
Abstract
Background
The innate immune regulation, especially by the type I IFN signature in the CD14+ monocytes, is known to be critical in the pathogenesis of autoimmune Sjögren’s syndrome (SjS) and systemic lupus erythematosus (SLE).
Objective
Since patients with one condition can be overlapped with another, this study is to identify shared differentially expressed genes (DEGs) in SjS and SLE compared to healthy controls (HCs) and refine transcriptomic profiles with the integrated Reactome and gene-drug network analysis for an anti-inflammation therapy.
Methods
CD14+ monocytes were purified from whole blood of SjS and SLE patients (females, ages from 32 to 62) and subject to bulk RNA-sequencing, followed by data analyses for comparison with HC monocytes (females, ages 30 and 33). Functional categorizations, using Gene Ontology (GO) and the Reactome pathway analysis, were performed and DEGs associated with therapeutic drugs were identified from the Drug Repurposing Hub (DHUB) database.
Results
The GO analysis revealed that DEGs in the inflammatory response and the cellular response to cytokine were highly enriched in both conditions. A propensity toward M1 macrophage differentiation appears to be prominent in SjS while the Response to Virus was significant in SLE monocytes. Through the Reactome pathway analysis, DEGs in the IFN signaling and the cytokine signaling in immune system were most significantly enriched in both. Upregulation of NGF-induced transcription activity in SjS and the complement cascade activity in SLE were also noted. Multiple anti-inflammatory drugs, such as prostaglandin-endoperoxide synthase and angiotensin-I-converting- enzyme were associated with the DEGs in these conditions.
Conclusions
Taken together, our analysis indicates distinct inflammatory transcriptomic profiles shared in SjS and SLE monocytes. Comprehensive characterizations of the data from these conditions will ultimately allow differential diagnosis of each condition and identification of therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sjögren’s Syndrome (SjS) is an autoimmune disease characterized by the inflammation of the salivary and lacrimal glands. Systemic lupus erythematosus (SLE) is also an autoimmune condition that causes widespread inflammation, affecting the kidneys, joints, skin, lungs, and blood vessels. It is estimated that around 20% of SLE patients are also affected by SjS and about 15–35% of SjS patients overlap with SLE (Ramos-Casals et al. 2007). The etiology is poorly understood, and many studies have investigated variety of factors, such as genetics, immune dysregulation, and epigenetics, to identify causes or regulators for these conditions. Monocytes have become an exciting cell type of interest as they act as a central player for the innate immune responses and serving as the foundation of the adaptive immune responses.
Monocytes have been closely studied in their roles in autoimmune diseases although their etiopathogenic roles remain undefined. Increased levels of non-classical monocytes have been observed in patients with SjS (Wildenberg et al. 2009). In addition, SjS monocytes have described dysfunctionality in the ability to engulf apoptotic epithelial cells and stimulate an immunosuppressant cytokine profile (Hauk et al. 2014). Instead, proinflammatory cytokine and chemokine secretion, such as IL-6, IL-18, type I IFN and BAFF, in SjS monocytes were up-regultaed while NF-κB inhibitor was down-regulated.
Unlike SjS, SLE has not demonstrated a clear distinction in the level or proportion of monocytes in patients. Results from one study showed an increased tendency for monocytes to undergo apoptosis with C5a complement involved (Chalmers et al. 2015). Monocytes also contribute to SLE pathogenesis through their regulation of the adaptive immune response. CD40L overexpression in SLE monocytes suggests a potential role in promoting B cell hyperactivity, which is observed in SLE patients, through its respective pathway. Furthermore, SLE monocytes tend to differentiate into dendritic cells with higher CD86 levels after IFN-α treatment, enhancing their capabilities to present autoantigens from autoreactive T and B cells (Blanco et al. 2001; Decker et al. 2006).
The transcriptional profile of SjS and SLE monocytes has been a topic of interest in elucidating the roles of monocytes in disease pathogenesis. Gene co-expression network analysis revealed four molecular signatures involved in process related with translation, IFN-signaling, and toll-like receptor signaling, which correlated with systemic and local inflammation. These gene signatures were partially inhibited by IFN-α and IFN-β receptor blockade, suggesting that circulating inflammatory mediators, including type I IFNs, contribute to the altered transcriptional profile of primary SjS monocytes (Lopes et al. 2021).
Analyses of these subsets revealed that “genes highly expressed in intermediate and nonclassical monocytes were significantly enriched in the transcriptional profile of primary SjS monocytes” (Lopes et al. 2021). Another study utilized single cell RNA sequencing (scRNA-seq) to identify 11 different monocyte subsets in SjS patients. The majority of these subsets had higher expression of TNF Superfamily Member 10 (TNFSF10) which is one of cytokine that belongs to the tumor necrosis factor (TNF) ligand family. This protein preferentially induces apoptosis in transformed and tumor cells but does not appear to kill normal cells although it is expressed at a significant level in most normal tissues and upregulation of interferon-related and neutrophil activation-associated pathways (He et al. 2022). Transcriptomic analysis of SLE immune cells belonging to a multiethnic cohort was performed with bulk RNA-seq. The researchers of this study utilized a four-tier approach, taking account a variety of factors, in order to identify various SLE subgroups, resulting in two subsets for NK and B cells, and three for CD4+ and CD14+ cells. From the data gathered, this study surveyed the variations in gene expression across different ethnic groups, bringing patient-specific care in SLE a step closer (Andreoletti et al. 2021).
The objective of this study is to identify global RNA expression profiles of CD14+ monocytes of SjS and SLE patients in an unbiased manner by bulk RNA-seq and utilize the transcriptome data for protein–protein interactions and gene-drug network analysis. Our study is the very first study to explore the disease-specific transcriptional change of monocytes in SjS and SLE patients, and by elucidating various biological roles and pathways, the ecology of immune responses common to monocytes in SjS and SLE and each specific immunity reaction, such as interferon alpha/beta/gamma signaling and cytokine signaling was announced.
Materials and methods
Ethics statement
This study was approved by the University of Florida Institutional Review Board, and a written permission was obtained from all who participated in the study. All clinical investigation was performed in accordance with the Declaration of Helsinki.
Study participants
The total of eight patients with SjS or systemic lupus erythematosus (SLE) diagnosis (n = 4/group) who were enrolled and donated their blood for the present study. The patients’ ages ranged from 47 to 56 for SjS and from 32 to 62 for SLE patients. They were all female patients. SjS diagnosis was made based on the 2012 American College of Rheumatology classification criteria (Shiboski et al. 2012). The 1997-updated criteria of the 1982-revised ACR criteria were used for SLE (Hochberg 1997). Two healthy controls (HC) include subjects without any signs and symptoms of dry mouth and/or dry eyes, or known autoimmune disorders. The possibility of bacterial or viral infection was evaluated by serology to rule out any primary and secondary infection.
CD14+ monocyte purification
Human whole blood samples were added with EDTA (1 mM final concentration) and RosetteSep™ Human Monocyte Enrichment Cocktail (STEMCELL Technologies, Vancouver, BC, Canada) and incubated for 20 min at room temperature. Blood samples were diluted with recommended medium (PBS containing 2% FBS and 1 mM EDTA), added to SepMate™-50 (STEMCELL Technologies, Vancouver, BC, Canada) containing Lymphoprep™ (STEMCELL Technologies, Vancouver, BC, Canada), and centrifuged at 1200×g for 10 min. Enriched cells were washed recommended medium and centrifuged at 500×g for 10 min. Recommended medium was aspirated, and remaining monocytes were collected for use.
RNA sample preparation and RNA-seq data production
The harvested monocytes isolated from patient whole blood were separated and homogenized in 500 ul of TRIzol reagent (Invitrogen, Carlsbad, CA, USA) using a micro-homogenizer following the manufacturer’s instructions. Ten RNA samples were purified using the RNeasy Mini Kit (Qiagen, Hilden, Germany), following the manufacturer’s instructions. Prior to constructing RNA-seq libraries, the quality of all RNA samples was checked by 28S/18S ratio and RNA integrity number (RIN) value using Agilent TapeStation 2100 system (Agilent Technologies, Santa Clara, CA, USA). All RNA samples show more than 8.0 RIN (RNA integrity number) values (Supplementary Fig. A). mRNA molecules were enriched and purified from 2 μg of the qualified RNA samples using oligo-dT magnetic beads. Double-stranded cDNA was immediately synthesized by SuperScript III reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA). According to the instruction of the NEBNext® Ultra™ RNA Library Prep Kit (Illumina, San Diego, CA, USA), a sequential process of end-repair, poly-A addition, and adaptor ligation on both ends was carried out. The final selected libraries were evaluated with Agilent TapeStation 2100 system and were 400–500 bp in size. The cDNA libraries were sequenced with an Illumina Novaseq 6000 (Illumina, San Diego, CA, USA), which generated paired-end (PE) reads of approximately 100 bp in size.
RNA-seq data analysis
Raw sequencing data were evaluated to discard low-quality reads by FAST-QC (https://www.bioinformatics.babraham.ac.uk/) as follows steps: Reads including more than 10% of skipped bases (marked as ‘N’s); sequencing reads including more than 40% of bases whose quality score is less than 20; their average quality score (< 20). Quality distributions of nucleotides, GC contents, the proportions of PCR duplication, and k-mer frequencies of sequencing data were also calculated (Martin and Wang 2011).
RNA-seq analysis and statistical comparison of differentially expressed genes (DEGs)
Only highly qualified reads to increase mapping quality were mapped to the human reference genome (Homo sapiens: GRCh38) using an accurate aligner TopHat2 v2.1.1 to identify express genes (Kim et al. 2013). We only used uniquely mapped read pairs for the analysis of downstream DEGs. Gene expression level was quantified by an R package, DESeq2 v1.26.0 (Love et al. 2014). DEGs for three groups were analyzed using DESeq2 methods in R. The DEGs with log2 fold-change (log2 FC) more than one and adjusted P-value (Q-value) less than 0.05 were considered statistically significant. The overall expression pattern between samples showed in pairwise correlation analysis and scatterplot, the hierarchical samples clustering heatmaps, and principal components analysis (PCA) plots. These general analyses for statistical validation were performed with the ggplot2 package using R (Steenwyk and Rokas 2021). The Heatmap clustering analysis of DEGs was performed based on the log2 FPKM values, and the heat map was generated using hclust2 package (v 3.6.2, available at https://github.com/SegataLab/hclust2) with the popular clustering distance (euclidean) and hierarchical clustering method (complete) functions. A Venn diagram was generated using jVenn (http://jvenn.toulouse.inra.fr/app/index.html) (Bardou et al. 2014).
Gene ontology (GO) function classification analysis for DEGs
The online functional prediction tool, Metascape (http://metascape.org/), was used for the biological interpretation of DEGs (Zhou et al. 2019). Functional enrichment analysis for DEGs was performed for three categories of GO terms: BP, MF, and CC. All DEGs were analyzed based on these three categories, focusing on the most significant enrichment terms. (i) A two-fold change (increase or decrease) between groups was considered significant. (ii) Terms with a BH-adjusted p-value (q-value) of < 10–4, minimum count of 3, and (iii) enrichment factor (ratio of the observed count to the count expected by chance) of > 1.5 were collected and clustered based on their membership similarities.
Integrative mining of biological pathways based on the Reactome database
To facilitate biological interpretation, the Reactome pathway database that interprets biological pathways is also used to identify the functional role of genes that show differences in gene expression depending on the patient types (Jassal et al. 2020). Reactome pathways were selected based on statistically significant differences (p < 0.05) (Szklarczyk et al. 2019).
Gene-drug network analysis
All DEGs, including genes with prominent promiscuous expression, were used to find potential drugs. We used the Drug Repurposing Hub (DHUB) dataset, available at https://clue.io/repurposing. The DHUB dataset is a suitable replacement for the results of our study as it provides information on comprehensive FDA-approved drugs, investigational drugs, and preclinical instrumental compounds for all diseases, not cancer-focused data. The key information introduces the communication of 2183 gene targets and 6798 drugs or compounds covering 24 disease domains. In addition, by providing a mechanism of action (MOA) for the drug, it was possible to differentiate compounds according to the antagonist and agonist potency in a DEG-dependent manner by SCI. In detail, as shown in Supplementary Table S5, we identified (i) genes, (ii) drugs, (iii) clinical stage, and (iv) disease area and its indications. Here, we constructed a drug-gene interaction network using Cytoscape (version: 3.9.1). As a result of the drug-gene network analysis, only effective drugs were included based on the number of evidence supporting the interaction with the gene.
Results
Transcriptome sequencing analysis
To identify transcriptional differences in innate immune responses between SS and SLE, a total of ten monocyte samples isolated from whole blood were subjected to bulk RNA-seq. The sequencing result revealed that an average of 66.8 million raw reads were produced, with a read length of 100 bp using an Illumina Nova-seq 6000 platform. Approximately 98.67% of the raw data was qualified through the quality control step for sequencing data, and the average number of 60.3 million reads, accounting for 91.58% of raw data, were uniquely mapped onto the human reference genome (Homo sapiens: GRCh38). After gene annotation using the Ensembl database (release 77), among the 23,362 reference genes, a total of 18,447 were detected in at least one sample, and 13,200 were commonly expressed in the entire group (Supplementary Table S1 and Supplementary Figure S1A). The distribution of individually expressed genes in the ten samples showed expression consistency among the samples, supporting no variability in the sample preparation and data generation. Uniform data conditions were confirmed in the gene expression distribution for each sample (Supplementary Fig. S1B).
To emphasize the association between the samples in each group, the reproducibility of technical replication was confirmed using the pairwise correlation analysis, based on the overall gene expression in each sample. As a result, similar distributions of gene expression were shown while confirming the distinct differences between HC and the patient groups (Supplementary Figure S1C). The log2 transformed FPKM was calculated to determine the similarity of gene expression among the samples. The three datasets demonstrated concordant transcriptome dynamics and high reproducibility between the samples among groups, as shown by the multidimensional scaling (MDS) plot (Supplementary Figure S1D). Moreover, the hierarchical clustering heatmap also showed the transcriptional concordance among the samples. The results showed that our clinical samples for SjS and SLE were separated from each other, which demonstrated concordant transcriptome alterations specific for each disease group (Supplementary Figure S1E).
Dynamic transcriptional changes in monocytes of SjS and SLE patients
To explore the transcriptional differences between the HC and the patient groups, we performed three pairwise comparisons as follows: group A (HC) vs. group B (SjS patients), group A (HC) vs. group C (SLE patients), and group B (SjS patients) vs. group C (SLE patients) (Supplementary Figure S2). In the comparison of DEGs for the SjS patient group compared to HC, we identified 291 genes (182 up-regulated and 109 down-regulated genes). The genes, such as interferon, alpha-inducible protein (IFI27), chemokine (C–C motif) ligand 2 (CCL2), early growth response 1 (EGR1), chemokine (C–C motif) ligand 3-like 1 (CCL3L1) and interferon-induced protein 44 (IFI44L) were most significantly up-regultaed in SjS with the expression log2 FC > 3. In contrast, genes, such as microtubule-associated tumor suppressor 1 (MTUS1), major histocompatibility complex, class II, DQ alpha 2 (HLA-DQA2), CD3e molecule, epsilon (CD3-TCR complex) (CD3E), interleukin 7 receptor (IL7R), matrix-remodeling associated 7 (MXRA7), were the most significantly down-regulated with the expression log2 FC < − 3 (Supplementary Figure S2).
As for SLE transcriptional differences, 209 up- and 99 down-regulated genes were identified in our analysis comparing the SLE data set with the HC data set. IFI27, EGR1 and CCL2 showed the highest expression difference, similar to DEGs between SjS and HC, and AXL receptor tyrosine kinase (AXL) and ubiquitin specific peptidase 18 (USP18), were also high in SLE. Similar to the DEGs between SjS compared to HC, MTUS1, HLA-DQA2, and IL7R, and trophoblast glycoprotein (TPBG) and poly(A)-specific ribonuclease (PARN)-like domain containing 1 (PNLDC1) were significantly down-regulated in SLE monocytes compared to HC monocytes.
In a comparative analysis between SjS and SLE, we identified 66 up-regulated genes in SjS and 105 up-regulated genes in SLE. ADAM metallopeptidase with thrombospondin type 1 motif, 2 (ADAMTS2), matrix-remodeling associated 7 (MXRA7), RecQ mediated genome instability 2 (RMI2), fms-related tyrosine kinase 3 (FLT3), and lipoma HMGIC fusion partner (LHFP) were detected to be the highest up-regulated genes in SLE patients. The genes, such as ankyrin repeat domain 24 (ANKRD24), C-type lectin domain family 18, member A (CLEC18A), thrombospondin 1 (THBS1), coiled-coil serine-rich protein 1 (CCSER1), and immunoglobulin heavy constant gamma 1 (IGHG1), were the most significantly up-regulated genes in SjS patient monocytes.
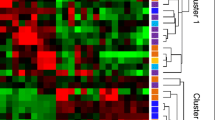
As shown in the unsupervised hierarchical clustering using z-scores for normalized values, the decisive shifts in DEGs have occurred in monocyte cells of SjS compared to HC and SLE compared to HC (Fig. 1). Therefore, we focused 267 up-regulated and 165 down-regulated gene sets, which was generated by comparing two sets of DEGs from the SjS vs. HC comparison and the SLE vs. HC comparison. Among them, 124 up-regulated and 43 down-regulated genes were shared within the autoimmune disease groups. The detailed information for DEGs is listed in Supplementary Table S2.

Summary of DEG analysis and significant aspects of transcriptional transition in SjS and SLE compared to HC. The number of A up- and B down-regulated genes identified in the two comparison sets (HC vs. SjS and HC vs. SLE). Overlapping areas in the Venn diagram represent genes common to both comparison groups. D Hierarchical clustering heatmap presents 291 DEGs in SjS and 308 in SLE. A histogram in the color key shows the number of expression values within each color bar
Classification of global gene ontology (GO) functions of DEGs
Predicting the functions of DEGs and their biological pathways involved is an important step to understand the impact of the DEGs on a particular disease. Therefore, we analyzed the biological functions of the DEGs identified in SjS and SLE monocytes through the GO classification (Fig. 2). Of the 267 up-regulated DEGs compared to HC (182 in SjS and 209 in SLE), 149 were significantly associated with 20 specific biological processes. A gene group showing SjS and SLE common, SjS-specific, and SLE-specific up-regulations was analyzed. Since SjS and SLE share many DEGs and pathways in our preliminary analysis when compared with HC, we described the comparison between the autoimmune disease (AID) group (SjS and SLE combined) with the HC group in the following sections and occasionally we applied GO to compare between SjS and SLE. Throughout SjS and SLE, the up-regulated DEG was intensively enriched to biological terms involved in the immune response containing "response to virus (GO:0009615)”, “cellular response to cytokine stimulus (GO:0071345)", and specifically "negative regulation of cell population proliferation (GO:0008285)" by accelerating the immune system also showed significant relevance (Fig. 2B). While, in aspects of down-regulation patterns, the genes related to major biological mechanisms required for normal cell activity and maintenance, such as "regulation of cell–cell adhesion (GO:0022407)" and "circadian regulation of gene expression (GO:0032922)", were down-expressed (Fig. 2D).

Function prediction analysis for DEG in SjS and SLE monocytes using Metascape. The bar chart of clustered enrichment ontology categories (GO) with a discrete color scale represents statistical significance. The function classification for A 266 up- and C 162 down-regulated DEGs, which were specific in three comparison sets (HC vs SjS, 85 up and 66 down; HC vs SLE, 58 up and 56 down; and common in both, 123 up and 43 down) are visualized and screened. The subset of representative GO terms from the full cluster of B up- and D down-DEGs were presented by network. Each term is represented by a circle node, where its size is proportional to the number of input genes falling into that term, and its color represents its cluster identity. The detailed results for the function classification of case-specific DEGs are listed in Supplementary Table S3. E Clustering heatmap of genes related to significant immune response function including the inflammatory response (GO:0006954) and cellular response to cytokine (GO:0071345). Within the DEG comparison group, DEG with up-regulation specific to SjS and SLE were expressed in pink and red letter, respectively, and down- regulation also were presented in blue and sky-blue letter. The gene marked in black is a gene that exhibits common expression changes in SjS and SLE
The result in Fig. 2E indicates that the inflammatory response (GO:0006954) and cellular response to cytokine (GO:0071345) were highly enriched in AID with the immunity-related genes including TNF, IL-10, IL-1B, CX3CR1, and AXL, which are well known as inflammatory cytokines or receptors. Among them, the representative molecules such as TNF, IL8, ZFP36, IFIT5, THBS1, and IL1B were up-regulated in SjS monocytes higher than in SLE monocytes. AXL, STAT1, CD40, NLRP6, CXCR4, and CD274 were significantly up-regulated in SLE monocytes higher than in SjS monocytes (Fig. 2E). The regulation of hemopoiesis (GO:1903706) process was prominent for the up-regultaed DEGs in SjS. This is because TNF, IL-1B, CCL2, and CX3CR1 cytokines are a set of marker molecule involved in various biological processes including hematopoiesis, cell survival, immunological functions, inflammation, apoptosis, and necrosis (Torkestani et al. 2021). A propensity toward M1 macrophage differentiation appears to be prominent based on the DEGs identified in SjS (Supplementary Table S2). In the SLE monocytes samples, the response to virus (GO:0009615) functional term was significant This is most likely due to up-regultaed genes involved in the pattern recognition receptor-mediated pathways such as STAT1 and TLR7, IFIH1, IFITM2, NLRP6, and ADARB1 in SLE. Activation of the complement cascade was also observed with C1qc and C1qb. Up-regultaed C1qa was detected in both AID (Supplementary Table S3).
Of the 165 down-regulated DEGs (109 in SjS and 99 in SLE), 86 were significantly associated with 20 specific biological process. The regulation of cell–cell adhesion (GO:0022407) process and positive regulation of immune response (GO:0050778) were most significantly enriched with ZFPM1, EP300, SMAD7, ZP3, RASGRP1, and CD3E. Especially, CD3E, CD247, HLA-DQA2, ZAP70, PLCG1, RHOH, and IGF1R which are the crucial factors associated with Th17 cell differentiation from peripheral blood were included in the functional classification of down-regulated DEGs (Fig. 2 and Supplementary Table S3).
Exploration of molecular biological pathway through the integrated Reactome analysis
In order to explore the biological pathway and immunological responses occurring in SjS and SLE monocytes in detail, we performed integrated analyses of genes with expression differences specific to both medical conditions using the Reactome Pathway Database version 77 (Croft et al. 2011). The 267 up-regulated (182 in SjS and 209 in SLE) and 165 down-regulated (109 in SjS and 99 in SLE) identifiers were subject to the Reactome pathway analyzer. For the up-regulated DEGs, we identified a network between transcriptomic changes that are specific for each condition or common to both conditions and crucial biological pathways. As a result, 92 genes were significantly accumulated in the 25 Reactome pathways (Fig. 3 and Supplementary Table S4).

Exploration of biological pathways for SjS-specific, SLE-specific, and common DEGs using the Reactome pathway analysis. The Reactome analysis mapped a network up- and down-regulated DEGs associated with biological pathway in detail. A large circular node indicates the significant Reactome pathway detected by integrated analysis with case-specific DEGs. The Reactome pathways were selected based on statistically significant differences (p < 0.05). The proportion of the pie reflected in the large circular node represents the ratio included in the corresponding Reactome pathway. A The red, orange, purple nodes are SjS-specific, SLE-specific, and up-regulated shared DEGs, respectively. B The sky blue, emerald, blue nodes are SjS-specific, SLE-specific, and down-regulated, shared DEGs, respectively
The interferon signaling (R-HSA:913531), including Interferon alpha/beta/gamma signaling (R-HSA:909733 and R-HSA:877300), was the most common significantly enriched pathway with 36 genes. The cytokine signaling in immune system (R-HSA:1280215) was also highly associated with 56 genes. In particular, guanylate-binding protein (GBP) 1, 3, 4, and 5 belonging to the superfamily of IFN-inducible guanosine triphosphate hydrolases (GTPases), which are known to control infection and autoimmunity(Wang et al. 2018), were remarkably enriched. We also found evidence that up-regulation of HIST1H1C, HIST1H1D, HIST1H1E, HIST1H4E, HIST2H2AC, and HIST2H2BE among 82 histone cluster genes (Histone cluster 1, H2A, B, and H4) were significantly associated with both autoimmune conditions. In addition, an integrated analysis confirmed noticeable differences in biological pathways between SjS and SLE. For SjS, the nerve growth factor (NGF)-induced transcription (R-HSA:9,031,628) activity of the EGR1, EGR2, EGR3, F3, FOSL1, JUNB, SGK1, TRIB1, and c-Jun was particularly noted in the SjS monocytes. The Complement cascade (R-HSA:166658) includes C1QA, C1QB, C1QC, C2, C3AR1, CFB, CLU, and SERPING1 was highly associated with SLE.
As for the down-regulated DEGs of AIDs, 17 genes among 165 DEG were significantly accumulated in the 17 Reactome pathways (Fig. 3 and Supplementary Table S4). The FCGR3A-mediated IL10 synthesis (R-HSA:9664323), generation of second messenger molecules (R-HSA:202433), and RORA activates gene expression (R-HSA:1368082) were most significantly enriched with CD247, ITPR3, PLCG1, PRKAR2B, CD3E, ZAP70, HLA-DQA2, EP300, and PPARA genes.
For SjS monocytes, ABLIM1 and PLCG1 related to the Netrin-1 signaling pathway (R-HSA:373752), which play an important role in neuronal movement and axon induction during the development of the nervous system, showed down-regulation. Only the pathway related to fertilization (R-HSA:1187000) involving CD9 and ZP3 was identified.
In the down-regulated genes of SLE monocytes, ‘the RORα activates gene expression’ pathway required for TH17 cell pathogenicity by regulating IL17 gene transcription was distinct in SLE patient monocytes with gene enrichment of EP300, PPARA, ARNTL2, ITPR3, and PRKAR2B.
Identification of potential drug or chemical candidates for SjS and SLE patient treatment
One of the aims of this study was to focus on improving the knowledge of drug therapy potential through understanding the transcriptomic profiles and interactions for AIDs. To explore the potential therapeutic drugs related to or may respond to transcriptional changes caused by both SjS and SLE, we performed gene-drug network analysis using the Broad’s Drug Repurposing Hub (DHUB) dataset (Corsello et al. 2017). A total of the 267 up-regulated (182 in SjS and 209 in SLE) and 165 down-regulated (109 in SjS and 99 in SLE) were implicated in the text mining step with Python.
Against the DHUB dataset, 436 interactions between 42 genes and 400 drugs were mined for up-regulated DEGs. Interactions of 208 between 42 genes and 400 drugs were mined for up-regulated DEGs. The data included a lot of information, such as preclinical, phased trials, and even withdrawn drugs. Therefore, we only focused on currently available drugs (launched), whose efficacy and use have already been clearly identified in the next step. As a result of comparing the up- and down-regulated genes in each period with the DHUB dataset, 207 interactions between 26 genes and 194 drugs and 98 interactions between 17 genes and 96 drugs were identified, respectively.
As a final step, considering the aberrantly biased change in gene expression by AIDs, drug antagonists were selected from gene-drug interactions focused on upregulation, and agonists were selected from downregulation focused on DEG. The results elicited 154 antagonistic interactions between 17 genes and 148 drugs (Supplementary Table S5). The representative connections with antagonistic drug were observed with prostaglandin-endoperoxide synthase (PTGS2), Complement C2 (C2), and angiotensin I converting enzyme (ACE). PTGS2, C2, and ACE genes showed the most association with 63, 21, and 20 drugs, respectively, and most of them were frequently used drugs in influential disease, rheumatology, endocrinology, cardiology, obstetrics, and nephrology, showing a close relationship with the autoimmune diseases.
In 37 agonistic interactions, four down-regulated genes along with 36 drugs. Estrogen receptor 1 (ESR1) and peroxisome proliferator activated receptor alpha (PPARA) were shown higher relations with 26 and nine agonist drugs, respectively. The ESR1 and PPARA gene were also screened for drugs aimed at treating hematologic diseases in the fields of endocrinology and hematology (Table 1).
Discussion
Monocytes play critical roles in activation of adaptive immune cells and maintenance of chronic inflammation in AID. Their diverse roles include phagocytosis, antigen presentation, cytokine and chemokine secretion, nitric oxide synthesis, and migration to the tissues with infection or injury (Lopes et al. 2021). A recent study with weighted gene co-expression network analysis identified four molecular signatures in monocytes from primary SjS patients, functionally annotated for processes related with translation, IFN-signaling, and toll-like receptor signaling (Lopes et al. 2021). Represented hub genes of all four signatures were partially inhibited by IFN-alpha/beta receptor blockade, indicating that the inflammatory mediators such as type I IFNs contribute to the altered transcriptional profile of primary SS-monocytes. In addition, a group led by Brkic et al. reported that the expression of the type I IFN signature of CD14+ monocytes in SjS compared to healthy controls (Brkic et al. 2013; Maria et al. 2014). The same group reported that hydroxychloroquine treatment down-regulated systemic interferon activation in primary SjS in the JOQUER randomized trial (Bodewes et al. 2020).
Our current GO analysis revealed that DEGs from bulk RNA-seq of AID monocytes were enriched in the inflammatory process and cellular response to cytokines. More specifically, TNF, IL8, ZFP36, IFIT5, THBS1, and IL1B were up-regulated in SjS monocytes to a greater degree than in SLE monocytes. We also observed enrichment of AXL, STAT1, CD40, NLRP6, CXCR4, and CD274 in SLE monocytes to a greater degree than in SjS monocytes. DEGs involved in M1 macrophage differentiation and viral response genes were also up-regultaed more in SjS and in SLE, respectively. These are consistent with published studies reporting that SjS and SLE have shown elevated inflammatory phenotype and/or the interferon signature (Brkic et al. 2013; Elkon and Wiedeman 2012; Maria et al. 2014).
In addition, an integrated analysis confirmed noticeable differences in biological pathways between SjS and SLE. In SjS monocytes, the nerve growth factor (NGF)-induced transcription (R-HSA:9031628) activity was particularly noted, whereas the complement cascade (R-HSA:166658) was highly associated with SLE. NGF levels are known to be increased in numerous inflammatory and autoimmune states, such as in multiple sclerosis, RA, SLE, and mastocytosis, along with increased accumulation of mast cells (Skaper 2017; Xu and Chen 2015). Mast cells can be activated and maturated by NGF to release potent mediators of inflammation, and in turn they can also produce and release NGF after degranulation. Moreover, NGF also exerts its proinflammatory action, not only on mast cells, but also in B and T cells, neutrophils and eosinophils (Kritas et al. 2014). Interestingly, our current analysis points out increased activity of various transcription factors involved in mast cell proliferation and hyperactivity in SjS monocytes.
As mentioned earlier, the activation of the complement system was characteristically identified in the transcriptional changes of SLE monocytes. The complement cascade is a rapid response system that can be activated either directly by a pathogen or indirectly by a pathogen-binding antibody, leading to a series of various effector functions (Dunkelberger and Song 2010). As a major feature of inflammatory lesions of SLE, immune complexes containing autoantigens and autoantibodies fix and activate complements, causing tissue damages such as in the kidney (Dunkelberger and Song 2010; Walport 2002a). Upregulation of DEGs associated with the activation of the complement system, therefore, may reflect activated classical pathway, triggered by the interaction of C1q with immune complexes as reported (Walport 2002b).
Our Reactome pathway analysis revealed that the Interferon Signaling (R-HSA:913531), including Interferon Alpha/Beta/Gamma Signaling (R-HSA:909733 and R-HSA:877300), was the most significantly enriched pathway with 36 genes. The analysis revealing 56 genes also supported the involvement of the Cytokine Signaling in Immune System. One interesting molecule, GBP, enrichment was noted in our analysis. The GBP family is mainly known for its various functions against invading microorganisms and pathogens as part of the innate immune response. The expression changes of interferon-α and GBP have been reported in rheumatic diseases such as RA, SLE, and SjS (Haque et al. 2021). Furthermore, GTPase of the immune-associated nucleotide-binding proteins (GIMAP) 4, 6, and 7, which are known to link to thymocyte development, apoptosis of peripheral lymphocytes, and T helper cell differentiation, were also included in the interferon signaling pathway (R-HSA:913531) (Filen and Lahesmaa 2010). Another interesting molecule was histone cluster genes, which were also enriched in the AID group. This result is consistent with the previous publications indicating that the epigenetic change and gene expression of the histone cluster are involved in the pathogenesis of AID, such as SLE (Doyle et al. 2013; Li et al. 2017).
As for down-regulated DEGs in AID by our Reactome pathway analysis, we found that the FCGR3A-mediated IL10 synthesis, Generation of Second Messenger Molecules, and RORA activates gene expression (R-HSA:1368082) were most significantly enriched. Of these, CD3E, CD247, ZAP70, PLCG1, and HLA-DQA2 are major molecules associated with the T-Cell Receptor (TCR) signaling pathway, mediated by CD3 phosphorylation/ZAP70 recruitment and TCR zeta chains. Thus, since both AID showed a significant decrease in the expression of CD3 and TCR zeta chains, particularly in monocytes from SjS patients, it is presumed that aberrant TCR signaling may play a role in impairing proper T cell development, activation, and immune tolerance as supported by a study (Lysechko and Ostergaard 2005).
Activation of Th17 cells by RORγt is an important biological pathway in AID (Hams et al. 2021; Nejati Moharrami et al. 2018). However, the expression of RORα was reduced in monocytes of SLE patients in our bulk RNA-seq analysis. RORγ and RORγt are two isoforms that are transcribed from the RORC gene, and four isoforms, RORα1–4, are produced from the corresponding RORA gene through alternative promoter usage and exon splicing (Castro et al. 2017). RORγt has been suggested to be important for Th17 differentiation by regulating the expression of Th17 genes. Mouse T cells lacking the expression of both RORγt and RORα showed a complete blockade in Th17 cell differentiation in contrast to the partial defects in T cells with a deficiency of either one, suggesting a complementary role for RORγt and RORα in Th17 commitment (Yang et al. 2008a). One interesting study demonstrated that RORα is a negative regulator of the inflammatory response in human primary smooth-muscle cells (Delerive et al. 2001). This action of RORα, especially RORα1 on NF-κB is associated with the induction of IκBα, the major inhibitory protein of the NF-κB signaling pathway, whose expression was found to be transcriptionally up-regultaed by RORα1 via a ROR response element in the IκBα promoter (Delerive et al. 2001). Therefore, down-regulated DEGs involved in the RORα-mediated pathway may suggest augmented inflammation via NF-κB in the monocytes of SLE.
Moreover, through the investigation of gene-drug interaction analysis, we selected and presented alternatives to various potent drugs that can be used for SjS or SLE symptom relief and treatment purposes (Table 1). Typically, non-steroidal anti-inflammatory (NSAIDs) drugs, such as indomethacin, aspirin, nabumetone, and celecoxib are useful drugs to alleviate some of the symptoms of autoimmune-activated patients by inhibiting cyclooxygenase activity (Bhunyakarnjanarat et al. 2021). Cox-2 (cyclooxygenase) enzyme coded by the PTGS2 gene is a major factor that can modify T cells and macrophages in several inflammatory and autoimmune diseases (Wise et al. 2022).
Furthermore, we presented potent drugs including amrinone, pentoxifylline, pirfenidone, pranlukast, and ibudilast, which are effective for inflammatory diseases including SjS and SLE by inhibiting phosphodiesterase, TGF beta receptor, leukotriene receptor activity (Kikuchi and Inagaki 2002; Li et al. 2018; Martin-Gomez et al. 2016). Phospodiesterase, especially type 4 is a cyclic adenosine monophosphate (cyclic AMP)-specific enzyme that hydrolyzes the cyclic AMP in cells into 5'-AMP. Phosphodiesterase inhibitor is known to inhibit TNF-alpha production and IL-1β and reduce inflammatory responses by increasing the intracellular cAMP concentration in monocyte blood cells (Brideau et al. 1999; Coon et al. 2014; Molnar-Kimber et al. 1992). Pharmacological studies also have shown that the signaling cascades by leukotriene receptor stimulates the degranulation, chemotaxis, and phagocytosis of neutrophils, leading to chronic inflammatory disorders (Sasaki and Yokomizo 2019).
As for potent agonist, PPAR is a type of steroid hormone nuclear receptor superfamily that plays an important role in lipid metabolism, energy balance, arteriosclerosis, and glucose regulation, and is an important factor in controlling inflammation activity (Choi and Bothwell 2012). Yang et al. study have shown that PPAR plays an important role in regulating immune responses by mechanisms, that involve transactivation or trans repression of gene expression through activation of transcription factors including NF-κB, AP1 and NFAT, and suggested that PPAR-α agonist will provide clues to the treatment of autoimmune diseases (Yang et al. 2008b).
The limitation of our current study includes a small sample number for each group. The recruitment of the patients is in progress and additional sets of the transcriptomic data are being accumulated for our upcoming studies. Nonetheless, the Reactomes and gene-drug target analyses provided novel findings regarding the inflammatory nature of AID monocytes. This information on gene-drug association may serve as a stepping stone to future drug design for SLE and/or SjS.
References
Andreoletti G, Lanata CM, Trupin L, Paranjpe I, Jain TS, Nititham J, Taylor KE, Combes AJ, Maliskova L, Ye CJ et al (2021) Transcriptomic analysis of immune cells in a multi-ethnic cohort of systemic lupus erythematosus patients identifies ethnicity- and disease-specific expression signatures. Commun Biol 4:1–13
Bardou P, Mariette J, Escudie F, Djemiel C, Klopp C (2014) jvenn: an interactive Venn diagram viewer. BMC Bioinformatics 15:293
Bhunyakarnjanarat T, Udompornpitak K, Saisorn W, Chantraprapawat B, Visitchanakun P, Dang CP, Issara-Amphorn J, Leelahavanichkul A (2021) Prominent indomethacin-induced enteropathy in Fcgriib defi-cient lupus mice: an impact of macrophage responses and immune deposition in gut. Int J Mol Sci 22:1377
Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J (2001) Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294:1540–1543
Bodewes ILA, Gottenberg JE, van Helden-Meeuwsen CG, Mariette X, Versnel MA (2020) Hydroxychloroquine treatment downregulates systemic interferon activation in primary Sjogren’s syndrome in the JOQUER randomized trial. Rheumatology 59:107–111
Brideau C, Van Staden C, Styhler A, Rodger IW, Chan CC (1999) The effects of phosphodiesterase type 4 inhibitors on tumour necrosis factor-alpha and leukotriene B4 in a novel human whole blood assay. Br J Pharmacol 126:979–988
Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA, Wildenberg ME, Beumer W, Drexhage HA, Versnel MA (2013) Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis 72:728–735
Castro G, Liu XJ, Ngo K, De Leon-Tabaldo A, Zhao SR, Luna-Roman R, Yu JX, Cao TH, Kuhn R, Wilkinson P et al (2017) ROR gamma t and ROR alpha signature genes in human Th17 cells. PLoS ONE 12:e0181868
Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C (2015) Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun 57:42–52
Choi JM, Bothwell ALM (2012) The nuclear receptor PPARs as important regulators of T cell functions and autoimmune diseases. Mol Cells 33:217–222
Coon TA, McKelvey AC, Weathington NM, Birru RL, Lear T, Leikauf GD, Chen BB (2014) Novel PDE4 inhibitors derived from Chinese medicine forsythia. PLoS ONE 9:e115937
Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M et al (2017) The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med 23:405–408
Croft D, O’Kelly G, Wu G, Haw R, Gillespie M, Matthews L, Caudy M, Garapati P, Gopinath G, Jassal B et al (2011) Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 39:D691-697
Decker P, Kotter I, Klein R, Berner B, Rammensee HG (2006) Monocyte-derived dendritic cells over-express CD86 in patients with systemic lupus erythematosus. Rheumatology 45:1087–1095
Delerive P, Monte D, Dubois G, Trottein F, Fruchart-Najib J, Mariani J, Fruchart JC, Staels B (2001) The orphan nuclear receptor ROR alpha is a negative regulator of the inflammatory response. EMBO Rep 2:42–48
Doyle HA, Aswad DW, Mamula MJ (2013) Autoimmunity to isomerized histone H2B in systemic lupus erythematosus. Autoimmunity 46:6–13
Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20:34–50
Elkon KB, Wiedeman A (2012) Type I IFN system in the development and manifestations of SLE. Curr Opin Rheumatol 24:499–505
Filen S, Lahesmaa R (2010) GIMAP proteins in T lymphocytes. J Signal Transduct 2010:268589
Hams E, Roberts J, Bermingham R, Fallon PG (2021) Functions for retinoic acid-related orphan receptor alpha (RORalpha) in the activation of macrophages during lipopolysaccharide-induced septic shock. Front Immunol 12:647329
Haque M, Siegel RJ, Fox DA, Ahmed S (2021) Interferon-stimulated GTPases in autoimmune and inflammatory diseases: promising role for the guanylate-binding protein (GBP) family. Rheumatology (oxford) 60:494–506
Hauk V, Fraccaroli L, Grasso E, Eimon A, Ramhorst R, Hubscher O, Leiros CP (2014) Monocytes from Sjogren’s syndrome patients display increased vasoactive intestinal peptide receptor 2 expression and impaired apoptotic cell phagocytosis. Clin Exp Immunol 177:662–670
He Y, Chen RJ, Zhang MQ, Wang B, Liao ZD, Shi GX, Li Y (2022) Abnormal changes of monocyte subsets in patients with Sjogren's syndrome. Front Immunol 13
Hochberg MC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40:1725
Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R et al (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48:D498–D503
Kikuchi M, Inagaki T (2002) A case of primary Sjogren’s syndrome associated with low titer cold agglutinin disease. Clin Exp Rheumatol 20:577–578
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14:R36
Kritas SK, Caraffa A, Antinolfi P, Saggini A, Pantalone A, Rosati M, Tei M, Speziali A, Saggini R, Pandolfi F et al (2014) Nerve growth factor interactions with mast cells. Int J Immunopathol Pharmacol 27:15–19
Li X, Tian R, Gao H, Yang Y, Williams BRG, Gantier MP, McMillan NAJ, Xu D, Hu Y, Gao Y (2017) Identification of a histone family gene signature for predicting the prognosis of cervical cancer patients. Sci Rep 7:16495
Li H, Zuo J, Tang W (2018) Phosphodiesterase-4 inhibitors for the treatment of inflammatory diseases. Front Pharmacol 9:1048
Lopes AP, Bekker CPJ, Hillen MR, Blokland SLM, Hinrichs AC, Pandit A, Kruize AA, Radstake TRDJ, van Roon JAG (2021) The transcriptomic profile of monocytes from patients with Sjogren's syndrome is associated with inflammatory parameters and is mimicked by circulating mediators. Front Immunol 12
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15:550
Lysechko TL, Ostergaard HL (2005) Differential Src family kinase activity requirements for CD3 xi phosphorylation/ZAP70 recruitment and CD3 epsilon phosphorylation. J Immunol 174:7807–7814
Maria NI, Brkic Z, Waris M, van Helden-Meeuwsen CG, Heezen K, van de Merwe JP, van Daele PL, Dalm VASH, Drexhage HA, Versnel MA (2014) MxA as a clinically applicable biomarker for identifying systemic interferon type I in primary Sjogren’s syndrome. Ann Rheum Dis 73:1052–1059
Martin JA, Wang Z (2011) Next-generation transcriptome assembly. Nat Rev Genet 12:671–682
Martin-Gomez MA, Caba Molina M, Cruz Caparros G, Munoz Vico J, Gomez Morales M (2016) Sjogren syndrom and mixed nephropathy. Significance of early kidney biopsy. Nefrologia 36:451–453
Molnar-Kimber KL, Yonno L, Heaslip RJ, Weichman BM (1992) Differential regulation of TNF-alpha and IL-1beta production from endotoxin stimulated human monocytes by phosphodiesterase inhibitors. Mediat Inflamm 1:411–417
Nejati Moharrami N, Bjorkoy Tande E, Ryan L, Espevik T, Boyartchuk V (2018) RORalpha controls inflammatory state of human macrophages. PLoS ONE 13:e0207374
Ramos-Casals M, Brito-Zeron P, Font J (2007) The overlap of Sjogren’s syndrome with other systemic autoimmune diseases. Semin Arthritis Rheum 36:246–255
Sasaki F, Yokomizo T (2019) The leukotriene receptors as therapeutic targets of inflammatory disease. Int Immunol 31:607–615
Shiboski SC, Shiboski CH, Criswell L, Baer A, Challacombe S, Lanfranchi H, Schiodt M, Umehara H, Vivino F, Zhao Y et al (2012) American College of Rheumatology classification criteria for Sjogren’s syndrome: a data-driven, expert consensus approach in the Sjogren’s International Collaborative Clinical Alliance cohort. Arthritis Care Res (hoboken) 64:475–487
Skaper SD (2017) Nerve growth factor: a neuroimmune crosstalk mediator for all seasons. Immunology 151:1–15
Steenwyk JL, Rokas A (2021) ggpubfigs: colorblind-friendly color palettes and ggplot2 graphic system extensions for publication-quality scientific figures. Microbiol Resour Announc 10:e0087121
Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P et al (2019) STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 47:D607–D613
Torkestani S, Moghimi H, Farsiabi R, Khazaei S, Eftekharian MM, Dalvand E (2021) Evaluation of serum levels of IL-6, IL-10, and TNIF-alpha in alopecia areata patients: a systematic review and meta-analysis. Biomed Res Therapy 8:4668–4678
Walport MJ (2002a) Complement and systemic lupus erythematosus. Arthritis Res 4(Suppl 3):S279-293
Walport MJ (2002b) Complement and systemic lupus erythematosus. Arthritis Res Ther 4:S279–S293
Wang Q, Wang X, Liang Q, Wang S, Xiwen L, Pan F, Chen H, Li D (2018) Distinct prognostic value of mRNA expression of guanylate-binding protein genes in skin cutaneous melanoma. Oncol Lett 15:7914–7922
Wildenberg ME, Welzen-Coppens JMC, van Helden-Meeuwsen CG, Bootsma H, Vissink A, van Rooijen N, de Merwe JPV, Drexhage HA, Versnel MA (2009) Increased frequency of CD16+monocytes and the presence of activated dendritic cells in salivary glands in primary Sjogren syndrome. Ann Rheum Dis 68:420–426
Wise RM, Al-Ghadban S, Harrison MAA, Sullivan BN, Monaco ER, Aleman SJ, Donato UM, Bunnell BA (2022) Short-term autophagy preconditioning upregulates the expression of COX2 and PGE2 and alters the immune phenotype of human adipose-derived stem cells in vitro. Cells 11:1376
Xu Y, Chen G (2015) Mast cell and autoimmune diseases. Mediat Inflamm 2015:246126
Yang XXO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS et al (2008a) T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28:29–39
Yang YH, Gocke AR, Lovett-Racke A, Drew PD, Racke MK (2008b) PPAR alpha regulation of the immune response and autoimmune encephalomyelitis. PPAR Res 2008:1–6
Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK (2019) Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun 10:1523
Acknowledgements
This paper was supported by Fund of Biomedical Research Institute, Jeonbuk National University Hospital (K.L.). This research was supported by Basic Science Research Capacity Enhancement Project through Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Ministry of Education (Grant No. 2019R1A6C1010033) (K. H.). It was also funded by the Sjögren’s Foundation (S.C.) and NIH/NIAMS/NIDCR (S.C.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
All authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
13258_2022_1308_MOESM1_ESM.pptx
Supplementary Figure S1. Bioinformatical observation for the expression data and sample correlation. (A) Venn diagram of total expressed gene in all cases. (B) The boxplot shows the gene expression of each sample subsequent to data normalization. The ordinate represents gene expression value, and the abscissa represents the HC, SjS, and SLE group with independent colors. The distribution of FPKM values for total expressed genes in the samples of each group is also shown. (C) The Euclidean distances based on all raw expression data were calculated between each sample, and the colors indicated the distances. The red mosaic color indicates that the gene expression between samples is identical. (C) The MDS plot shows how similar and close the transcriptome changes of each sample are based on the global gene expression level. (D) The hierarchical clustering analysis of total expressed genes is shown. A histogram in the top color key shows the number of expression values (PPTX 500 KB)
13258_2022_1308_MOESM2_ESM.pptx
Figure S2. Statistical summary of DEGs identified from three comparison sets (HC vs. SjS, HC vs. SLE, and SjS vs. SLE). The number of up- and down-regulated genes identified in the three comparison sets are separately shown. (A) HC vs SjS, (B) HC vs SLE, and (C) SjS vs SLE. Overlapping areas in the Venn diagram represent the number of genes that do not change. The size of the circle is proportional to the number of DEGs in each comparison set. The FPKM volcano, scatter, and MA plots were constructed by pairwise comparison between both cases. In this statistical analysis, red and blue dots represent the statistically significant up- and down-regulated DEGs. The middle line in black dots indicated no difference in the mean expression values between samples (PPTX 1023 KB)
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, K.E., Mun, S., Kim, Sm. et al. The inflammatory signature in monocytes of Sjögren’s syndrome and systemic lupus erythematosus, revealed by the integrated Reactome and drug target analysis. Genes Genom 44, 1215–1229 (2022). https://doi.org/10.1007/s13258-022-01308-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-022-01308-y