
Abstract
Background
Carbon monoxide (CO) poisoning affects several vital organs; however, the long-term damage to lungs has not been well investigated.
Objective
This study investigated the morphological changes and the associated molecules 6 weeks after CO poisoning in rat models. Rats in the CO poisoning group were exposed to 1500 ppm CO in a gas chamber for 3 h. After 6 weeks, the rats in the control and CO poisoning groups were sacrificed. Pathological changes were assessed using hematoxylin and eosin staining. Western blotting and antibody arrays for inflammatory signaling molecules were performed using lung homogenates. Complete blood counts and Krebs Von Den Lungen (KL)-6 levels were compared among rats in the control group and those immediately and 6 weeks after CO poisoning using blood sampling from an indwelling catheter.
Results
CO-poisoned lungs showed emphysematous changes with increased mean linear intercept (p = 0.0007). SERPINA1, IL-13, CD44, and GDF7 levels were significantly decreased, whereas TGFβ, ɑ-SMA, MMP9, and NF-κB levels were significantly increased in the CO-poisoned lungs. KL-6 serum levels were significantly higher in the CO-poisoned rats (p < 0.0001).
Conclusion
This is the first study to demonstrate the long-term effects and emphysematous changes in CO-poisoned lung models. We recommend long-term monitoring following CO poisoning.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Carbon monoxide (CO) is a toxic gas produced by fire, engine exhaust, and faulty furnaces (Rose et al. 2017). CO binds to hemoglobin (Hb) with high affinity and disrupts the binding of oxygen to Hb. Consequently, carboxyhemoglobin (COHb) inhibits oxygen delivery and damages organs under hypoxic conditions. The vulnerable organs are the heart and brain; clinically, intoxicated patients often present with myocardial injury and neurocognitive sequelae after brain injury (Hampson et al. 2012; Lippi et al. 2012). The effects of CO on other organs are insignificant compared to those in the two major organs. However, CO poisoning is associated with the development and progression of acute kidney injury (AKI) (Kim et al. 2018) and adrenal insufficiency (Huang et al. 2022). These organs, which are indirectly affected by CO, might have injury mechanisms similar to those of the heart and brain.
The lungs and bronchial trees are directly exposed to CO via gas instillation. However, the clinical evidence for lung damage caused by CO is unclear. An experimental study reported the effects of CO on alveolar epithelial permeability in the lungs (Fein et al. 1980). Tracheally instilled isotopes, which are poorly diffusible tracers, were present in large quantities in rabbits exposed to CO. Another study assessed the effect of CO on alveolar epithelial cells and revealed that CO interrupted alveolar fluid clearance in isolated, ventilated, and perfused rabbit lungs (Althaus et al. 2009). These investigators suggested that the deleterious effects of CO on alveolar fluid reabsorption should be considered when using CO as a therapeutic modality, especially in acute respiratory distress syndrome (ARDS). CO has recently been used as an anti-inflammatory agent in many diseases, such as sepsis (Hoetzel et al. 2007), chronic obstructive pulmonary disease (COPD) (Bathoorn et al. 2007), and acute lung injury (Hoetzel et al. 2009; Ryter and Choi 2006; Wilson et al. 2010). However, the toxic gas has variable characteristics. Compared with low-dose therapeutic CO exposure, high-dose intoxicating CO can damage the alveolar and bronchial epithelia. However, only a few studies have investigated the effects of CO on the alveoli or lungs (Althaus et al. 2009; Fein et al. 1980), and to date, the toxic effects of CO on lungs have not been fully demonstrated in human or experimental studies. This study aimed to investigate the effects of CO on lung tissues using pathological findings and biomarkers of lung damage.
Materials and methods
Animals
All animal experiments and surgical procedures were approved by the Institutional Animal Care and Use Committee of Yonsei University Wonju College of Medicine (YWC-210308-1) and conducted according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Twelve adult male Sprague–Dawley rats (250 g) were obtained from Dae Han Bio Link (Eumseong, Korea) and housed under a 12 h light/12 h dark cycle at a constant room temperature (20–22 ℃), with free access to food and water.
Exposure of rats to CO
The acute CO poisoning model was established according to published protocols (Thom et al. 2004). Briefly, the six rats were placed in an acrylic chamber with a 53 cm × 38 cm × 27 cm. The CO and O2 concentrations and humidity, pressure, and temperature inside the chamber were measured using a Gastiger 2000 (Wandi, China) or PO2-250 (Lutron, USA) and MHB-382SD (Lutron, USA), respectively. The rats inhaled 1500 ppm CO gas for 3 h and were then exposed to fresh air until they regained consciousness. Approximately 0.2 mL of whole blood was drawn from the right atrium through a catheter for the COHb assay using a GEM Premier 5000 (Werfen, MA, USA). Rats with high COHb concentrations (> 50%) were considered successful models of severe acute CO poisoning. All 12 animals were sacrificed 6 weeks after CO poisoning (Fig. 1).

Schematic diagram of the experimental schedule. Successful induction of carbon monoxide (CO) poisoning was screened by carboxyhemoglobin (COHb) concentration in blood immediately after CO exposure. Rats were examined on the day of poisoning and at 6 weeks
Histology
After 6 weeks, all 12 rats, including 6 CO-poisoned rats, were deeply anesthetized with 3% isoflurane (Hana Pharm, Seoul, Korea) in a mixture of 80% N2O and 20% O2. Their blood was then collected from the abdominal aorta, centrifuged, and stored at − 80 °C until use. After ligation of the hilum of the right lung, the right lungs were removed and stored at − 80 °C until use.
The left lungs were removed and perfused with a fixative solution containing 4% paraformaldehyde in 0.1 M phosphate-buffered saline (pH 7.0). The samples were then processed in preparation for paraffin embedding, and 4-μM-thick sections were cut. Pathological changes in the lung samples were examined using hematoxylin and eosin (H&E) staining (Thermo, USA). The mean linear intercept (MLI) of the lung tissue was calculated using morphological analysis. MLI was evaluated to determine the degradation and consequent air space enlargement within the lung parenchyma. The MLI was determined for each region on an overlay comprising horizontal and vertical lines. All intercepts with an alveolar septal number (ASN) were counted under a microscope at the intersection point of the two lines in the central field of view. The total length (L) of all lines divided by the number of intercepts provided the MLI for the region studied. The formula used to estimate the average diameter of a single alveolus is MLI = L/ASN (μm) (Yu et al. 2018).
Complete blood count (CBC)
For the CBC assay, 0.2 mL of blood was anticoagulated with ethylenediaminetetraacetic acid. Hematological parameters, including white blood cells (WBC), red blood cells (RBC), hemoglobin (Hb), platelets (PLT), mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), neutrophils, lymphocytes, monocytes, eosinophils, and basophils, were measured using an Automatic Blood Analyzer (XN-V, SYSMEX, Japan).
Preparation of the lung and Western blot
The right lung was lysed in 500 μL of cold radioimmunoprecipitation assay buffer (RIPA) solution, which contains 50 mM Tris–HCl, pH 7.5, 1% Triton X-100, 150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], and 1% sodium deoxycholate, with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). Tissue lysate was centrifuged at 13,000 × g for 15 min at 4 °C. After collecting the supernatant, the protein concentration was analyzed using a Bicinchoninic acid (BCA) assay kit (Molecular Probes, Eugene, Oregon, USA). For electrophoresis, 50 μg of protein was diluted in sample buffer (60 mM Tris–HCl, pH 6.8; 14.4 mM β-mercaptoethanol; 25% glycerol; 2% SDS; and 0.1% bromophenol blue), heated for 10 min, and then separated on a 10% SDS reducing gel. Using a trans-blot system, the separated proteins were transferred onto polyvinylidene difluoride (PVDF) membranes (Invitrogen, Carlsbad, CA, USA). Blots were blocked for 1 h at room temperature in Tris-buffered saline (TBS) (10 mM Tris–HCl, pH 7.5 and 150 mM NaCl) containing 5% nonfat dry milk, then washed three times with TBS before being incubated at 4 °C overnight with anti-rabbit tumor growth factor (TGF) β (1:1000, ABclonal, MA, USA), anti-rabbit alpha-smooth muscle actin (ɑSMA) (1:1000, ABclonal, MA, USA), anti-rabbit macrophage metalloproteinase (MMP) 9 (1:1000, ABclonal MA, USA), anti-rabbit nuclear factor (NF)-κB (1:1000, ABclonal, MA, USA), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:3000, Cell signaling, MA, USA) antibodies in TBST (10 mM Tris, pH 7.5; 150 mM NaCl; and 0.02% Tween 20) containing 5% nonfat dry milk. After being washed three times with TBST, the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (1:3000, Santa Cruz Biotech, Santa Cruz, CA, U.S.A.) for 1 h in TBST containing 3% nonfat dry milk at room temperature. After being washed three times with TBST, the proteins were detected using an ECL detection system (Amersham Pharmacia Biotech, Piscataway, NJ, USA). The protein bands on the blots were seen using an enhanced chemiluminescence detection system (Bio-Rad, Hercules, CA, USA). Scanning densitometry and computer-assisted image analysis software (Image Lab 6.1, Bio-Rad) were used to quantify the signals. Protein levels were expressed as ratio of the values of the detected protein bands to those of the GAPDH band.
Antibody array
To identify several anti- and pro-inflammatory cytokines related to lung disease modulated by CO poisoning, 1500 proteins were analyzed in lungs from CO-poisoned rats using an array-based antibody array. An antibody-based cytokine array system (L Series 1500, RayBio® Label-Based Rat Antibody Array 1, RayBiotech, Norcross, GA) was used to detect the cytokine and growth factor levels in normal and CO-poisoned rats. Briefly, normal and CO-poisoned lung extract samples were incubated with a labeling reagent and washed according to the manufacturer's instructions. Samples were diluted 1:50 (v/v) in blocking buffer and incubated on pre-blocked arrays overnight at 4 °C. Membranes were then incubated with streptavidin-conjugated peroxidase for 2 h and exposed to a peroxidase substrate for 5 min before development on an X-ray film. Densitometric analysis was performed on a Kodak ImageStation 4000 M (Eastman Kodak Company, Rochester, NY, USA) with background subtraction from spot edges. The spot data were normalized to the positive control spot on each array.
Enzyme-linked immunosorbent assay (ELISA)
Serum Krebs Von Den Lungen-6 (KL-6, MyBioSource, San Diego, CA, USA) protein levels were measured using ELISA. The BCA assay kit was used to measure and standardize the total protein concentration (Molecular Probes, Eugene, Oregon, USA). The standard was diluted, and then the samples, standards, and blank were added to the plate wells and incubated for 1 h at 37 °C. The liquid was discarded, and the plate was washed five times and patted dry. Chromogenic reaction reagent was added and incubated in the dark for 15 min at 37 °C. Finally, the stop solution was added, and the absorbance at 450 nm was measured within 10 min.
Statistical analysis
Data are expressed as mean ± standard error of the mean (SEM) or mean ± standard deviation (SD). A t-test was used to compare the differences between the two groups. Analysis of variance (ANOVA) was used to compare differences between three or more groups. Data were analyzed using PRISM 5.01 (GrapPad Software, Boston, MA, USA). Differences with p < 0.05 were considered statistically significant.
Results
CO poisoning model
The free-moving rats in the chamber became slightly drowsy after 20 min of exposure to 1500 ppm CO and reached a semi-comatose state approximately 10–15 min later. The vein blood analysis measured before and immediately after CO poisoning showed that the COHb level increased to a near-lethal level (50.50% ± 4.38%) in the CO-poisoned rats and only 0.30% ± 0.42% in the control group. Elevated COHb values after 6 weeks of intoxication were normalized (0.40% ± 0.21%) (Table 1).
Emphysematous changes in CO-poisoned lungs
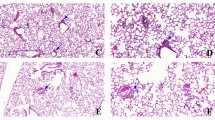
The structural features of CO-poisoned lungs were examined using the MLI measurement technique, and the results are shown in Fig. 2. Morphologically, after 6 weeks of CO poisoning, the lungs showed enlargement of the alveolar airspaces and destruction of the septal walls of alveoli in the field of view under a microscope (× 200 magnification). The MLI was significantly increased in the lungs of the CO-poisoned group compared with in those of the control group (69.42 ± 3.33 μm vs. 42.95 ± 1.16 μm, p < 0.0001) (Fig. 2). An increased MLI value indicates that the inner diameter of the alveoli has been enlarged.

Representative histological pictures of emphysematous changes to lung architecture. A Hematoxylin and eosin-stained lung tissue image from a healthy control rat displays normal architecture with appropriate airway size and morphology (Scale bar is 50 μm). B Hematoxylin and eosin-stained lung tissue image from a rat that poisoned by carbon monoxide (CO) exhibits distorted architecture with enlarged alveolar (scale bar is 50 μm). CO-poisoned rat lung showed emphysematous change at 6 weeks after exposure to CO at 1500 ppm for 3 h. C and D Lower magnification microscope image (scale bar is 200 μm). E The mean linear intersect (MLI) quantified from tissue sections demonstrates significantly increased airway space in CO-poisoned lung. The morphometric analysis of the MLI showed a significant difference between the control group (42.95 ± 1.16 μm) and CO-poisoned group (69.42 ± 3.33 μm) (p < 0.0001). Data are presented as the mean ± standard error of mean for each group
Changes in cell differentiation after CO poisoning
Total WBC counts did not differ according to the blood sampling time. However, compared with those of the control rats, neutrophil counts increased immediately after CO poisoning (1718 ± 762 × 106/L vs. 683 ± 253 × 106/L, p < 0.01) and decreased 6 weeks after CO poisoning, although still higher than that of the control rats (1531 ± 543 × 106/L vs. 683 ± 253 × 106/L, p < 0.05). On the other hand, lymphocyte (4810 ± 628 × 106/L vs. 6584 ± 630 × 106/L, p < 0.01) and monocyte (320 ± 51 × 106/L vs. 653 ± 258 × 106/L, p < 0.01) decreased significantly immediately after CO poisoning and recovered to a level similar to that of the normal group after 6 weeks. Their decrease appears to be a transient phenomenon following CO poisoning. (Table 2, Fig. 3).

Changes in cell differentiation after carbon monoxide (CO) Poisoning. Immediately after CO poisoning, the neutrophil count increased (1718 ± 762 × 106/L vs 683 ± 253 × 106/L, p < 0.01) and lymphocyte and monocyte count decreased. At 6 weeks after CO poisoning, cell counts showed recovery but neutrophil count was still higher than the count before CO poisoning (1531 ± 543 vs 683 ± 253 × 106/L, p < 0.05). *, ** denote significant differences (*p < 0.05; **p < 0.01) compared to the control levels. †, †† denote significant differences (†p < 0.05; ††p < 0.0001) between six weeks after CO poisoning and post-CO poisoning. Data are presented as the mean ± standard deviation for each group
Affected expression of genes related to alveolar destruction in CO-poisoned lungs
We performed antibody array to find out what changes carbon monoxide poisoning causes in various proteins in tissues, and among them, SERPINA1, IL-13, CD44, and growth differentiation factor (GDF) 7 decreased in association with lung emphysema. The levels of expression SERPINA1, IL-13, CD44, and GDF 7 were significantly decreased in the CO poisoning group than in the control group. (Fig. 4).

Screening of protein expression by carbon monoxide (CO) poisoning in lung tissue. A The level of SERPINA1 expression was significantly decreased in the CO-poisoned group compared to the control group at 6 weeks after CO poisoning (p = 0.0053). B The level of IL-13 was significantly decreased in the CO-poisoned group (p < 0.0001). C The level of CD44 was significantly decreased in the CO-poisoned group (p = 0.0004). D the level of GDF7 was significantly decreased in the CO-poisoned group (p = 0.0079). E Protein levels were quantified by antibody array. Data are presented as the mean ± standard error of mean for each group
Modulation of tissue remodeling protein in CO-poisoned lungs
To identify the protein associated with the alveolar and lung damages induced by CO poisoning, the expressions of various factors, including TGFβ, ɑ-SMA, MMP9, and NF-κB, were investigated using Western blot (Fig. 5A). TGFβ, ɑ-SMA, MMP9, and NF-κB levels were significantly elevated in the CO-poisoned group compared with the control group (Fig. 5B–E) (p = 0.0194, 0.0409, 0.0321, 0.0325, respectively).

Expression of proteins contributing to tissue remodeling and destruction. A Western blot analysis in the CO-poisoned lung. B–E Representative Western blot analysis of TGFβ, ɑ-SMA, MMP9, and NF-κB. Western blots were quantified using densitometry analysis, normalized to GAPDH, and graphed as mean ± SEM. TGFβ (B), ɑ-SMA (C), MMP9 (D), and NF-κB (E) was significantly increased in the CO-poisoned group compared to the control group at 6 weeks after CO poisoning. * denotes difference (p < 0.05) compared to the control group. Data are presented as the mean ± standard error of mean for each group
Increased KL-6 levels in the serum of CO-poisoned rats
To examine the effect of CO poisoning on the expression of KL-6, ELISA using serum was performed 6 weeks after CO poisoning. The levels of serum KL-6 were significantly higher in the CO-poisoned rats than those in the control group (1607 ± 336.1 U/mL vs. 231 ± 26.56 U/mL, p < 0.0001) (Fig. 6).

Serum concentration of KL-6 in control and CO-poisoned rats. Serum levels of KL-6 were not changed on the day of CO poisoning, but the levels increased explosively at 6 weeks after poisoning (1607 ± 336.1 U/mL vs. 231 ± 26.56 U/mL, p < 0.0001). Data are presented as the mean ± standard error of mean for each group
Discussion
This study investigated the effects of CO on lung tissues using pathological findings and biomarkers of lung damage, and we observed emphysematous changes in the alveoli of rat lungs after CO poisoning. Additionally, biomarkers of inflammatory activity that mediate the pathogenesis of emphysema were also altered in the serum and lungs of CO-poisoned rat models.
Although the lung airway is the organ directly exposed to inhaled gas, the effects of CO on the lungs are not established; furthermore, the information on the chronic effects is minimal. The emphysematous changes caused by CO poisoning in this study can be explained by a mechanism similar to that in tobacco smoking, a well-known risk factor of emphysema (Global Initiative for Chronic Obstructive Lung Disease 2021). Smoking-induced oxidative stress alters neutrophil elastase and anti-elastase homeostasis, destroying alveolar structure and leading to emphysema (Lungarella et al. 2008). Increased oxidative stress in airway epithelial cells alters cell differentiation, followed by airway remodeling (Wohnhaas et al. 2021). The major mechanisms of damage from CO intoxication include oxidative stress and free radicals. Oxidative stress from CO intoxication damages mitochondrial function and causes cellular inflammation in vulnerable organs (Rose et al. 2017). These changes may cause tissue damage in various organs, including the lung, as smoking does. However, unlike exposure to smoking, CO poisoning is a one-time acute episode of significant exposure to toxic gas. Therefore, structural alveolar damage, such as emphysema caused by CO poisoning, has not been investigated. Long-term damage and development of chronic diseases in nonpulmonary organs after CO poisoning have been reported previously. CO poisoning was associated with the development of chronic kidney disease (Wei et al. 2021), a high incidence of ischemic stroke (Kwak et al. 2021), and a higher long-term mortality rate of patients with myocardial injury due to CO poisoning (Henry et al. 2006) in long-term follow-up data.
In this study, the lungs at 6 weeks after CO poisoning showed decreased alpha1-antitrypsin (AAT) and IL-13 levels. SERPINA1 encodes AAT, which protects against alveolar structural destruction caused by neutrophil elastase. Mutations in several alleles of SERPINA1 result in AAT deficiency of varying severity. Clinically, AAT deficiency presents as emphysema in the lungs and is the major genetic pathophysiology of the disease (Strnad et al. 2020). IL-13 is a pleiotropic cytokine primarily produced by activated T cells (McKenzie et al. 1993). IL-13 overexpression stimulates and activates TGF-ß1, which plays a role in lung injury and repair, including emphysema and fibrosis (Lee et al. 2001; Morris et al. 2003; Warburton et al. 2013). IL-13 also stimulates MMP and cathepsin-based pathways in the lungs, causing emphysema and COPD (Zheng et al. 2000). Conversely, some investigators have suggested the protective role of IL-13 against the development of emphysema (Boutten et al. 2004). In their study using human lung samples, IL-13 mRNA and protein levels decreased in the severe emphysema group. They explained that decreased IL-13 levels contributed to the development of emphysema through protease imbalance, oxidant injury, apoptosis, and repair function defects. However, our results, as in a previous study, cannot explain whether decreased IL-13 levels developed during emphysematous changes or was the mediator of disease development. In addition to AAT and IL-13, some molecules with protective roles against inflammation, oxidative stress, and tissue damage, such as CD44 and GDF7, are decreased in CO-poisoned lungs (Dong et al. 2022; Hodge et al. 2007; Teder et al. 2002).
On the other hand, major epithelial–mesenchymal transition (EMT) molecules, such as TGF-ß1, MMP, NF-κB, and α-SMA, are increased in the lungs of the CO-poisoned group. These molecules lead to improper synthesis and deposition of extracellular matrix and alveolar epithelial damage (Holm Nielsen et al. 2019; Park et al. 2019). Damage to various activated signaling pathways results in fibrosis and emphysema of the lungs (Morris et al. 2003; Schuliga 2015; Warburton et al. 2013).
KL-6 has been suggested as an indicator of the activity of interstitial lung disease (ILD) (Kohno et al. 1989). Especially in idiopathic pulmonary fibrosis (IPF), KL-6 is associated with a risk of acute exacerbation (Aloisio et al. 2021). In addition to IPF, other respiratory diseases show increased levels of KL-6 (Lederer et al. 2009; Xu et al. 2017). Increased KL-6 in at-risk patients was associated with the diagnosis of ARDS. KL-6 is expressed in type II pneumocytes and becomes stronger when alveolar epithelial cells are damaged (Kohno et al. 1993). The increased KL-6 levels observed in the CO-intoxicated model can be explained by two mechanisms. First, KL-6 levels reflect alveolar capillary permeability (Sakai et al. 2013). These experimental results were represented in studies of patients with ARDS defined by increased alveolar capillary permeability (Briassoulis et al. 2006; Sato et al. 2004). CO intoxication also increases permeability, according to a study using animal models (Fein et al. 1980). Second, KL-6 levels are increased in patients with emphysema (Kokuho et al. 2015). COPD biomarkers are not easily interpreted and have very low specificity (Stockley et al. 2019). Although KL-6 is not specific for emphysema or COPD, previous studies have revealed that its concentration is elevated in emphysema compared with in smokers or healthy controls (Kohno et al. 1989; Kokuho et al. 2015). Moreover, serum KL-6 levels were higher in patients with combined pulmonary fibrosis and emphysema than in those with IPF only (Demirdöğen 2022). These findings are inconsistent, but emphysematous changes in the alveoli may be related to increased KL-6 level, as an indicator of epithelial cell damage.
Based on the results of our study, the implications of structural changes on long-term respiratory-related prognoses have not yet been fully estimated. However, according to clinical studies on emphysema patients, emphysema is not a negligible, simple scar. Emphysematous changes in the lung are unnoticeable until the damage becomes severe (Wewers 1989). Early emphysematous damage can lead to the development of emphysema once it is initiated and is a predictor of forced expiratory volume decline (Bhatt et al. 2017; Bodduluri et al. 2017). Individuals with emphysema may have normal spirometric findings and are therefore regarded as a subtype of pre-COPD (Agustí and Hogg 2019; Martinez et al. 2022). Therefore, the effects of CO intoxication do not end in a single episode. Instead, it represents the beginning of future damage, including structural and functional damage. Ultimately, CO-intoxicated patients should be monitored for the development and progression of emphysema and decline in lung function.
Our study had some limitations. The use of CO as a novel therapy for several refractory diseases has received increasing attention (Bathoorn et al. 2007; Hoetzel et al. 2007; Hoetzel et al. 2009). Treatment is usually performed using low-dose CO. However, our acute CO poisoning models could not explain the effects of CO in various levels. Nevertheless, without the knowledge on long-term and occult damage, therapeutic trials should be started very carefully.
To our knowledge, this is the first study to demonstrate the long-term (6 weeks) effects of CO poisoning in a lung model. This study is also the first to report the morphological changes with emphysema in this lung model. Although more information on the link between CO poisoning and emphysema is needed, our study suggests the need for proper long-term monitoring following CO poisoning and the need for clinical trials. In conclusion, this study confirms emphysematous changes in the alveoli of rat lungs and alterations in biomarkers of inflammatory activity that mediate the pathogenesis of emphysema after CO poisoning.
Data availability
The authors confirm that the data supporting the findings of this study are available within this article.
References
Agustí A, Hogg JC (2019) Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med 381:1248–1256. https://doi.org/10.1056/NEJMra1900475
Aloisio E, Braga F, Puricelli C, Panteghini M (2021) Prognostic role of Krebs von den Lungen-6 (KL-6) measurement in idiopathic pulmonary fibrosis: a systematic review and meta-analysis. Clin Chem Lab Med 59:1400–1408. https://doi.org/10.1515/cclm-2021-0199
Althaus M et al (2009) Carbon monoxide rapidly impairs alveolar fluid clearance by inhibiting epithelial sodium channels. Am J Respir Cell Mol Biol 41:639–650. https://doi.org/10.1165/rcmb.2008-0458OC
Bathoorn E et al (2007) Anti-inflammatory effects of inhaled carbon monoxide in patients with COPD: a pilot study. Eur Respir J 30:1131–1137. https://doi.org/10.1183/09031936.00163206
Bhatt SP et al (2017) Computed tomography measure of lung at risk and lung function decline in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 196:569–576. https://doi.org/10.1164/rccm.201701-0050OC
Bodduluri S et al (2017) Biomechanical CT metrics are associated with patient outcomes in COPD. Thorax 72:409–414. https://doi.org/10.1136/thoraxjnl-2016-209544
Boutten A et al (2004) Decreased expression of interleukin 13 in human lung emphysema. Thorax 59:850–854. https://doi.org/10.1136/thx.2004.025247
Briassoulis G et al (2006) Circulating levels of KL-6 in acute respiratory distress syndrome sepsis or traumatic brain injury in critically ill children. Pediatr Pulmonol 41:790–795. https://doi.org/10.1002/ppul.20465
Demirdöğen E et al (2022) Serum Krebs von den Lungen-6: promising biomarker to differentiate CPFE from IPF. Sarcoidosis Vasc Diffuse Lung Dis. 39:e2022035. https://doi.org/10.36141/svdld.v39i4.11344
Dong P et al (2022) Growth differentiation factor 7 prevents sepsis-induced acute lung injury in mice. Evid Based Complement Alternat Med 2022:3676444. https://doi.org/10.1155/2022/3676444
Fein A et al (1980) Carbon monoxide effect on alveolar epithelial permeability. Chest 78:726–731. https://doi.org/10.1378/chest.78.5.726
Global Initiative for Chronic Obstructive Lung Disease (2021) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease 2022 report. https://goldcopd.org/archived-reports/
Hampson NB, Piantadosi CA, Thom SR, Weaver LK (2012) Practice recommendations in the diagnosis, management, and prevention of carbon monoxide poisoning. Am J Respir Crit Care Med 186:1095–1101. https://doi.org/10.1164/rccm.201207-1284CI
Henry CR et al (2006) Myocardial injury and long-term mortality following moderate to severe carbon monoxide poisoning. JAMA 295:398–402. https://doi.org/10.1001/jama.295.4.398
Hodge S et al (2007) Smoking alters alveolar macrophage recognition and phagocytic ability: implications in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 37:748–755. https://doi.org/10.1165/rcmb.2007-0025OC
Hoetzel A et al (2007) Carbon monoxide in sepsis. Antioxid Redox Signal 9:2013–2026. https://doi.org/10.1089/ars.2007.1762
Hoetzel A et al (2009) Carbon monoxide prevents ventilator-induced lung injury via caveolin-1. Crit Care Med 37:1708–1715. https://doi.org/10.1097/CCM.0b013e31819efa31
Holm Nielsen S et al (2019) Serological assessment of activated fibroblasts by alpha-smooth muscle actin (α-SMA): a noninvasive biomarker of activated fibroblasts in lung disorders. Transl Oncol 12:368–374. https://doi.org/10.1016/j.tranon.2018.11.004
Huang CC et al (2022) Association between carbon monoxide poisoning and adrenal insufficiency: a nationwide cohort study. Sci Rep 12:16219. https://doi.org/10.1038/s41598-022-20584-y
Kim YJ et al (2018) Analysis of the development and progression of carbon monoxide poisoning-related acute kidney injury according to the Kidney Disease Improving Global Outcomes (KDIGO) criteria. Clin Toxicol (phila) 56:759–764. https://doi.org/10.1080/15563650.2018.1424890
Kohno N et al (1989) New serum indicator of interstitial pneumonitis activity. sialylated carbohydrate antigen KL-6. Chest 96:68–73. https://doi.org/10.1378/chest.96.1.68
Kohno N et al (1993) KL-6, a mucin-like glycoprotein, in bronchoalveolar lavage fluid from patients with interstitial lung disease. Am Rev Respir Dis 148:637–642. https://doi.org/10.1164/ajrccm/148.3.637
Kokuho N et al (2015) Diagnostic values for club cell secretory protein (CC16) in serum of patients of combined pulmonary fibrosis and emphysema. COPD 12:347–354. https://doi.org/10.3109/15412555.2014.948994
Kwak K et al (2021) Association between carbon monoxide intoxication and incidence of ischemic stroke: a retrospective nested case-control study in South Korea. J Stroke Cerebrovasc Dis 30:105496. https://doi.org/10.1016/j.jstrokecerebrovasdis.2020.105496
Lederer DJ et al (2009) Circulating KL-6, a biomarker of lung injury, in obstructive sleep apnoea. Eur Respir J 33:793–796. https://doi.org/10.1183/09031936.00150708
Lee CG et al (2001) Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 194:809–821. https://doi.org/10.1084/jem.194.6.809
Lippi G et al (2012) Pathophysiology, clinics, diagnosis and treatment of heart involvement in carbon monoxide poisoning. Clin Biochem 45:1278–1285. https://doi.org/10.1016/j.clinbiochem.2012.06.004
Lungarella G, Cavarra E, Lucattelli M, Martorana PA (2008) The dual role of neutrophil elastase in lung destruction and repair. Int J Biochem Cell Biol 40:1287–1296. https://doi.org/10.1016/j.biocel.2007.12.008
Martinez FJ et al (2022) Treatment trials in young patients with chronic obstructive pulmonary disease and pre–chronic obstructive pulmonary disease patients: time to move forward. Am J Respir Crit Care Med 205:275–287. https://doi.org/10.1164/rccm.202107-1663SO
McKenzie AN et al (1993) Interleukin 13, a T-cell-derived cytokine that regulates human monocyte and B-cell function. Proc Natl Acad Sci USA 90:3735–3739. https://doi.org/10.1073/pnas.90.8.3735
Morris DG et al (2003) Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 422:169–173. https://doi.org/10.1038/nature01413
Park YJ et al (2019) Effects of β-Sitosterol from corn silk on TGF-β1-induced epithelial–mesenchymal transition in lung alveolar epithelial cells. J Agric Food Chem 67:9789–9795. https://doi.org/10.1021/acs.jafc.9b02730
Rose JJ et al (2017) Carbon monoxide poisoning: pathogenesis, management, and future directions of therapy. Am J Respir Crit Care Med 195:596–606. https://doi.org/10.1164/rccm.201606-1275CI
Ryter SW, Choi AM (2006) Therapeutic applications of carbon monoxide in lung disease. Curr Opin Pharmacol 6:257–262. https://doi.org/10.1016/j.coph.2006.03.002
Sakai M, Kubota T, Ohnishi H, Yokoyama A (2013) A novel lung injury animal model using KL-6-measurable human MUC1-expressing mice. Biochem Biophys Res Commun 432:460–465. https://doi.org/10.1016/j.bbrc.2013.01.123
Sato H et al (2004) KL-6 levels are elevated in plasma from patients with acute respiratory distress syndrome. Eur Respir J 23:142–145. https://doi.org/10.1183/09031936.03.00070303
Schuliga M (2015) NF-kappaB signaling in chronic inflammatory airway disease. Biomolecules 5:1266–1283. https://doi.org/10.3390/biom5031266
Stockley RA, Halpin DMG, Celli BR, Singh D (2019) Chronic obstructive pulmonary disease biomarkers and their interpretation. Am J Respir Crit Care Med 199:1195–1204. https://doi.org/10.1164/rccm.201810-1860SO
Strnad P, McElvaney NG, Lomas DA (2020) Alpha(1)-antitrypsin deficiency. N Engl J Med 382:1443–1455. https://doi.org/10.1056/NEJMra1910234
Teder P et al (2002) Resolution of lung inflammation by CD44. Science (1979) 296:155–158. https://doi.org/10.1126/science.1069659
Thom SR et al (2004) Delayed neuropathology after carbon monoxide poisoning is immune-mediated. Proc Natl Acad Sci USA 101:13660–13665. https://doi.org/10.1073/pnas.0405642101
Warburton D, Shi W, Xu B (2013) TGF-β-Smad3 signaling in emphysema and pulmonary fibrosis: an epigenetic aberration of normal development? Am J Physiol Lung Cell Mol Physiol 304:L83-85. https://doi.org/10.1152/ajplung.00258.2012
Wei KY et al (2021) Carbon monoxide poisoning and chronic kidney disease risk: a nationwide, population-based study. Am J Nephrol 52:292–303. https://doi.org/10.1159/000515383
Wewers M (1989) Pathogenesis of emphysema: assessment of basic science concepts through clinical investigation. Chest 95:190–195. https://doi.org/10.1378/chest.95.1.190
Wilson MR et al (2010) Efficacy and safety of inhaled carbon monoxide during pulmonary inflammation in mice. PLoS ONE 5:e11565. https://doi.org/10.1371/journal.pone.0011565
Wohnhaas CT et al (2021) Cigarette smoke specifically affects small airway epithelial cell populations and triggers the expansion of inflammatory and squamous differentiation associated basal cells. Int J Mol Sci 22:7646. https://doi.org/10.3390/ijms22147646
Xu L, Bian W, Gu XH, Shen C (2017) Differing expression of cytokines and tumor markers in combined pulmonary fibrosis and emphysema compared to emphysema and pulmonary fibrosis. COPD 14:245–250. https://doi.org/10.1080/15412555.2017.1278753
Yu N et al (2018) Treatment with eucalyptol mitigates cigarette smoke-induced lung injury through suppressing ICAM-1 gene expression. Biosci Rep 38:20171636. https://doi.org/10.1042/bsr20171636
Zheng T et al (2000) Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 106:1081–1093. https://doi.org/10.1172/jci10458
Acknowledgement
We thank Prof. Hyun Kim and Prof. Sang-Ha Kim for supporting this study.
Funding
This work was supported by the National Research Foundation of Korea, which is funded by the Korean government (Ministry of Science and Information and Communications Technology, grant no. NRF-2021R1A2C200492211).
Author information
Authors and Affiliations
Contributions
SJL, JYL, and WYL: contributed to conceptualization; JYL: was responsible for methodology, software, and formal analysis; JYL and MK: were responsible for validation, resources, and data curation; SJL, TK, MK, and JYL: did investigation; SJL and JYL: were involved in visualization and writing—original draft preparation; TK, YSC, MK, and WYL: contributed to writing—review and editing; WYL: was involved in supervision and project administration; YSC: performed funding acquisition. All the authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interests
Seok Jeong Lee declares that she has no conflict of interest. Taeyeong Kim declares that he has no conflict of interest. Yong Sung Cha declares that he has no conflict of interest. Min Kim declares that she has no conflict of interest. Ji Yong Lee declares that she has no conflict of interest. Won-Yeon Lee declares that he has no conflict of interest.
Ethical approval
The animal study protocol was approved by the Institutional Animal Care and Use Committee of Yonsei University Wonju College of Medicine (YWC-210308-1, 7-Apr-2022) and conducted according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, S.J., Kim, T., Cha, Y.S. et al. Alveolar damage and development of emphysema in rats with carbon monoxide poisoning. Mol. Cell. Toxicol. (2023). https://doi.org/10.1007/s13273-023-00405-7
Accepted:
Published:
DOI: https://doi.org/10.1007/s13273-023-00405-7