
Abstract
Background
Elymus atratus (Nevski) Hand.-Mazz. is perennial hexaploid wheatgrass. It was assigned to the genus Elymus L. sensu stricto based on morphological characters. Its genome constitution has not been disentangled yet.
Objective
To identify the genome constitution and origin of E. atratus.
Methods
In this study, genomic in situ hybridization and fluorescence in situ hybridization, and phylogenetic analysis based on the Acc1, DMC1 and matK sequences were performed.
Results
Genomic in situ hybridization and fluorescence in situ hybridization results reveal that E. atratus 2n = 6x = 42 is composed of 14 St genome chromosomes, 14 H genome chromosomes, and 14 Y genome chromosomes including two H-Y type translocation chromosomes, suggesting that the genome formula of E. atratus is StStYYHH. The phylogenetic analysis based on Acc1 and DMC1 sequences not only shows that the Y genome originated in a separate diploid, but also suggests that Pseudoroegneria (St), Hordeum (H), and a diploid species with Y genome were the potential donors of E. atratus. Data from chloroplast DNA showed that the maternal donor of E. atratus contains the St genome.
Conclusion
Elymus atratus is an allohexaploid species with StYH genome, which may have originated through the hybridization between an allotetraploid Roegneria (StY) species as the maternal donor and a diploid Hordeum (H) species as the paternal donor.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Traditionally, species in Triticeae with the same genome or genome combinations were classified into the same genus (Löve 1984; Lu 1993; Yen et al. 2005; Zhang and Zhou 2007; Baum et al. 2011; Lucía et al. 2019). Existing studies suggested that Elymus sensu lato included combinations of seven different basic genomes: St, H, P, W, Ns, Y, and Xm (Baum et al. 1995; Zhang and Zhou 2007; Dong et al. 2015). Based on the genome combinations, Elymus s.l. was further divided into ten genera, including the Elymus s.s. (StH), Roegneria K. Koch (StY), Douglasdeweya C. Yen, J. L. Yang & B. R. Baum (StP), Stenostachys Turcz. (HW), Hystrix Moench (NsXm), Campeiostachys Drobov (StYH), Kengyilia C. Yen & J. L. Yang (StYP), Anthosachne Steudel (StYW), Pascopyrum Á. Löve (StHNsXm), and Connorochloa Barkworth, S. W. L. Jacobs & H. Q. Zhang (StYWH) (Yen and Yang 1990; Cai 1997; Yen et al. 2005, 2006, 2011, 2013; Barkworth et al. 2009; Baum et al. 2011). As cryptic genera, Elymus and Campeiostachys are difficult to distinguish based on their morphology. In recent years, several suspected species in Elymus have been identified with the StYH genome and were classified into the genus Campeiostachys (Yang et al. 2015, 2016; Tan et al. 2021, 2022). At present, the genome composition of many species in Elymus has not been identified.
Campeiostachys is an allohexaploid perennial genus of the Triticeae (Poaceae) with StYH genome, Campeiostachys schrenkiana (Fisch. & C. A. Mey.) Drobov as the type species (Baum et al. 2011; Yen et al. 2013). The genus was established by Drobov in 1941 and revised by Baum et al. (2011) and Yen et al. (2013), with 14 species and 13 varieties at present (Baum et al. 2011; Yen et al. 2013; Yang et al. 2015, 2016; Tan et al. 2021, 2022). The majority of the species in Campeiostachys are from Asia, particularly western China, but the genus extends from the Baltic to Japan (Yen et al. 2013). It has been proved that the St sub-genome of Campeiostachys originated from Pseudoroegneria (Nevski) Á. Löve, the H sub-genome originated from Hordeum L., and the diploid donor of the Y sub-genome is still under controversy and unknown (Jensen 1990; Kellogg et al. 1996; Adderley and Sun 2014). Due to the dominant effect of the genes of the St and H genomes, although Elymus (StH) and Campeiostachys (StYH) have different genome combinations, it is almost impossible to distinguish between them depending only on morphological characters (Assadi and Runemark 1995; Baum et al. 2011; Yen et al. 2013). Nevertheless, they differ in genome composition and can be easily distinguished by cytogenetic and molecular methods (Sakamoto and Muramatsu 1966; Zhang et al. 2006; Lei et al. 2016; Tang et al. 2017; Wang et al. 2019).
Genomic in situ hybridization (GISH) and genome-specific fluorescence in situ hybridization (FISH) can effectively detect the genome composition and chromosomal rearrangement of polyploid species in Triticeae (Ørgaard and Heslop-Harrison 1994; Li et al. 2001; Yu et al. 2010; Lucía et al. 2019). Dou et al. (2011) determined the genome composition of six Triticeae species using GISH. Wang et al. (2017) developed a FISH marker St2-80 that can easily distinguish the St genome from other basic genomes in Triticeae. Based on GISH, Lucía et al. (2019) discovered a species in Triticeae whose genome composition is EbP and established a new genus Pauneroa V. Lucía, E. Rico, K. Anamth.-Jon. & M. M. Mart.Ort.
Phylogenetic analysis has been a successful tool for investigating the genome composition, phylogenetic relationship, and parental donor of Triticeae species (Huang et al. 2002; Petersen and Seberg 2002; Nasernakhaei et al. 2015; Tang et al. 2017). The single- or low-copy nuclear genes are less susceptible to concerted evolution and can generate handy markers for polyploid phylogeny (Rauscher et al. 2004; Soltis et al. 2004; Sha et al. 2010). Chloroplast DNA (cpDNA) is inherited from the maternal donor in grasses, so it can provide useful DNA markers for identifying the maternal donor (Mason-Gamer et al. 2002; Liu et al. 2006; Dong et al. 2015; Wang et al. 2019). Tang et al. (2017) determined the genomic constitution of Elymus villosus Muhl. ex Willd, which is StH based on single-copy nuclear sequences. Wang et al. (2019) explored the origin of Elytrigia lolioides (Kar. et Kir.) Nevski is based on two single-copy nuclear genes, one mult-copy nuclear DNA region and one cpDNA region. In summary, GISH and FISH, in combination with molecular phylogenetic analyses based on single-copy nuclear genes and cpDNA regions are efficient to explore the genome constitution and origin of target species in Triticeae.
Elymus atratus (Nevski) Hand.-Mazz. is a perennial wheatgrass. Based on the morphological characteristics, it was assigned to the genus Elymus (Kuo 1987; Yen et al. 2013). At present, its genome composition is still unknown. In the present study, we performed GISH, FISH, and phylogenetic analyses based on two single-copy nuclear genes and one cpDNA region, aiming to: (1) confirm the genome composition of E. atratus; (2) investigate the origin of E. atratus, and provide insight into its taxonomic treatments.
Materials and methods
Plant materials
In this study, samples of E. atratus were collected from Sichuan, China, with accession number ZY15003. The voucher specimens of E. atratus were deposited in the Herbarium of Triticeae Research Institute of Sichuan Agricultural University, China (SAUTI). St2-80 (St genome-specific probe) (Wang et al. 2017), the genomic DNA of Hordeum bogdanii Wilensky (H, 2n = 14) and the genomic DNA of Roegneria ciliaris (Trin.) Nevski (StY, 2n = 28) were used as probes.
E. atratus was also used for phylogenetic analyses. For the phylogenetic analyses, the Acc1, DMC1, and matK accessions of diploid species and polyploid species with three different genome combinations (StY, StH, StYH) from Triticeae were downloaded from GenBank (http://www.ncbi.nlm.nih.gov). The basic information about these sequences is listed in Tables S1, S2, and S3.
Chromosome preparation and in situ hybridization
The roots meristems were collected from adult plants, further treated with nitrous oxide at 1mpa for three hours, then fixed with 90% glacial acetic acid, and kept with 70% alcohol. The chromosome plates were prepared using drop methods (Zhang et al. 2006). The CTAB method (Doyle and Doyle 1990) was used to extract the total genomic DNA from fresh leaves. Plasmid St2-80 was extracted using EndoFree Maxi Plasmid kit (TANGEN BIOTECH, Beijing, China). DNA was labeled using DIG-Nick Translation Kit (Roche, Indianapolis, IN, USA). The hybridization procedure was followed as described by Han et al. (2009). The concentration ratio of the labeling probe genomic DNA and the blocking genomic DNA was 1:100 (ng/uL). Images of GISH and FISH were captured by Olympus BX61 fluorescence microscopy (Japan). Fifteen metaphase cells for E. atratus were observed and images were processed using Adobe Photoshop CS6.
Sequence amplification and phylogenetic analyses
The Acc1, DMC1, and matK genes were amplified with the primers listed in Table 1. The 50 uL reaction mixture was used for PCR amplification, containing 25 uL 1 × phanta mix buffer, 1 mM dNTP mix, 1uL DNA polymerase (Vazyme, Nanjing, China), 10 μm of each primer, 200 ng of template DNA, and distilled deionized water to the 50 uL. PCR products were cloned into the 007VS vector (TSINGKE Biological Technology, Beijing, China). Selected independent clones for sequencing by Sangon Biological Engineering and Technology Service Ltd. (Shanghai, China).
DNA sequences were confirmed through BLAST nucleotide alignment on the NCBI database. The multiple sequences were aligned using MAFFT. jModelTest 3.0 (Posada and Crandall 1998) was used to determine appropriate DNA substitution models and gamma rate heterogeneity. Phylogenetic analyses were conducted using the maximum-likelihood method in PhyML 3.0 (Guindon et al. 2009) and Bayesian inference (BI) in MrBayes version 3.1.2 (Huelsenbeck and Ronquist 2001). Statistical support for nodes in ML analysis was estimated by using 1000 fast bootstrap replicates.
Results
GISH and FISH analyses
We conducted monochrome GISH by using the H genome (from H. bogdanii) as probe and StY genome (from R. ciliaris) as block. The results showed that E. atratus contains 2n = 42 chromosomes (Fig. 1a, d). Fourteen of 42 chromosomes displayed strong H signal on the entire length (Fig. 1b, c). In addition, the two other chromosomes also showed a bright H hybridization signal but only in part of the long arms of the chromosomes (Fig. 1c). In contrast, 28 chromosomes displayed a StY signal when using the StY genome as probe and the H genome as block (Fig. 1e, f). Specifically, twenty-six chromosomes exhibited the entire chromosome signal while two chromosomes showed a partly bright hybridization signal (Fig. 1f). The results of double-color GISH are consistent with monochrome GISH (Fig. 1g–i). Thus, the genomic constitution of E. atratus is StYH with one pair of chromosomes showing translocations between H and St/Y subgenomes.
To determine the translocation type in the two translocated chromosomes, we performed GISH and FISH using the H genome (from H. bogdanii) and St2-80 (St genome-specific FISH probe) as probes to distinguish H and St genomes, respectively (Fig. 2). The GISH results showed that 14 chromosomes of E. atratus displayed H signals on the entire length, and part of the long arms of two chromosomes also showed H genome signals (Fig. 2b, d). The FISH results showed that 14 chromosomes displayed St2-80 signal and no St-type signal was observed on the two translocated chromosomes (Fig. 1c, d). It is no doubt that the translocation type in the two translocated chromosomes is between the subgenome H and the subgenome Y.

GISH on somatic metaphase cells from root tips of Elymus atratus. a chromosomes of E. atratus counterstained by DAPI. b, c 14 chromosomes showing H signal when probed with H-genome DNA of H. bogdanii and blocked with StY-genome DNA of R. ciliaris, in addition, two translocation chromosomes are observed (indicated by arrows). d chromosomes of E. atratus counterstained by DAPI. e, f 28 chromosomes showing red fluorescence (StY) when probed with StY-genome DNA of R. ciliaris and blocked with H-genome DNA of H. bogdanii. Two translocated chromosomes are observed (indicated by arrows). g–i 14 chromosomes showing H signal and 28 chromosomes showing StY signal using H-genome DNA and StY-genome DNA as probes. Two translocated chromosomes are observed (indicated by arrows). Bar = 10 μm

GISH and FISH on somatic metaphase cells from root tips of Elymus atratus. a chromosomes of E. atratus counterstained by DAPI. b H genome DNA as probe to generate hybridization signals on chromosomes of E. atratus in GISH. c St2-80 as probe to generate hybridization signals on chromosomes of E. atratus in FISH. d, 14 chromosomes of E. atratus showing H genome signals on the entire length and part of the long arms of two chromosomes also showing H genome signals (indicated by arrows), 14 chromosomes of E. atratus showing St type St2-80 signals. Bar = 10 μm
Phylogenetic analyses
Phylogenetic analysis of Acc1 gene sequences
The Acc1 sequence length of Elymus atratus ranged from 1436 to 1440 bp. The Acc1 data matrix contained 1617 characters, of which 261 were variable characters and 130 were informative. The dataset has 71 sequences for phylogenetic analysis, using the species Bromus inermis L. as the outgroup. A single phylogenetic tree was generated using TIM + F + I + G4 which is the best-fitted model (− Ln likelihood = 6929.715).

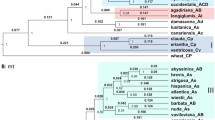
Maximum likelihood tree based on Acc1 gene sequences of Elymus atratus and its related species using Bromus inermis as outgroup. The capital letters in brackets after the species name indicate the genome composition of the species. The numbers above and below the branches indicate bootstrap values > 50% and Bayesian posterior probability values > 90%, respectively. Vertical bars indicate the subgenome that characterize each of the three main clades of the phylogeny
Both ML and BI analyses with the Acc1 sequences of E. atratus were divided into three clades, namely St, Y, and H clades (Fig. 3). In the St clade, five diploid species of Pseudoroegneria (St genome donor) plus five tetraploid species of Elymus with the StH genomes, five tetraploid species of Roegneria with the StY genomes, and E. atratus (BS = 85%, PP = 1.00) were clustered. The Y clade contained Peridictyon sanctum (Boiss.) Nevski with the Xp genome, two diploid species of Dasypyrum (Coss. & Durieu) T. Durand with the V genome, five Roegneria species (StY), and E. atratus (BS = 96%, PP = 1.00). The H clade included three diploid Hordeum species (H genome donor), five Elymus species (StH), and E. atratus (BS = 98%, PP = 1.00). Thus, the phylogenetic analysis of Acc1 sequences illustrated that E. atratus contains St, Y, and H sub-genomes.
Phylogenetic analysis of DMC1 gene sequences
The DMC1 sequence length of E. atratus ranged from 978 to 981 bp. The DMC1 data matrix contained 1121 characters, 860 of which were constant, 270 were variable, and 113 were informative. The dataset included 62 sequences for phylogenetic analysis, using the species Bromus inermis L. as the outgroup. We inferred a phylogenetic tree based on HKY + F + G4 which is the best-fitted model (− Ln likelihood = 4813.056).
The DMC1 sequences from E. atratus were divided into three clades, named St, Y, and H clades, respectively (Fig. 4). The St clade included five diploid species in Pseudoroegneria (St genome donor), five tetraploid species in Elymus (StH), and six tetraploid species in Roegneria (StY), and E. atratus (BS = 96%, PP = 1.00). The Y clade only contained Roegneria (StY) and E. atratus (BS = 96%, PP = 1.00). The H clade contained five diploid species in Hordeum (H genome donor), five Elymus species (StH), and E. atratus (BS = 91%, PP = 1.00). The phylogenetic analysis of DMC1 sequences suggested that E. atratus is an allopolyploid species containing St, Y, and H genomes.

Maximum likelihood tree based on DMC1 gene sequences of Elymus atratus and its related species using Bromus sterilis as outgroup. The capital letters in brackets after the species name indicate the genome composition of the species. The numbers above and below the branches indicate bootstrap values > 50% and Bayesian posterior probability values > 90%, respectively. Vertical bars indicate the subgenome that characterize each of the three main clades of the phylogeny
Phylogenetic analysis of matK gene sequences
The matK sequences length of E. atratus was 844 bp. The matK data matrix contained 844 characters, 714 of which were constant, 98 were variable, and 61 were informative. This dataset contained 42 sequences for phylogenetic analysis, using the species Bromus tectorum L. as the outgroup. We inferred a phylogeny based on TIM + F + G4, which is the best-fitted model (− Ln likelihood = 2092.347).

Maximum likelihood tree based on matK gene sequences of Elymus atratus and its related species using Bromus tectorum as outgroup. The capital letters in brackets after the species name indicate the genome composition of the species. The numbers above and below the branches indicate bootstrap values > 50% and Bayesian posterior probability values > 90%, respectively. Vertical bars indicate the subgenome that characterize each of the twoe main clades of the phylogeny
The matK sequence from E. atratus was divided into two clades, named St + V + E + D and H (Fig. 5). The St + V + E + D clade included five diploid species in Pseudoroegneria (St genome donor), Dasypyrum villosum (V genome donor), Thinopyrum bessarabicum (Eb genome donor), Lophopyrum elongatum (Ee genome donor), one species in Elymus (StH), and 12 tetraploid species in Roegneria (StY), and E. atratus (BS = 63%). E. atratus formed a subclade with Pseudoroegneria strigosa (St), Pseudoroegneria spicata (St), and Roegneria dura (StY) (BS = 78%). The H clade only contained five diploid species in Hordeum (H and I genome donor) (BS = 100%, PP = 1.00). Other diploid species in Triticeae clustered into other clades with high support. The matK sequences suggest that the maternal donor of E. atratus contains the St genome.
Discussion
Genome constitution of Elymus atratus
According to the flora of China, E. atratus is a hexaploid wheatgrass and is mainly distributed in China (Sichuan, Qinghai, Gansu, Xinjiang, and Tibet). Liu (1985) conducted a karyotype analysis of E. atratus, which was collected in the Aba Tibetan Autonomous Prefecture of Sichuan, and the results showed that it contained 42 chromosomes. Surprisingly, cytological observations by Lu et al. (1990) not only identified the hexaploid karyotype but also reported a tetraploid karyotype of E. atratus. Whether tetraploid or hexaploidy, the genome composition of E. atratus has not been reported in previous studies. In the present study, the results of GISH and FISH detected that E. atratus has 14 St genome chromosomes, 14 Y genome chromosomes, and 14 H genome chromosomes. The allopolyploid species in Triticeae contains a corresponding copy of each basic genome, and these copies are clustered separately with the diploid donor (Petersen et al. 2006; Lei et al. 2022). The phylogenetic analysis based on Acc1 and DMC1 sequences showed that E. atratus have St, Y, and H subgenome copies, suggesting that E. atratus is an allohexaploid species. The results of the cytological and phylogenetic analysis proved that E. atratus is an allohexaploid, and the genome formula is StStYYHH.
Origin of the Y genome
The origin of the Y genome remains enigmatic. Some researchers believe that it is derived from the St genome, and others think its origin is from an unknown donor but has not been discovered or extinct (Jensen 1990; Kellogg et al. 1996; Mason-Gamer et al. 2005). Liu et al. (2006) based on sequences from the ITS nuclear DNA region concluded that the Y genome was gradually differentiated from the St genome. Okito et al. (2009) based on RAPD results also supported the Y genome differentiation from the St genome. Based on five gene sequences, Liu et al. (2022) suggested that allotetraploid species with the StY genome may have originated from the autotetraploid (StSt) of Pseudoroegneria. Alternatively, additional research results support the independent origin of the Y genome from a diploid species (Dewey 1984; Sun et al. 2008; Sun and Komatsuda 2010; Lei et al. 2022). Sun and Komatsuda (2010) inferred that the Y genome originated independently from a diploid species based on sequences of the EF-G nuclear single-copy gene and that Y genome was most similar to the W genome than to the St genome. Adderley and Sun (2014) based on phylogenetic analyses of sequences of the Pgk1 single-copy gene, inferred that the Y genome originated independently, and the Y genome donor was closely related to the V genome and Xp genome. Lei et al. (2022) also supported that the Y and St genomes were of different origins and closely related to the V and Xp genomes. In our study, phylogenetic analysis results based on Acc1 and DMC1 sequences showed that the St clade and Y clade were separated with high support. In conclusion, we suggest that the origin of the Y genome and the St genome is independent.
Origin of Elymus atratus
Allopolyploids of Triticae are produced by interspecific hybridization of different genera with different sets of genomes (Sears 1954; Kimber and Alonso 1981; Dewey 1984; Heslop-Harrison 1992; Petersen et al. 2011). In the present study, the results of GISH, FISH, and phylogenetic analyses based on the single-copy genes Acc1 and DMC1 confirmed that the genome composition of E. atratus is StYH. These results indicate that the parents of E. atratus are species with St, Y, or H genomes. There are several possibilities for the origin of E. atratus: St × HY, Y × StH, or H × StY. On the one hand, the Y genome originated independently from a diploid species with the Y genome which have not been identified yet (Torabinejad and Mueller 1993; Gao et al. 2014). On the other hand, the known genera with Y genome are Roegneria (StY), Campeiostachys (StYH), Kengyilia (StYP), Anthosachne (StYW), and Connorochloa (StYWH). None species with the genome combination HY has ever been reported. In conclusion, E. atratus is probably the results of the natural cross between a tetraploid Roegneria (StY) and a diploid of Hordeum (H).
The cpDNA is maternally inherited in grasses (Middleton et al. 2014). Therefore, sequences from cpDNA regions have been widely used to identify the maternal donor of allopolyploid species or genera in Triticeae (Yan et al. 2014; Dong et al. 2015; Sha et al. 2017; Wang et al. 2019). Lei et al. (2018) based on the analyses of sequences of ndhF and trnH-psbA cpDNA reginos speculated that St or StY might be the maternal donor of the species of Campeiostachys. In this study, phylogenetic analysis based on matK gene sequences revealed that E. atratus formed a subclade with Pseudoroegneria strigose (St), Pseudoroegneria spicata (St), and Roegneria dura (StY), this suggests that the maternal donor of E. atratus contains the St genome. In summary, we believe that E. atratus may have been originated from a natural cross between a tetraploid species of Roegneria as a maternal donor and a diploid species of Hordeum as a paternal donor.
Taxonomic treatment of Elymus atratus
The main morphological characteristics of Elymus atratus are as follows: leaf sheaths glabrous; leaf blades involute, often coiled; spikes flexuous and nodding, with dense spikelets; two spikelets on each rachis node, spikelets are black and purple after maturity; glumes oblong or lanceolate, apex acuminate; lemmas lanceolate, lemma awned (10–25 mm); palea equal to lemma (Yen et al. 2013). Based on the morphological characteristics, Elymus atratus belongs to the Elymus (Yen et al. 2013). But phenotype is the co-consequence of genetics and environments. Some studies have shown that there are cryptic species (such as Roegneria panormitana (Parl.) Nevski and R. heterophylla (Bornm. ex Melderis & Rech. f.) C. Yen, J. L. Yang & B. R. Baum) and cryptic genera (such as Elymus and Campeiostachys) in Triticeae (Baum et al. 2011; Yen et al. 2013). As cryptic genera, although Elymus and Campeiostachys are morphologically similar, their genome composition is different, Elymus contains the StH genome and Campeiostachys contains the StYH (Yen et al. 2013). Therefore, we can distinguish between those two genera by determining the genome composition of the species. The GISH, FISH, and phylogenetic analyses results demonstrated that the genome constitution of Elymus atratus is StYH. According to the genome classification systems in Triticeae, E. atratus should be transferred to the genus Campeiostachys and renamed Campeiostachys atrata (Nevski) Y. H. Zhou, H. Q. Zhang & L. Tan.
References
Adderley S, Sun GL (2014) Molecular evolution and nucleotide diversity of nuclear plastid phosphoglycerate kinase (PGK) gene in Triticeae (Poaceae). Gene 533:142–148
Assadi M, Runemark H (1995) Hybridisation, genomic constitution and generic delimitation in Elymus s. l. (Poaceae: Triticeae). Plant Syst Evol 194:189–205
Barkworth ME, Jacobs SWL (2009) Connorochloa: a new genus in Triticeae. Breed Sci 59:685–686
Baum BR, Yang JL, Yen C (1995) Taxonomic separation of Kengyilia (Poaceae: Triticeae) in relation to nearest related Roegneria, Elymus, and Agropyron, based on some morphological characters. Plant Syst Evol 194:123–132
Baum BR, Yang JL, Yen C, Agafonov AV (2011) A taxonomic synopsis of the genus Campeiostachys Drobov. J Syst Evol 49:146–159
Cai LB (1997) A taxonomical study on the genus Roegneria C. Koch from China. Acta Phytotax Sin 35:148–177
Dewey DR (1984) The genomic system of classification as a guide to intergeneric hybridization with the perennial Triticeae. In: Gustafson JP (ed) Gene manipulation in plant improvement: 16th Stadler genetics symposium. Springer US, Boston, pp 209–279
Dong ZZ, Fan X, Sha LN, Wang Y, Zeng J, Kang HY, Zhang HQ, Wang XL, Zhang L, Ding CB (2015) Phylogeny and differentiation of the St genome in Elymus L. sensu lato (Triticeae; Poaceae) based on one nuclear DNA and two chloroplast genes. BMC Plant Biol 15:179
Dou QW, Zhang TL, Tsujimoto H (2011) Physical mapping of repetitive sequences and genome analysis in six Elymus species by in situ hybridization. J Syst Evol 49:347–352
Doyle JJT, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Gao G, Gou XM, Wang Q, Zhang Y, Deng JB, Ding CB, Zhang L, Zhou YH, Yang RW (2014) Phylogenetic relationships and Y genome origin in Chinese Elymus (Triticeae: Poaceae) based on single copy gene DMC1. Biochem Syst Ecol 57:420–426
Guindon S, Delsuc F, Dufayard JF, Gascuel O (2009) Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137
Han FP, Gao Z, Birchler JA (2009) Reactivation of an inactive centromere reveals epigenetic and structural components for centromere specification in Maize. Plant Cell 21:1929–1939
Heslop-Harrison JS (1992) Molecular cytogenetics, cytology and genomic comparisons in the Triticeae. Hereditas 116:93–99
Huang SX, Sirikhachornkit A, Su XJ, Faris J, Gill B, Haselkorn R, Gornicki P (2002) Genes encoding plastid acetyl-CoA carboxylase and 3-phosphoglycerate kinase of the Triticum/Aegilops complex and the evolutionary history of polyploid wheat. Proc Natl Acad Sci 99:8133–8138
Huelsenbeck JP, Ronquist F (2001) MrBayes: bayesian inference of phylogenetic trees. Bioinformatics 17:754–755
Jensen KB (1990) Cytology, fertility, and morphology of Elymus kengii (Keng) Tzvelev and E. grandiglumis (Keng) A. Lve (Triticeae: Poaceae). Genome 33:563–570
Kellogg EA, Appels R, Mason-Gamer RJ (1996) When genes tell different stories: the diploid genera of Triticeae (Gramineae). Syst Bot 21:321–347
Kimber G, Alonso LC (1981) The analysis of meiosis in hybrids. III. Tetraploid hybrids. Can J Genet Cytol 23:235–254
Kuo PC (1987) Flora republicae popularis sinicae. Science Press, Beijing
Lei YX, Fan X, Sha LN, Wang Y, Kang HY, Zhou YH, Zhang HQ (2016) Phylogenetic relationships and the maternal donor of Roegneria (Triticeae: Poaceae) based on three nuclear DNA sequences (ITS, Acc1, and Pgk1) and one chloroplast region (trnL-F). J Syst Evol 65:185–191
Lei YX, Liu J, Fan X, Sha LN, Wang Y, Kang HY, Zhou YH, Zhang HQ (2018) Phylogeny and maternal donor of Roegneria and its affinitive genera (Poaceae: Triticeae) based on sequence data for two chloroplast DNA regions (ndhF and trnH–psbA). J Syst Evol 56:105–119
Lei YX, Fan X, Sha LN, Wang Y, Kang HY, Zhou YH, Zhang HQ (2022) Phylogenetic relationships and the maternal donor of Roegneria (Triticeae: Poaceae) based on three nuclear DNA sequences (ITS, Acc1, and Pgk1) and one chloroplast region (trnL-F). J Syst Evol 60:305–318
Li CB, Zhang DM, Ge S, Lu BR, Hong DY (2001) Identification of genome constitution of Oryza malampuzhaensis, O. minuta, and O. punctata by multicolor genomic in situ hybridization. Theor Appl Genet 103:204–211
Liu YH (1985) Studies on the karyotypes of 11 species of Elymus from China. Plant Sci J 3:325–330
Liu QL, Ge S, Tang HB, Zhang XL, Zhu GF, Lu BR (2006) Phylogenetic relationships in Elymus (Poaceae: Triticeae) based on the nuclear ribosomal internal transcribed spacer and chloroplast trnL-F sequences. New Phytol 170:411–420
Liu QL, Liu L, Ge S, Fu LP, Bai SQ, Lv X, Wang QK, Chen W, Wang FY, Wang LH et al (2022) Endo-allopolyploidy of autopolyploids and recurrent hybridization—A possible mechanism to explain the unresolved Y-genome donor in polyploid Elymus species (Triticeae: Poaceae). J Syst Evol 60:344–360
Löve Á (1984) Conspectus of the Triticeae. Feddes Repertorium 95:425–521
Lu BR (1993) Meiotic studies of Elymus nutans and E. jacquemontii (Poaceae, Triticeae) and their hybrids with Pseudoroegneria spicata and seventeen Elymus species. Plant Syst Evol 186:193–212
Lu BR, Yan J, Yang JL (1990) Cytological observations on Triticeae materials from Xinjiang. Acta Bot Yunnan 12:57–66
Lucía V, Enrique R, Anamthawat-Jónsson K, Martínez-Ortega MM (2019) Cytogenetic evidence for a new genus of Triticeae (Poaceae) endemic to the Iberian Peninsula: description and comparison with related genera. Bot J Linn Soc 4:523–546
Mason-Gamer RJ, Orme NL, Anderson CM (2002) Phylogenetic analysis of north American Elymus and the monogenomic Triticeae (Poaceae) using three chloroplast DNA data sets. Genome 45:991–1002
Mason-Gamer RJ, Burns MM, Naum M (2005) Polyploidy, introgression, and complex phylogenetic patterns within Elymus. Czech J Genet Plant Breed 41:21–26
Middleton CP, Senerchia N, Stein N, Akhunov ED, Keller B, Wicker T, Kilian B (2014) Sequencing of chloroplast genomes from wheat, barley, rye and their relatives provides a detailed insight into the evolution of the Triticeae tribe. PLoS ONE 9:e85761
Nasernakhaei F, Rahiminejad MR, Saeidi H, Tavassoli M (2015) Phylogenetic relationships among the Iranian Triticum diploid gene pool as inferred from the loci Acc1 and Pgk1. Phytotaxa 201:111–121
Okito P, Mott IW, Wu YJ, Wang RC (2009) A Y genome specific STS marker in Pseudoroegneria and Elymus species (Triticeae: Gramineae). Genome 52:391–400
Ørgaard M, Heslop-Harrison JS (1994) Investigation of genome relationships between Leymus psathyrostachys and Hordeum inferred from genomic DNA: DNA in situ hybridization. Ann Botany 73:195–203
Petersen G, Seberg O (2002) Molecular evolution and phylogenetic application of DMC1. Mol Phylogenet Evol 22:43–50
Petersen G, Seberg O, Yde M, Berthelsen K (2006) Phylogenetic relationships of Triticum and Aegilops and evidence for the origin of the A, B, and D genomes of common wheat (Triticum aestivum). Mol Phylogenet Evol 39:70–82
Petersen G, Seberg O, Salomon B (2011) The origin of the H, St, W, and Y genomes in allotetraploid species of Elymus L. and Stenostachys Turcz. (Poaceae: Triticeae). Plant Syst Evol 291:197–210
Posada D, Crandall K (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Rauscher JT, Doyle JJ, Brown AH (2004) Multiple origins and nrDNA internal transcribed spacer homeologue evolution in the Glycine tomentella (Leguminosae) allopolyploid complex. Genetics 166:987–998
Sakamoto S, Muramatsu M (1966) Cytogenetic studies in the tribe Triticeae. II. Tetraploid and hexaploid hybrids of Agropyron. Japanese J Genet 41:155–168
Sears ER (1954) The aneuploids of common wheat. In, pp 1–59
Sha LN, Fan X, Yang RW, Kang HY, Ding CB, Zhang L, Zheng YL, Zhou YH (2010) Phylogenetic relationships between Hystrix and its closely related genera (Triticeae; Poaceae) based on nuclear Acc1, DMC1 and chloroplast trnL-F sequences. Mol Phylogenet Evol 54:327–335
Sha LN, Fan X, Wang XL, Dong ZZ, Zeng J, Zhang HQ, Kang HY, Wang Y, Liao JQ, Zhou YH (2017) Genome origin, historical hybridization and genetic differentiation in Anthosachne australasica (Triticeae; Poaceae), inferred from chloroplast rbc L, trn H- psb A and nuclear Acc1 gene sequences. Ann Botany 119:95–107
Soltis DE, Soltis PS, Tate JA (2004) Advances in the study of polyploidy since plant speciation. New Phytol 161:173–191
Sun G, Komatsuda T (2010) Origin of the Y genome in Elymus and its relationship to other genomes in Triticeae based on evidence from elongation factor G (EF-G) gene sequences. Mol Phylogenet Evol 56:727–733
Sun G, Ni Y, Daley T (2008) Molecular phylogeny of RPB2 gene reveals multiple origin, geographic differentiation of H genome, and the relationship of the Y genome to other genomes in Elymus species. Mol Phylogenet Evol 46:897–907
Tan L, Zhang HQ, Chen WH, Deng MQ, Zhou YH (2021) Genome composition and taxonomic revision of Elymus purpuraristatus and Roegneria calcicola (Poaceae: Triticeae) based on cytogenetic and phylogenetic analyses. Bot J Linn Soc 196:245–255
Tan L, Huang QX, Song Y, Wu DD, Cheng YR, Zhang CB, Sha LN, Fan X, Kang HY, Wang Y et al (2022) Biosystematics studies on Elymus breviaristatus and Elymus sinosubmuticus (Poaceae: Triticeae). BMC Plant Biol 22:57
Tang C, Qi J, Chen N, Sha LN, Wang Y, Zeng J, Kang HY, Zhang HQ, Zhou YH, Fan X (2017) Genome origin and phylogenetic relationships of Elymus villosus (Triticeae: Poaceae) based on single-copy nuclear Acc1, Pgk1, DMC1 and chloroplast trnL-F sequences. Biochem Syst Ecol 70:168–176
Torabinejad J, Mueller RJ (1993) Genome analysis of intergeneric hybrids of apomictic and sexual Australian Elymus species with wheat, barley and rye: implication for the transfer of apomixis to cereals. Theor Appl Genet 86:288–294
Wang L, Shi QH, Su HD, Wang Y, Sha LN, Fan X, Kang HY, Zhang HQ, Zhou YH (2017) St2-80 - a new FISH marker for St genome and genome analysis in Triticeae. Genome 60:553–563
Wang L, Jiang YY, Shi QH, Wang Y, Sha LN, Fan X, Kang HY, Zhang HQ, Sun GL, Zhang L et al (2019) Genome constitution and evolution of Elytrigia lolioides inferred from Acc1, EF-G, ITS, TrnL-F sequences and GISH. BMC Plant Biol 19:158
Yan C, Hu Q, Sun G (2014) Nuclear and chloroplast DNA phylogeny reveals complex evolutionary history of Elymus pendulinus. Genome 57:97–109
Yang CR, Zhang HQ, Zhao FQ, Liu XY, Fan X, Sha LN, Kang HY, Wang Y, Zhou YH (2015) Genome constitution of Elymus tangutorum (Poaceae: Triticeae)inferred from meiotic pairing behavior and genomic in situ hybridization. J Syst Evol 53:529–534
Yang C-R, Baum B-R, Chen W-H, Zhang H-Q, Liu X-Y, Fan X, Sha L-N, Kang H-Y, Wang Y, Zhou Y-H (2016) Genomic constitution and taxonomy of the Chinese hexaploids Elymus cylindricus and E. breviaristatus (Poaceae: Triticeae). Bot J Linn Soc 182:650–657
Yen C, Yang JL (1990) Kengyilia gobicola, a new taxon from West China. Can J Bot 68:1894–1897
Yen C, Yang JL, Baum BR (2005) Douglasdeweya: a new genus, with a new species and a new combination (Triticeae: Poaceae). Can J Bot 83:413–419
Yen C, Yang JL, Baum BR (2006) Biosystematics of Triticeae: volume I. China Agricultural Press, Beijing
Yen C, Yang JL, Baum BR (2011) Biosystematics of Triticeae: volume III. China Agricultural Press, Beijing
Yen C, Yang JL, Baum BR (2013) Biosystematics of Triticeae: volume V. China Agricultural Press, Beijing
Yu HQ, Zhang C, Ding CB, Zhang HQ, Zhou YH (2010) Genome constitutions of Roegneria alashanica, R. elytrigioides, R. magnicaespes and R. grandis (Poaceae: Triticeae) via genomic in-situ hybridization. Nord J Bot 28:1–6
Zhang HQ, Zhou YH (2007) Meiotic analysis of the interspecific and intergeneric hybrids between Hystrix patula Moench and H. Duthiei ssp. longearistata, Pseudoroegneria, Elymus, Roegneria, and Psathyrostachys species (Poaceae, Triticeae). Bot J Linn Soc 153:213–219
Zhang HQ, Yang RW, Dou QW, Tsujimoto H, Zhou YH (2006) Genome constitutions of Hystrix patula, H. duthiei ssp. duthiei and H. duthiei ssp. longearistata (Poaceae: Triticeae) revealed by meiotic pairing behavior and genomic in-situ hybridization. Chromosome Res 14:595–604
Acknowledgements
We would like to express our appreciation to Xuehua Deng for her management of the experimental plants. This study was supported by the National Natural Science Foundation of China (Grant Nos. 32270388 and 32200180) and the Science and Technology Bureau of Sichuan Province (23NSFSC1995).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Table S1 The accession numbers of Acc1 gene sequences used in phylogenetic analysis; Table S2 The accession numbers of DMC1 gene sequences used in phylogenetic analysis; Table S3 The accession numbers of matK gene sequences used in phylogenetic analysis. Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tan, L., Wu, DD., Zhang, CB. et al. Genome constitution and evolution of Elymus atratus (Poaceae: Triticeae) inferred from cytogenetic and phylogenetic analysis. Genes Genom 46, 589–599 (2024). https://doi.org/10.1007/s13258-024-01496-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-024-01496-9