
Abstract
Background
NR4A family genes play crucial roles in cancers. However, the role of NR4A family genes in cancers remains paradoxical as they promote or suppress tumorigenesis.
Objective
We aimed to conduct comprehensive analyses of the association between the expression of NR4A family genes and tumor microenvironment (TME) based on bioinformatics methods.
Methods
We collected RNA-seq data from 33 cancer types and 20 normal tissue sites from the TCGA and GTEx databases. Expression patterns of NR4A family genes and their associations with DNA methylation, miRNA, overall survival, drug responses, and tumor microenvironment were investigated.
Results
Significant downregulation of all NR4A family genes was observed in 15 cancer types. DNA promoter methylation and expression of NR4A family genes were negatively correlated in five cancers. The expression of 10 miRNAs targeting NR4A family genes was negatively correlated with the expression of NR4A family genes. High expression of all NR4A family genes was associated with poor prognosis in stomach adenocarcinoma and increased expressions of NR4A2 and NR4A3 were associated with poor prognosis in adrenocortical carcinoma. In addition, we found an elevated expression of NR4A2, which enhances the response to various chemotherapeutic drugs, whereas NR4A3 decreases drug sensitivity. Interestingly, in breast cancer, NR4A3 was significantly associated with C2 (IFN-γ dominant), C3 (inflammatory), and C6 (TGF-β dominant) immune subtypes and infiltrated immune cell types, implying both oncogenic and tumor-suppressive functions of NR4A3 in breast cancer.
Conclusion
The NR4A family genes have the potential to serve as a diagnostic, prognostic, and immunological marker of human cancers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is the leading cause of early mortality in several countries (CP et al. 2020). The incidence and mortality rates of cancer are rapidly increasing worldwide (Sung et al. 2021). There were 10.3 million cancer deaths and 19.3 million new cancer cases in 2020. Thus, it is important to enhance our comprehensive understanding of tumor suppressors and oncogenes.
In previous studies, nuclear receptor subfamily 4 group A (NR4A) family genes were found to be significantly dysregulated in various types of cancers (Mohan et al. 2012). NR4A is a subfamily of transcription factors consisting of three members: NR4A1, NR4A2, and NR4A3 (Beard et al. 2015). They have similar domain structures, including N- and C-terminal domains containing activation function domains, and ligand-binding domain (LBD), which surround a DNA-binding domain, and a hinge region (Safe and Karki 2021). These transcription factors respond to various signals and have various biological functions in humans by regulating the pathways involved in homeostasis, proliferation, cell migration, apoptosis, metabolism, DNA repair, and glucose utilization (Mohan et al. 2012). NR4A family genes are known to promote or suppress tumorigenesis based on these regulations (Mohan et al. 2012).
The role of these genes has mainly been investigated in blood-derived and solid tumors (Yousefi et al. 2022). Studies on blood-derived cancers have shown that NR4A family genes act as tumor suppressors, suggesting that repression of NR4A1 and NR4A3 contributes to the progression of leukemia and lymphoma (Wenzl et al.). In solid tumors, including breast and colon cancers, the upregulation of NR4A family genes has been shown to be pro-oncogenic and is often associated with poor prognosis and inhibition of apoptosis (Safe et al. 2016).
However, the role of NR4A family genes in solid tumors remains controversial. The induced expression of NR4A2 promotes apoptosis while inhibiting the growth of bladder cancer cell lines (Inamoto et al. 2008). Some in-vitro studies have shown that NR4A3 is downregulated in breast and lung cancer cell lines (Ohkubo et al. 2000; Fedorova et al. 2019). In silico study using breast, lung, prostate, colorectal, uterine, and ovarian cancer tissues showed that NR4A1 was downregulated in metastatic cancer compared to that in primary tumors (Ramaswamy et al. 2003). Another study using The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) breast cancer data showed that the expression levels of NR4A family genes were significantly lower in tumor samples than in normal samples (Yousefi et al. 2022). Although several studies have demonstrated the importance of the NR4A family genes in cancer, the role of these genes remains paradoxical, and an association between NR4A family genes and the tumor microenvironment (TME) has not been reported. Therefore, it is necessary to conduct a comprehensive pan-cancer analysis of NR4A family genes to assess the role of potential therapeutic targets in clinical research.
In this study, we analyzed the expression profiles of NR4A family genes in 33 cancer types using TCGA pan-cancer data and a Genotype-Tissue Expression (GTEx) dataset. Associations between the expression of NR4A family genes and DNA methylation, miRNAs, immune subtype, overall survival, drug sensitivity, and TME were also evaluated. Our results suggest the possibility for the clinical application of NR4A family genes as prognostic and therapeutic targets in patients with cancer.
Materials and methods
Pan-cancer data collection
RNA-seq data in the fragments per kilobase of transcript per million reads (FPKM) format of TCGA and GTEx, which were processed and integrated uniformly by the Toil process, were downloaded from UCSC Xena (https://xenabrowser.net/datapages/, accessed on November 8 2023) (Vivian et al. 2017). Mature miRNA expression data, Illumina 450k DNA methylation beta-value data, clinical data, survival data, and immune subtype data were downloaded from UCSC Xena. A total of 9,807 tumor samples and 5,973 normal samples were used in this study. Detailed information on the samples is shown in Supplementary Table 1.
Gene expression analysis of NR4A family genes in pan-cancer
The difference in the overall expression levels of NR4A family genes across TCGA 33 cancers was analyzed using the Kruskal–Wallis test, and Spearman’s correlations between NR4A family genes were investigated. We then performed differential expression analysis between tumor and adjacent normal samples using the Wilcoxon signed-rank test. A heatmap of the log2 (fold change) values was plotted.
Correlation between the expression of NR4A family genes and DNA methylation
Using the 450k array methylation beta-value data in TCGA database, the mean beta values of CpG sites in the promoter regions (TSS200, TSS1500, 5’ UTR) were calculated. Associations between the mean beta values and the expression of NR4A family genes were calculated using Spearman’s correlation.
Correlation between the expression of NR4A family genes and miRNA
We downloaded miRNA regulation data of NR4A family genes from miRWalk, an open-source platform that incorporates TargetScan, miRDB, and miRTarBase datasets and provides predicted and validated miRNA target data of known genes (Sticht et al. 2018). Spearman’s correlation between mature miRNA expression and the mRNA expression of NR4A family genes was calculated. Only miRNAs predicted to target the NR4A family genes from at least two databases were used.
Survival analysis
Patients were divided into high- and low-expression groups based on the median expression levels of the NR4A family genes. Overall survival was assessed using the Kaplan–Meier method. Intergroup differences were evaluated using a log-rank test, and statistical significance was set at p < 0.05. We then performed Univariate Cox proportional hazard regression to identify the association between the expression levels of NR4A family genes and pan-cancer prognosis. The hazard ratios (HR) were plotted as forest plots.
Correlation between the expression of NR4A family genes and drug sensitivity
The National Cancer Institute (NCI)-60 dataset was downloaded from the CellMiner version 2.9 database (https://discover.nci.nih.gov/cellminer/), which included mRNA expression levels of NR4A family genes and z-scores of drug sensitivity in 60 different tumor cell lines from nine different cancer types (Shankavaram et al. 2009). Spearman’s correlation coefficient was used to test the association between the expression of NR4A family genes and drug sensitivity. The sensitivities of 248 FDA-approved drugs and 519 drugs in clinical trials were selected and used in the correlation analysis.
Expression of NR4A family genes by different immune subtypes
Six immune subtypes, including wound healing (C1), IFN-γ dominant (C2), inflammatory (C3), lymphocyte depleted (C4), immunologically quiet (C5), and TGF-β dominant (C6), were defined regarding five representative immunological signatures (Thorsson et al. 2018). As each immune subtype has different biological and clinical features that determine the effectiveness of anticancer therapy, we analyzed the association between the expression levels of NR4A family genes using the Kruskal–Wallis test. The distribution of immune subtypes is shown in Supplementary Table 2.
Correlation between the expression of NR4A family genes and TME
ESTIMATE is an algorithm that produces stromal, immune, and ESTIMATE scores that infer the stromal cell infiltration level, immune cell infiltration level, and tumor purity, respectively (Yoshihara et al. 2013). Spearman’s correlation was used to measure the association between the expression of NR4A family genes and the three scores.
Immune deconvolution analysis for breast cancer
Each fraction of immune cell infiltration in BRCA was estimated using CIBERSORT, a linear support vector regression-based algorithm for quantifying the infiltration of 22 immune cells from gene expression profiles (Newman et al. 2015). Patients were divided into high- and low-expression groups based on the median expression levels of NR4A3. We investigated the differentially infiltrated immune cells between the high- and low-expression groups using the Wilcoxon signed-rank test.
Statistical analysis
Statistical significance of the comparison between tumor and normal samples was tested using the Wilcoxon signed-rank test. Correlation analysis among the NR4A family genes was performed using the corrplot version 0.92 R package. The Kruskal–Wallis test was used to evaluate the association between the expression of NR4A family genes and tumor immune subtype. Spearman’s correlation was used to test the association between gene expression and degree of DNA methylation, stromal score, immune score, ESTIMATE score, and drug sensitivity. All tests were selected based on normality tests in our datasets, and a Benjamini-Hochberg corrected p (FDR) < 0.05 was considered statistically significant. All statistical analyses were performed using R software version 4.2.3, and visualizations were conducted using ggplot2 version 3.4.2, ggpubr version 0.6.0, pheatmap version 1.0.12, survival version 3.5.5, and survminer version 0.4.9 R packages (R Core Team 2023).
Results
Gene expression analysis of NR4A family genes in pan-cancer
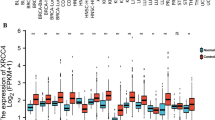
To investigate the expression profiles of NR4A family genes in cancer, the expression levels of NR4A family genes across 33 types of cancers in TCGA database were examined. Our results showed that the expression levels of the NR4A family genes were significantly different, with NR4A1 showing the highest expression level (Fig. 1A). NR4A1 and NR4A3 showed the most significant positive correlation, suggesting that they may have common biological roles in cancer (Fig. 1B). Differential expression was analyzed using the Wilcoxon signed-rank test. For the differential expression analysis, two cancers (MESO and UVM) with no normal samples were excluded. A heatmap was drawn based on the log2(fold change) of NR4A family gene’s median expression value in tumor and normal samples (Fig. 1C). Our differential expression analysis showed that all members of the NR4A family genes were differentially expressed in most cancers, including BLCA, BRCA, ESCA, KIRC, KIRP, LAML, LGG, LIHC, LUAD, LUSC, OV, PAAD, PRAD, SKCM, TGCT, THCA, THYM, UCEC, and UCS (FDR < 0.05; Fig. 2). NR4A1 expression was significantly lower in tumor samples than in normal samples in 24 cancers (ACC, BLCA, BRCA, CESC, COAD, ESCA, GBM, HNSC, KIRC, KIRP, LGG, LIHC, LUAD, LUSC, OV, PRAD, READ, SKCM, STAD, TGCT, THCA, THYM, UCEC, and UCS) and was significantly overexpressed in LAML and PAAD (Fig. 2A). Similar downregulation trends were observed for NR4A2 except for its upregulation in ESCA, LAML, PAAD, and SKCM (Fig. 2B). NR4A3 was significantly downregulated in 18 cancers (BLCA, BRCA, CESC, COAD, KIRC, KIRP, LGG, LIHC, LUAD, LUSC, OV, PRAD, READ, TGCT, THCA, THYM, UCEC, and UCS), and significantly upregulated in ESCA, LAML, PAAD, and SKCM (Fig. 2C). These results suggest that NR4A1, NR4A2, and NR4A3 were associated with tumor-suppressive functions in most cancers.

(A) Violin plots represent the expression levels of NR4A family genes across each cancer (* : p < 0.05; ** : p < 0.01; *** : p < 0.001; **** : p < 0.0001). (B) Spearman’s correlation between the NR4A family genes. (C) Heatmap showing differential expression of the NR4A family genes across each cancer

Violin plots demonstrate differential expression of NR4A family genes between tumor and normal samples (* : FDR < 0.05; ** : FDR < 0.01; *** : FDR < 0.001; **** : FDR < 0.0001)
Correlation between the expression of NR4A family genes and DNA methylation
In theory, DNA methylation of the promoter region regulates mRNA expression by recruiting proteins involved in gene suppression or by interrupting the binding of transcription factors. To measure the effect of DNA methylation on gene expression, Spearman’s correlations between the mRNA expression data of the NR4A family genes and the mean beta values of their corresponding CpG probes were computed. We found significant correlations in five cancer types: DLBC, KICH, PAAD, TGCT, and THYM (absolute correlation coefficient > 0.3, FDR < 0.05; Fig. 3, Supplementary Table 3). NR4A1 in TGCT, NR4A2 in DLBC, and NR4A3 in KICH, PAAD, and THYM were negatively correlated, while only NR4A2 in THYM was positively correlated. Thus, we speculate that epigenetic alterations in NR4A family genes may cause tumorigenesis in several cancers.

Spearman’s correlations between mRNA expression of NR4A family genes and methylation across each cancer. Red and blue dots indicate positive and negative correlations, respectively
Correlation between the expression of NR4A family genes and miRNA
Micro RNAs (miRNAs) are key regulators of mRNA expression. To clarify the interactions between miRNAs and NR4A family genes in human cancers, we calculated Spearman’s correlations between miRNAs targeting NR4A family genes and their expression across 33 cancer types. In total, 26 miRNAs that were predicted to target NR4A family genes in at least two databases were used. Among these, 12 miRNAs showed significant correlations with TGCT, SARC, ESCA, KIRC, LIHC, and THCA (absolute correlation coefficient > 0.3, FDR < 0.05; Fig. 4, Supplementary Table 4). In detail, miR-200b-5p expression was negatively correlated with NR4A1 expression in TGCT (Fig. 4A). Similar trends were observed between miR-409-3p and NR4A2 expression in SARC and TGCT (Fig. 4B). In case of NR4A3, the expression of seven miRNAs (miR-103a-3p, miR-20b-5p, miR-3154, miR-455-5p, miR-494-3p, miR-501-5p, and miR-675-3p) and miR-136-5p were negatively correlated with NR4A3 expression in TGCT and SARC, respectively. Notably, all NR4A family genes in TGCT showed significant negative correlations with at least one miRNA. Furthermore, miR-10a-5p in ESCA, KIRC, and LIHC, miR-10b-5p in KIRC, and miR-20b-5p in THCA were positively correlated with NR4A3 expression. These results indicate that regulation of the expression of NR4A family genes via miRNAs may be associated with cancer progression.

Spearman’s correlations between mRNA expression of NR4A family genes and target miRNA expression across each cancer (A–C). Red and blue dots indicate positive and negative correlations, respectively
Association between the expression of NR4A family genes and patients’ overall survival
We investigated the association between the expression of NR4A family genes and overall survival (OS) using the Kaplan–Meier method. We found that the expression of NR4A family genes was significantly associated with patient outcomes and that the direction of this association varied (Fig. 5, p < 0.05). NR4A1 is a favorable prognostic indicator for KICH and KIRC and plays a damaging role in STAD. NR4A2 was a positive prognostic factor for CHOL, PCPG, and SKCM and a negative prognostic factor for ACC and STAD. Similarly, NR4A3 is a positive prognostic factor for CHOL and plays a damaging role in ACC, LGG, and STAD. Particularly, all NR4A family genes play damaging roles in STAD. In addition, the hazard ratios (HR) of the NR4A family genes were calculated using univariate Cox regression (Supplementary Fig. 1, Supplementary Table 5).

Kaplan–Meier overall survival curves comparing groups in high and low expression of NR4A family genes (A–L). Only plots with significance (p < 0.05) were displayed
Correlation between the expression of NR4A family genes and drug sensitivity
To analyze the potential association between the expression of NR4A family genes and drug sensitivity, we performed correlation analysis using data from the CellMiner database (Shankavaram et al. 2009). We found that the expression of NR4A family genes was significantly correlated with 197 (25.7%) of the 767 drugs under FDA approval or clinical trials (absolute correlation coefficient > 0.3, FDR < 0.05, Fig. 6, Supplementary Table 6). The expression of NR4A1 was positively correlated with seliciclib and hydrastinine HCl and negatively correlated with SGX-523. The expression of NR4A2 was positively correlated with the sensitivity to dabrafenib, AZ-628, cobimetinib, alvespimycin, and PF-03758309 and negatively correlated with EC-330. The expression of NR4A3 was negatively correlated with TPX-0005, ON-123,300, and SAR-20,347 sensitivity. These results suggest that dysregulated expression of NR4A family genes may be involved in resistance to chemotherapy and targeted drug therapy.

Spearman’s correlations between the mRNA expression of NR4A family genes and drug sensitivity. The plots were sorted by FDR
Expression of NR4A family genes using different immune subtypes
To investigate the potential association between the expression levels of NR4A family genes and the six different immune subtypes of TCGA cancers, differential expression analysis was performed using the Kruskal–Wallis test. We found that the expression of NR4A family genes across the immune subtypes was significantly different in BRCA, KIRC, and LUAD (Fig. 7, FDR < 0.05 for all NR4A family genes). In these cancers, the high expression of NR4A family genes was mainly associated with the C3 subtype. In BRCA, NR4A1 was highly expressed in C3, NR4A2 was highly expressed in C3 and C4, and NR4A3 was highly expressed in C2, C3, and C6 (Fig. 7A). In KIRC, NR4A1 and NR4A3 were highly expressed at C5, while NR4A2 showed high expression at C3 (Fig. 7B). In LUAD, all NR4A family genes showed the highest expression at C3 (Fig. 7C). These results suggest that NR4A family genes may be related to tumor immunity in BRCA, KIRC, and LUAD.

Violin plots showing the differential expression of NR4A family between C1–C6 immune subtypes in (A) BRCA, (B) KIRC, (C) LUAD, and (D) PRAD (* : FDR < 0.05; ** : FDR < 0.01; *** : FDR < 0.001; **** : FDR < 0.0001)
Association between the expression of NR4A family genes and TME
To explore the association between the expression of NR4A family genes and the TME, the immune score of TCGA tumor samples was computed using the ESTIMATE algorithm. Spearman’s correlation between the immune score and the expression of NR4A family genes was examined. We found that the expression of all NR4A family genes was significantly correlated with immune infiltration in different cancer types (absolute correlation coefficient > 0.3, FDR < 0.05; Fig. 8A, Supplementary Table 7). The expression of NR4A1 and NR4A2 was significantly and positively correlated with immune scores in LAML, DLBC, and TGCT. Particularly, the expression of NR4A3 was significantly correlated with the immune score in multiple cancer types. For instance, the expression of NR4A3 was positively correlated with the immune score in BLCA, BRCA, CESC, CHOL, COAD, DLBC, ESCA, HNSC, LUSC, PAAD, READ, STAD, THYM, and UCS and negatively correlated with ACC and UVM. Furthermore, we analyzed the relationship between the NR4A family genes and stromal and ESTIMATE scores using the ESTIMATE algorithm to infer the ratio of stromal cell infiltration to tumor purity (Fig. 8B–C). In most cases, NR4A3 was positively correlated with stromal, immune, and ESTIMATE scores, indicating that NR4A3 may be associated with high stromal cell infiltration, high immune cell infiltration, and low tumor purity. The correlation coefficients are shown in Supplementary Table 7.

(A ~ C) Spearman’s correlations between the mRNA expression of NR4A family genes and (A) immune score, (B) stromal score, and (C) ESTIMATE score across each cancer. Red and blue dots indicate positive and negative correlations, respectively. (D) The association between the fraction of immune cell infiltration and the expression of NR4A3 in BRCA (* : FDR < 0.05; ** : FDR < 0.01; *** : FDR < 0.001; **** : FDR < 0.0001)
Immune deconvolution analysis for breast cancer
As the expression of NR4A3 in BRCA showed the most significant positive correlation with the immune score and the most significant association with immune subtypes, we further analyzed immune cell infiltration in BRCA. Twelve types of immune cells (naïve B cells, resting dendritic cells, M1 macrophages, activated mast cells, monocytes, neutrophils, activated NK cells, activated CD4 memory T cells, resting CD4 memory T cells, CD8 T cells, follicular helper T cells, and gamma delta T cells) were significantly enriched in NR4A3 high group (Fig. 7D). NR4A3 low group showed high fractions of M0/M2 macrophages, resting mast cells, resting NK cells, plasma cells, and naïve CD4 + T cells (Fig. 7D). These findings suggest that the expression level of NR4A3 could be a relevant marker of the infiltration level of immune cells in breast cancer and could provide options for breast cancer immunotherapy. The different immune cell fractions based on the expression levels of NR4A1 and NR4A2 are shown in Supplementary Fig. 2.
Discussion
Since NR4A family genes are known to function as both pro-oncogenes and tumor suppressors, their biological functions are ambiguous. To address this, we focused on clarifying the characteristics of the NR4A family genes across 33 types of cancers.
We found that NR4A family genes were downregulated in most cancers. First, NR4A1 was found to be downregulated in 24 cancers, including BRCA and COAD. Low expression of NR4A1 in triple-negative breast cancer has been associated with lymph node metastasis, advanced tumor stage, and poor relapse-free survival (Wu et al. 2017). A previous study demonstrated that NR4A1 inhibited Wnt signaling and retarded tumorigenesis in a mouse model of colorectal cancer (Chen et al. 2012). Second, NR4A2 was downregulated in 20 cancers, including BLCA. NR4A2 activation via DIM-C-pPhCl induces apoptosis and inhibits growth of bladder cancer cell lines (Inamoto et al. 2008). Third, NR4A3 was downregulated in 18 cancers including BRCA, LUAD, and LUSC. Previously, NR4A3 played a tumor-suppressive role in breast and lung cancers by triggering apoptosis (Fedorova et al. 2019). In summary, consistent with previous findings, we confirmed the tumor-suppressive role of the NR4A family genes in certain types of cancer.
Correlation analysis between DNA methylation and mRNA expression showed that NR4A family genes are regulated by promoter methylation in several cancers. The mRNA expression levels of NR4A1 in TGCT, NR4A2 in DLBC, and NR4A3 in THYM were downregulated, and their methylation levels were negatively correlated with mRNA expression. NR4A2 is methylated in B-cell lymphoma cell lines and unmethylated in healthy controls (Bethge et al. 2013). Regarding NR4A3, promoter hypermethylation and mRNA down-regulation were reported in patients with gastric cancer (Yeh et al. 2016).
We also investigated the association between the expression of NR4A family genes and miRNAs targeting them. We found significant correlations between the expression of NR4A family genes and miRNAs in several cancer types. Interestingly, all NR4A family genes exhibited significant negative correlations with at least one miRNA in TGCT, suggesting their role as oncogenes: miR-200b-5p targeted NR4A1 in TGCT, miR-409-3p targeted NR4A2 in SARC and TGCT, and miR-455-5p and miR-675-3p targeted NR4A3 in TGCT. Although our miRNA findings are novel for TGCT and SARC, their oncogenic role in other cancers has been previously reported in emerging studies. Evidence was found in studies on miR-200b-5p and miR-409-3p in prostate cancer (Lin et al. 2014; Josson et al. 2014). In contrast, two miRNAs (miR-10a-5p in ESCA, KIRC, and LIHC and miR-10b-5p in KIRC) were positively correlated with the expression of NR4A3, suggesting a tumor-suppressive role in cancer. Notably, both miR-10a-5p and miR-10b-5p were found in KIRC, and these results were supported by previous studies in which they were significantly downregulated as tumor suppressors in kidney renal cell carcinoma (Khella et al. 2017; Tan et al. 2021).
The aberrant expression of NR4A family genes was significantly associated with OS and drug responses in several cancers. In particular, increased expression of all NR4A family genes was associated with a poor prognosis in STAD, which is consistent with a previous study (Han et al. 2013). In addition, we are the first to report that increased expression of NR4A2 and NR4A3 is associated with a poor prognosis in ACC. We also revealed an elevated expression of NR4A2, which enhances responses to various chemotherapeutic drugs, whereas NR4A3 decreases drug sensitivity. Further studies are necessary to elucidate the biological interactions between NR4A family genes and drugs.
We identified significant associations between the expression of NR4A family genes and immune subtypes in BRCA, KIRC, and LUAD. In BRCA and LUAD, NR4A1 showed the highest expression at C3 (inflammatory). In KIRC, the expression of NR4A1 was the highest in C3 followed by C5 (immunologically quiet), while high expression of NR4A2 was associated with C3 and C6 (TGF-β dominant). In BRCA, high expression of NR4A2 was associated with C3 and C4 (lymphocyte-depleted), whereas NR4A3 was highly expressed in C2, C3, and C6. Interestingly, as the C3 subtype is known to have the most favorable prognosis and the C6 subtype is related to the worst prognosis in a pan-cancer study, NR4A3 may have both oncogenic and tumor-suppressive roles (Thorsson et al. 2018). These characteristics imply a potentially crucial role for the NR4A family genes in the immune system.
Our TME analysis revealed the most significant association between NR4A3 and immune infiltration score in BRCA. Further investigation of the effect of NR4A3 on BRCA revealed 12 higher fractions and six lower fractions of immune cell types in NR4A3 high group than in the NR4A3 low group. Certain cell types with higher proportions (e.g., M1 macrophages, activated NK cells, activated CD4 memory T cells, CD8 T cells, follicular helper T cells, and gamma delta T cells) have anti-tumor functions (Lei et al. 2020). In contrast, among the lower fractions of immune cells, M2 macrophages and resting NK cells are mainly associated with pro-tumor roles (Lei et al. 2020). The roles of neutrophils (lower fraction) and monocytes (higher fraction) can be either anti-tumor or pro-tumor, depending on the interactions among neighboring cells in the TME (Ugel et al. 2021; Que et al. 2022). Previous studies have reported an association between NR4A3 and the tumor immune microenvironment. NR4A3 inhibits the memory potential and effector functions of CD8 + T cells (Odagiu et al. 2020). Moreover, double knockout of PRDM1 and NR4A3 transformed CAR-T cell phenotypes from TIM-3 + CD8 + to TCF1 + CD8 + cells to counter the exhaustion of tumor-infiltrated CAR T cells and improve antitumor responses, whereas this effect was not achieved with a single knockout of PRDM1 or NR4A3 alone (Jung et al. 2022). Based on our findings and previous reports, NR4A3 may have both oncogenic and tumor-suppressive functions under specific immune cell conditions, independent of its downregulation in breast tumor tissues. Although the effects of NR4A3 on the tumor immune system are debatable, our results may help in understanding the role of NR4A3 in selecting appropriate breast cancer immunotherapies.
Our study has some limitations. Further validation is required because our findings have not been validated using independent datasets or in vitro/in vivo experiments. For TME analysis, immune deconvolution results should be investigated using single-cell sequencing or immunological experiments. Future studies focusing on the biological mechanisms of NR4A family genes at both the cellular and molecular levels are necessary to confirm our findings.
Our study provides the first comprehensive pan-cancer analysis of NR4A family genes which may be candidate diagnostic and prognostic factors for various cancers. Their expression is significantly downregulated in most cancers and is associated with methylation levels, miRNA expression, immune subtype, overall survival, drug sensitivity, and TME. In particular, NR4A3 was significantly associated with the immune subtypes and infiltrating immune cell types in BRCA. Although these findings need to be validated experimentally, we have provided the transcriptomic features and biological functions of NR4A family genes in pan-cancer. Thus, we suggest blueprints for further studies on pan-cancer function and the use of NR4A family genes as new diagnostic and prognostic markers and therapeutic targets.
References
Beard JA, Tenga A, Chen T (2015) The interplay of NR4A receptors and the oncogene–tumor suppressor networks in cancer. Cell Signal 27:257–266. https://doi.org/10.1016/j.cellsig.2014.11.009
Bethge N, Honne H, Hilden V et al (2013) Identification of highly methylated genes across various types of B-Cell Non-hodgkin Lymphoma. PLoS ONE 8:e79602. https://doi.org/10.1371/journal.pone.0079602
Chen H-Z, Liu Q-F, Li L et al (2012) The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by downregulating wnt signalling. Gut 61:714–724. https://doi.org/10.1136/gutjnl-2011-300783
CP W, E W, BW S (2020) World Cancer Report. Cancer Research for Cancer Prevention
Fedorova O, Petukhov A, Daks A et al (2019) Orphan receptor NR4A3 is a novel target of p53 that contributes to apoptosis. Oncogene 38:2108–2122. https://doi.org/10.1038/s41388-018-0566-8
Han Y, Cai H, Ma L et al (2013) Expression of orphan nuclear receptor NR4A2 in gastric cancer cells confers chemoresistance and predicts an unfavorable postoperative survival of gastric cancer patients with chemotherapy. Cancer 119:3436–3445. https://doi.org/10.1002/cncr.28228
Inamoto T, Papineni S, Chintharlapalli S et al (2008) 1,1-Bis(3′-indolyl)-1-(p-chlorophenyl)methane activates the orphan nuclear receptor Nurr1 and inhibits bladder cancer growth. Mol Cancer Ther 7:3825–3833. https://doi.org/10.1158/1535-7163.MCT-08-0730
Josson S, Gururajan M, Hu P et al (2014) miR-409-3p/-5p promotes tumorigenesis, Epithelial-To-Mesenchymal Transition, and bone metastasis of human prostate Cancer. Clin Cancer Res 20:4636–4646. https://doi.org/10.1158/1078-0432.CCR-14-0305
Jung I-Y, Narayan V, McDonald S et al (2022) BLIMP1 and NR4A3 transcription factors reciprocally regulate antitumor CAR T cell stemness and exhaustion. Sci Transl Med 14:eabn7336. https://doi.org/10.1126/scitranslmed.abn7336
Khella HWZ, Daniel N, Youssef L et al (2017) miR-10b is a prognostic marker in clear cell renal cell carcinoma. J Clin Pathol 70:854–859. https://doi.org/10.1136/jclinpath-2017-204341
Lei X, Lei Y, Li J-K et al (2020) Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett 470:126–133. https://doi.org/10.1016/j.canlet.2019.11.009
Lin H-M, Castillo L, Mahon KL et al (2014) Circulating microRNAs are associated with docetaxel chemotherapy outcome in castration-resistant prostate cancer. Br J Cancer 110:2462–2471. https://doi.org/10.1038/bjc.2014.181
Mohan HM, Aherne CM, Rogers AC et al (2012) Molecular pathways: the role of NR4A Orphan Nuclear receptors in Cancer. Clin Cancer Res 18:3223–3228. https://doi.org/10.1158/1078-0432.CCR-11-2953
Newman AM, Liu CL, Green MR et al (2015) Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12:453–457. https://doi.org/10.1038/nmeth.3337
Odagiu L, Boulet S, De Maurice D et al (2020) Early programming of CD8 + T cell response by the orphan nuclear receptor NR4A3. Proc Natl Acad Sci U S A 117:24392–24402. https://doi.org/10.1073/pnas.2007224117
Ohkubo T, Ohkura N, Maruyama K et al (2000) Early induction of the orphan nuclear receptor NOR-1 during cell death of the human breast cancer cell line MCF-7. Mol Cell Endocrinol 162:151–156. https://doi.org/10.1016/S0303-7207(00)00222-7
Que H, Fu Q, Lan T et al (2022) Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochimica et Biophysica Acta (BBA) -. Reviews Cancer 1877:188762. https://doi.org/10.1016/j.bbcan.2022.188762
R Core Team (2023) R: a Language and. Environment for Statistical Computing
Ramaswamy S, Ross KN, Lander ES, Golub TR (2003) A molecular signature of metastasis in primary solid tumors. Nat Genet 33:49. https://doi.org/10.1038/ng1060
Safe S, Karki K (2021) The paradoxical roles of Orphan Nuclear receptor 4A (NR4A) in Cancer. Mol Cancer Res 19:180–191. https://doi.org/10.1158/1541-7786.MCR-20-0707
Safe S, Jin U-H, Morpurgo B et al (2016) Nuclear receptor 4A (NR4A) family - orphans no more. J Steroid Biochem Mol Biol 157:48–60. https://doi.org/10.1016/j.jsbmb.2015.04.016
Shankavaram UT, Varma S, Kane D et al (2009) CellMiner: a relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics 10:277. https://doi.org/10.1186/1471-2164-10-277
Sticht C, Torre CDL, Parveen A, Gretz N (2018) miRWalk: an online resource for prediction of microRNA binding sites. PLoS ONE 13:e0206239. https://doi.org/10.1371/journal.pone.0206239
Sung H, Ferlay J, Siegel RL et al (2021) Global Cancer statistics 2020: GLOBOCAN estimates of incidence and Mortality Worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71:209–249. https://doi.org/10.3322/caac.21660
Tan P, Chen H, Huang Z et al (2021) MMP25-AS1/hsa-miR-10a-5p/SERPINE1 axis as a novel prognostic biomarker associated with immune cell infiltration in KIRC. Mol Therapy - Oncolytics 22:307–325. https://doi.org/10.1016/j.omto.2021.07.008
Thorsson V, Gibbs DL, Brown SD et al (2018) The Immune Landscape of Cancer. Immunity 48:812–830. e14
Ugel S, Canè S, De Sanctis F, Bronte V (2021) Monocytes in the Tumor Microenvironment. Annu Rev Pathol 16:93–122. https://doi.org/10.1146/annurev-pathmechdis-012418-013058
Vivian J, Rao AA, Nothaft FA et al (2017) Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 35:314–316. https://doi.org/10.1038/nbt.3772
Wenzl K, Troppan K, Neumeister P, Deutsch AJA The nuclear orphan receptor NR4A1 and NR4A3 as Tumor suppressors in Hematologic Neoplasms. Curr Drug Targets 16:38–46
Wu H, Bi J, Peng Y et al (2017) Nuclear receptor NR4A1 is a tumor suppressor down-regulated in triple-negative breast cancer. Oncotarget 8:54364–54377. https://doi.org/10.18632/oncotarget.17532
Yeh C-M, Chang L-Y, Lin S-H et al (2016) Epigenetic silencing of the NR4A3 tumor suppressor, by aberrant JAK/STAT signaling, predicts prognosis in gastric cancer. Sci Rep 6:31690. https://doi.org/10.1038/srep31690
Yoshihara K, Shahmoradgoli M, Martínez E et al (2013) Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 4:2612. https://doi.org/10.1038/ncomms3612
Yousefi H, Fong J, Alahari SK (2022) NR4A family genes: a review of Comprehensive Prognostic and Gene expression Profile analysis in breast Cancer. Front Oncol 12:777824. https://doi.org/10.3389/fonc.2022.777824
Acknowledgements
The authors gratefully acknowledge the collaboration of the study participants and research staff included in this study.
Funding
This research was supported by Incheon National University Research Grant in 2022.
Author information
Authors and Affiliations
Contributions
Conceptualization: Mi-Ryung Han; Methodology: Mi-Ryung Han; Data curation: Seong-Woo Park; Formal analysis and investigation: Seong-Woo Park; Writing – original draft preparation: Seong-Woo Park, Mi-Ryung Han; Writing – review and editing: Mi-Ryung Han; Funding acquisition: Mi-Ryung Han.
Corresponding author
Ethics declarations
Conflict of interest
No potential conflicts of interest relevant to this article are reported.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Park, SW., Han, MR. A pan-cancer analysis unveiling the function of NR4A family genes in tumor immune microenvironment, prognosis, and drug response. Genes Genom 46, 977–990 (2024). https://doi.org/10.1007/s13258-024-01539-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-024-01539-1