Exposure to lonidamine [600 μM] induced a significant decrease in ΔΨm in LNCaP and PC3 cells (a). Effects of etomoxir [100 μM] on mitochondrial membrane potential in LNCaP (b) and PC3 (c) cell lines were not significant. Data represent the average values from triplicate experiments ± SD. P < 0.05 (*), P < 0.01 (**), and P < 0.001 (***) were considered significant
As shown in Fig. 6b and c, a time-dependent decline in the red/green ratio was observed in PC3 and LNCaP cells after exposure to etomoxir [100 μM] in the presence of glucose or palmitate, but these decreases were not significant.
Analysis of nuclear morphology
In order to confirm that lonidamine or etomoxir treatment induces apoptosis in PC3 and LNCaP cells, the apoptotic changes in the cell morphology were detected using Hoechst 33,258 staining. Exposure of PC3 and LNCaP cells to the lonidamine [600 μM] induced distinct changes in cell morphology, i.e., increase in the chromatin condensation and fragmentation, cytoplasmic shrinkage, and plasma membrane blebbing compared with that of controls (Fig. 7a–d). Instead, in response to exposure of the cells with etomoxir [100 μM], morphological features of apoptosis were not marked.

Detection of typical features of apoptosis nuclear condensation by Hoechst 33258 staining. PC3 control (a) and treatment with lonidamine[600 μM] (b), LNCaP control (c) and treatment (d). Consider changes in cell morphology, i.e., increase in the chromatin condensation or fragmentation (dashed arrow) and plasma membrane blebbing (arrow) compared with that of controls
Discussion
According to Warburg phenomenon, cancer cells depend largely on glycolysis and preferentially convert pyruvate to lactate, even in the presence of oxygen [5]. However, there are some contradictions regarding glucose or fatty acid utilization by prostate cancer cells [6, 7, 10, 16, 30, 31]. Therefore, the present study was designed to evaluate the effect of hexokinase and CPT-1 inhibition on the cell viability and growth of PC3 and LNCaP cell lines.
HK and CPT-1 activities were significantly suppressed by lonidamine and etomoxir in a dose-dependent manner, in LNCaP and PC3 cells. HK inhibition induced a significant cell death and apoptosis. However, in contrast to HK inhibition, CPT-1 inhibition had no significant effect on these cell lines.
Induction of apoptosis was caspase-3-dependent and through alterations in ΔΨm. In addition, inhibition of HK and CPT-1 culminated in a significant decline in ATP levels.
Through dose-dependent inhibition by lonidamine and etomoxir, hexokinase and CPT-1 activities were suppressed approximately by 50 % in both cell lines. The source of another 50 % activity may be due to the function of enzymes that catalyze similar reactions which are not responsive to lonidamine or etomoxir. For example, lonidamine interferes with the function of mitochondrial bound hexokinase (hexokinase 2) [19, 32] but with no effect on the other HK isozymes [33]. Apart from mitochondria, other subcellular organelles, such as peroxisomes, also contain CPT-like enzyme activities [34, 35] which etomoxir are unable to inhibit.
According to the results of current study, there is a connection between CPT-1 activity and palmitate. Our data have shown that CPT-1 activity is significantly higher in the presence of palmitate than that in the presence of glucose. It was previously confirmed that exposure to palmitate, at low concentrations (50–100 μM), resulted in a significant increase in CPT-1 activity [36]. Therefore, it can be inferred that these prostate cell lines not only have the ability to use palmitate but can also promote CPT-1 activity in response to palmitate and, probably, other long-chain fatty acid uptake. Liu et al. showed that fatty acid uptake is dominated over glucose by PC3 and LNCaP [10]. Therefore, some of the palmitate may be used for energy production via β-oxidation.
Our findings state that in the presence of palmitate, CPT-1 activity is higher in LNCaP cells than that of PC3 cells. Indeed, OCR was also lower for LNCaP cells following exposure to etomoxir. Previous data also showed that LNCaP cells express much higher CPT1 when compared with PC3 cells [37]. These data imply that metabolic properties of LNCaP cells differ from that of PC3 cells. In line with these findings, Higgins et al. showed that LNCaP cells had more oxidative phenotype than PC3 and DU145 cells, based upon respiration, lactate production, and ATP levels [38].
The fact that some prostate cancer cell lines grow in low levels of glucose [8] and inhibition of glucose uptake [7] or inhibition of hexokinase activity) our findings) resulted in a cell death implies that glucose and glycolysis had some indispensable roles in the prostate cell growth.
In the presence of glucose, lower CPT-1 activity is reasonable because of probable increased in malonyl-CoA as a result of active glucose metabolism [39, 40]. However, our findings propose two lines of evidences suggesting that consumption of glucose is, to some extent, preferred by PC3 and LNCaP cells over palmitate. First, in the presence of glucose, loss of ATP content was observed in both cell lines following HK inhibition (approximately 60 %) while ATP levels dropped upon CPT-1 inhibition about 18–27 % in LNCaP and PC3 cells. Second, falling in ATP levels, following CPT-1 inhibition, was lowered in the presence of glucose versus palmitate, which suggests that glucose is the primary source for energy production. ATP depletion by palmitate inaccessibility could be compensated by an increase in the glycolysis flow [41, 42].
On the other hand, following exposure of the cells to etomoxir or lonidamine for more than 1 h, the level of ATP remains relatively unchanged. Therefore, either ATP consumption is decreased by the cells or these cells may use some alternative pathways, other than glycolysis, to supply ATP, such as glutaminolysis [43] or short- and medium-chain fatty acid oxidation which cross the mitochondrial membrane, independent of CPT-1.
In the presence of palmitate (culture medium without glucose), O2 consumption has been decreased by both cell lines which can be inferred as β-oxidation inhibition by etomoxir.
Viability assay revealed that inhibition of HK lead to a significant increase in the cell death in a time-dependent manner. However, CPT-1 inhibition could not exert significant cytotoxic effect on LNCaP and PC3 cells.
Considering the rate of cell death and reduction in the ATP level in response to HK and CPT-1 inhibition, it could be concluded that long-chain fatty acids such as palmitate may be used to supply energy for prostate cancer cells, though less than glucose. However, the role of glucose is not limited to the energy production; it may also undertake some important and indispensable responsibility in overall growth and survival of prostate cancer cells. glucose serves as a precursor for DNA synthesis via pentose phosphate pathway [44, 45] and diacylglycerol, a ligand for protein kinase C, which suggest that glucose had an additional role in signal transduction [46, 47]. In addition, glucose has a regulatory effect on the cholesterol biosynthesis through HMG-CoA reductase activation [48].
Dominant uptake of palmitate over glucose [10] could be probably used for biogenesis of new cellular membranes in rapidly dividing cancer cells. Increased de novo fatty acid synthesis in prostate cancer cells through over expression of fatty acid synthase was reported [30, 31] which request glucose as a major lipogenic substrate [49]. Use of glucose as lipogenic substrate and consumption of produced fatty-acid for energy production is not acceptable since the consequence of the resultant futile cycle is only loss of energy.
The results of present study indicated that the mechanism of induced cell death following hexokinase inhibition is apoptosis which lead to a significant increase in the overall cell death (early and late apoptosis). However, CPT-1 inhibition had not induced significant apoptosis in both cell lines which seems logical since it did not confer marked cell cytotoxicity on LNCaP and PC3 cell lines.
To evaluate the molecular mechanisms underlying the cell death induced by Lon, caspase-3 activities and ΔΨm were assayed. Following treatment by Lon, the result demonstrated that a peak in caspase-3 activity achieved by 48 h which followed by the maximum cell death at 72 h. It was shown that activation of caspases-3 ensures the completion of apoptotic process [50].
On the other hand, perturbance of ΔΨm, following hexokinase inhibition, occurred after 24 h which maximized at 36 and 48 h. This finding is in accordance with the previous reports denoting that ΔΨm may be an early event in the apoptotic process [31] and caspase-3 activation occurred following ΔΨm disruption [49] by the release of hydrolases and some enzymes activators including caspases, from mitochondria [50].
Conclusion
The results of present study demonstrated that glucose is not only the preferred substrate for ATP production but it may also confers some other advantages for prostate cancer cells, as hexokinase inhibition results in a decline in ATP levels and cell viability. However, palmitate and probably other long-chain fatty acids can be used for energy production and inhibition of CPT-1 has minimal effect on the viability of prostate cancer cells. Apparently, in condition of CPT-1 inhibition in the presence of palmitate, alternative pathway(s) take the responsibility of palmitate, a replacement that glucose is deprived of it. Our results represented direct evidence on the importance of glucose for prostate cancer cell growth. However, evidences on the dominant role of fatty acids rely on the peroxisomal β-oxidation which does not degrade fatty acids completely and acts as a chain-shortening system which does not contribute to the energy production [8, 44].
In conclusion, we only blocked the transport of long-chain fatty acid to the mitochondria, but further evaluation of other enzymes involved in the fatty acid β-oxidation may open a new approach to the prostate cancer biology. Using other hexokinase inhibitors such as 3-bromopyruvate or 2-deoxy glucose may be informative regarding to cancer cell metabolism.
References
Warburg O. Über den stoffwechsel menschlicher tumorzellen. J Mol Med. 1925;4:2396–7.
Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–43.
Koukourakis MI, Giatromanolaki A, Harris AL, Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: a metabolic survival role for tumor-associated stroma. Cancer Res. 2006;66:632–7.
Swietach P, Vaughan-Jones RD, Harris AL. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev. 2007;26:299–310.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9.
Ben Sahra I, Laurent K, Giuliano S, Larbret F, Ponzio G, Gounon P, et al. Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells. Cancer Res. 2010;70:2465–75.
Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, et al. A novel inhibitor of glucose uptake sensitizes cells to FAS-induced cell death. Mol Cancer Ther. 2008;7:3546–55.
Singh G, Lakkis CL, Laucirica R, Epner DE. Regulation of prostate cancer cell division by glucose. J Cell Physiol. 1999;180:431–8.
Liu Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006;9:230–4.
Liu Y, Zuckier LS, Ghesani NV. Dominant uptake of fatty acid over glucose by prostate cells: a potential new diagnostic and therapeutic approach. Anticancer Res. 2010;30:369–74.
Sinha K. Elevated α-methylacyl-CoA racemase enzymatic activity. Am J Pathol2004;164:787-793
Rubin M, Zhou M, Dhanasekaran SM. α-Methylacyl-CoA racemase as tissue biomarker of PC. JAMA 2002;287:1662-1670
Luo J, Zha S, Gage WR, Dunn TA, et al. α-Methylacyl-CoA racemase; a new molecular marker for prostate cancer. Cancer Res 2002; 62:2220–2226.
Jiang Z, W.B.A., Rock K L, Xu Y, Savas L, Khan A, et al., P504S: a new molecular marker for the detection of prostate carcinoma. Am J Surg Pathol 2001; 25:13971404.
Zhou M, Chinnaiyan AM, Kleer CG, Lucas PC, Rubin MA. Alpha-methylacyl-CoA racemase: a novel tumor marker over-expressed in several human cancers and their precursor lesions. Am J Surg Pathol 2002; 26:926-931.
Zha S, Ferdinandusse S, Hicks JL, Denis S, Dunn TA, Wanders RJ, et al. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate. 2005;63:316–23.
Brawer MK. Lonidamine: basic science and rationale for treatment of prostatic proliferative disorders. Rev Urol. 2005;7(7):S21–6.
Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, de Martino C. Lonidamine, a selective inhibitor of aerobic glycolysis of murine tumor cells. J Natl Cancer Inst. 1981;66:497–9.
Floridi A, Paggi MG, D'Atri S, De Martino C, Marcante ML, Silvestrini B, et al. Effect of lonidamine on the energy metabolism of Ehrlich ascites tumor cells. Cancer Res. 1981;41:4661–6.
Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc Natl Acad Sci. 1977;74:3735–9.
Xu FY, Taylor WA, Hurd JA, Hatch GM. Etomoxir mediates differential metabolic channeling of fatty acid and glycerol precursors into cardiolipin in H9c2 cells. J Lipid Res. 2003;44:415–23.
Morillas M, Clotet J, Rubí B, Serra D, Arino J, Hegardt F, et al. Inhibition by etomoxir of rat liver carnitine octanoyltransferase is produced through the co-ordinate interaction with two histidine residues. Biochem J. 2000;351:495–502.
Gerondaes P, Alberti K, Agius L. Interactions of inhibitors of carnitine palmitoyltransferase I and fibrates in cultured hepatocytes. Biochem J. 1988;253:169–73.
Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. 2001;276:14890–5.
Robey RB, Raval BJ, Ma J, Santos AV. Thrombin is a novel regulator of hexokinase activity in mesangial cells. Kidney Int. 2000;57:2308–18.
Karlic H, Lohninger S, Koeck T, Lohninger A. Dietary l-carnitine stimulates carnitine acyltransferases in the liver of aged rats. J Histochem Cytochem. 2002;50:205–12.
Salami S, Karami-Tehrani F. Biochemical studies of apoptosis induced by tamoxifen in estrogen receptor positive and negative breast cancer cell lines. Clin Biochem. 2003;36:247–53.
Perry SW, Norman JP, Barbieri J, Brown EB, Gelbard HA. Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. Biotechniques. 2011;50:98.
Srinivasan S, Stevens M, Wiley JW. Diabetic peripheral neuropathy: evidence for apoptosis and associated mitochondrial dysfunction. Diabetes. 2000;49:1932–8.
Epstein JI, Carmichael M, Partin AW. OA-519 (fatty acid synthase) as an independent predictor of pathologic state in adenocarcinoma of the prostate. Urology. 1995;45:81–6.
Rossi S, Graner E, Febbo P, Weinstein L, Bhattacharya N, Onody T, et al. Fatty acid synthase expression defines distinct molecular signatures in prostate cancer. Mol Cancer Res. 2003;1:707–15.
Oudard S, Poirson F, Miccoli L, Bourgeois Y, Vassault A, Poisson M, et al. Mitochondria-bound hexokinase as target for therapy of malignant gliomas. Int J Cancer. 1995;62:216–22.
Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–57.
Kumaravel G, Gandour R, Krueger M, Ramsay R. Comparison of the active sites of the purified carnitine acyltransferases from peroxisomes and mitochondria by using a reaction-intermediate analogue. Biochem J. 1993;294:645–51.
Murthy M, Pande SV. Malonyl-CoA binding site and the overt carnitine palmitoyltransferase activity reside on the opposite sides of the outer mitochondrial membrane. Proc Natl Acad Sci. 1987;84:378–82.
Pimenta AS, Gaidhu MP, Habib S, So M, Fediuc S, Mirpourian M, et al. Prolonged exposure to palmitate impairs fatty acid oxidation despite activation of AMP-activated protein kinase in skeletal muscle cells. J Cell Physiol. 2008;217:478–85.
Nomura DK, Lombardi DP, Chang JW, Niessen S, Ward AM, Long JZ, et al. Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem Biol. 2011;18:846–56.
Higgins L, Withers H, Garbens A, Love H, Magnoni L, Hayward S, et al. Hypoxia and the metabolic phenotype of prostate cancer cells. Biochim Biophys Acta (BBA)-Bioenerg. 2009;1787:1433–43.
Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011;25:1041–51.
Ruderman NB, Dean D. Malonyl CoA, long chain fatty acyl CoA and insulin resistance in skeletal muscle. J Basic Clin Physiol Pharmacol. 1998;9:295–308.
Pike LS, Smift AL, Croteau NJ, Ferrick DA, Wu M. Inhibition of fatty acid oxidation by etomoxir impairs NADPH production and increases reactive oxygen species resulting in ATP depletion and cell death in human glioblastoma cells. Biochim Biophys Acta (BBA)-Bioenerg. 2011;1807:726–34.
Kim C, Wong J, Wen J, Wang S, Wang C, Spiering S, et al. Studying arrhythmogenic right ventricular dysplasia with patient-specific iPSCs. Nature. 2013;105–10.
Lu W, Pelicano H, Huang P. Cancer metabolism: is glutamine sweeter than glucose? Cancer Cell. 2010;18:199–200.
Pouyssegur J, Franchi A, Silvestre P. Relationship between increased aerobic glycolysis and DNA synthesis initiation studied using glycolytic mutant fibroblasts. Nature. 1980;287:445–7.
Jannière L, Canceill D, Suski C, Kanga S, Dalmais B, Lestini R, et al. Genetic evidence for a link between glycolysis and DNA replication. PLoS One. 2007;2:e447.
Farese RV, Standaert ML, Arnold TP, Yamada K, Musunuru K, Hernandez H, et al. Preferential activation of microsomal diacylglycerol/protein kinase C signaling during glucose treatment (de novo phospholipid synthesis) of rat adipocytes. J Clin Investig. 1994;93:1894–9.
Miele C, Paturzo F, Teperino R, Sakane F, Fiory F, Oriente F, et al. Glucose regulates diacylglycerol intracellular levels and protein kinase C activity by modulating diacylglycerol kinase subcellular localization. J Biol Chem. 2007;282:31835–43.
Burg JS, Espenshade PJ. Glucose controls phosphoregulation of HMG-CoA reductase through the PP2A-related phosphatase Ppe1 and Insig in fission yeast. J Biol Chem. 2011;111:233452.
Mathews EH, Liebenberg L, Pelzer R. High-glycolytic cancers and their interplay with the body’s glucose demand and supply cycle. Med Hypotheses. 2011;76:157–65.
Zhang A, Wu Y, Lai H, Yew D. Apoptosis—a brief review. Neuroembryol Aging. 2005;3:47–59.
Acknowledgments
Part of this work was supported by a Ph.D. grant (Rouhallah Najjar Sadeghi) from Tarbiat Modares University and also by the Iranian National Science Foundation (INSF Code: 91046780). The authors thank them for their supports.
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sadeghi, R.N., Karami-Tehrani, F. & Salami, S. Targeting prostate cancer cell metabolism: impact of hexokinase and CPT-1 enzymes. Tumor Biol. 36, 2893–2905 (2015). https://doi.org/10.1007/s13277-014-2919-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-014-2919-4