
Abstract
We assessed genetic and phenotypic variation in 105 maize germplasm accessions from RDA-Genebank of Korea and performed association analyses for 11 agronomical traits and 100 simple sequence repeats (SSR). Genetic diversity (GD) analysis revealed a total of 1104 alleles at the 100 SSR loci. The average number of alleles per locus was 11.0. The average GD and polymorphic information content values were 0.73 and 0.70, respectively. The average major allele frequency was 0.41. Population structure analysis indicated that these maize accessions comprised two major groups and one admixed group based on a membership probability threshold of 0.80. The two major groups contained 35 and 46 maize accessions. A mixed linear model of association analysis revealed five marker-trait associations with a significance level of P ≤ 0.01 involving five SSR markers. A general linear model showed 72 marker-trait associations involving 42 SSR markers. We confirmed the presence in the general linear model associations of the five significant marker-trait associations (SMTAs) identified in the mixed linear model. For these SMTAs, two loci were associated with stem diameter and one locus each was associated with ear row number, leaf width, and leaf length. These results should prove useful for breeding new inbred lines by selecting parental lines using molecular markers and will help to preserve maize genetic resources in Korea.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maize (Zea mays L.) is one of the most important crops in the world for food, livestock feed, and biofuel because it has excellent adaptability to a wide range of environments in tropical and temperate regions (Steinhoff et al. 2012). Researchers in many countries are interested in the collection and preservation of maize germplasms, and maize genetic resources are stored in gene banks throughout the world (Li et al. 2004). Today, maize consumption in Korea is increasing as the population transitions from a traditional diet based on rice to a western diet based on meat. Thus, the country needs new high-performing maize cultivars. RDA-Genebank maintains 8472 genetic variants of maize (http://www.genebank.go.kr/). Although many RDA-Genebank maize PGRs (Plant Genetic Resources) have already been evaluated in the field for population diversity and phenotypic characteristics, there has been a lack of analysis of genetic diversity (GD) and relationships at the molecular level. In addition, most of the archived accessions have been utilized rarely or not at all in maize breeding programs. In order to improve breeding material selection and development of core germplasms, we selected 105 accessions from 1000 maize accessions from the Korean RDA-Genebank for further analysis based on their morphological characteristics, such as silking stage, plant height, and ear length (Kim et al. 2016).
Understanding the GD and population structure is helpful for germplasm preservation and utilization for crop improvement. GD information provides breeders with tools to develop new and improved cultivars with better traits (Govindaraj et al. 2015). However, utilization of PGR is lower than it could be because of inadequate information for breeders and lack of genetic information about cultivar collections (Vančetović et al. 2010). Molecular markers are used to assess GD and population structure to predict hybrid performance and heterosis because these markers are inherited by Mendelian rules and are not influenced by environmental factors (Kashiani et al. 2012; Legesse et al. 2007; Solomon et al. 2012). A remarkable variety of molecular marker techniques have developed in recent years. Among different molecular marker systems, simple sequence repeats (SSRs) or microsatellites, which are short sequences containing tandemly repeated copies of 1–6 nucleotides, are the most suitable markers for assessing GD and population structure among PGR of maize. SSR analysis has advantages for identifying high levels of allelic variation and is reliable, reproducible, and discriminating (Rafalski et al. 1996; Akagi et al. 1997; Smith et al. 1997; Enoki et al. 2002). Confirming the genetic basis of agronomically valuable traits is very important for crop improvement in plant breeding programs. Association analysis based on linkage disequilibrium was first introduced in human genetics to identify genes that control traits (Khoury et al. 2009). This method has since been successfully applied to analysis of genetic markers and agronomic traits of interest in many crops (Flint-Garcia et al. 2005; Yu and Buckler 2006), such as rice (Borba et al. 2010), maize (Mezmouk et al. 2011), and barley (Lorenz et al. 2010). Furthermore, association analysis provides some advantages over quantitative trait locus (QTL) mapping, including reduced time and costs, evaluation of more than two alleles per locus, and increased mapping resolution (Flint-Garcia et al. 2005; Zhu et al. 2008).
Genetic analysis is very important to ensure the long-term success of preservation and utilization of PGR in the RDA-Genebank. Therefore, our objective was to investigate the GD of 105 maize accessions from the RDA-Genebank using SSR markers, and to evaluate their population structure and clustering patterns. We also attempted to elucidate the genetic bases of agronomic traits by analyzing the associations of 100 SSR markers with 11 agronomic traits. These results will help to improve maize breeding programs and will inform the preservation and application of maize genetic resources in Korea.
Materials and methods
Plant materials and phenotypic evaluation
The accession numbers and sources of 105 maize accessions are listed Table 1. All maize accessions were obtained from the Genebank of National Agrobiodiversity Center (NAAS) of the Rural Development Administration (RDA) of Korea. The 30 seeds for each maize accession were sowed in plastic plug tray at 25 April 2015 and keep in greenhouse to 3–4 leaf stage for germination and cultivation. And then seedling plants transferred to field at the College of Agriculture and Life Sciences, Kangwon National University, Chuncheon, Gangwon-do. The experimental design was a randomized complete block with three replicates. Each replication was planted as a block design with seven plants per each accession, and the distance between rows and plants was 70 and 25 cm, respectively. A total of 11 agronomic traits were evaluated in a field: percent germination rate (GR), tasseling stage (DT), silking stage (DS), stem diameter (SD), plant height (PH), ear height (EH), leaf width (LW), leaf length (LL), ear length (EL), ear row number (ER), 100 kernel weight (100 KW). Basic statistics were performed using Microsoft Office Excel 2010.
DNA extraction and SSR analysis
Genomic DNA was extracted from young maize leaves as described by Dellaporta et al. (1983), with minor modifications. One hundred SSR markers, distributed across the ten maize chromosomes (ten loci per chromosome), were used to evaluate genetic variation in 105 maize accessions from RDA Genebank. The SSR markers used in this study were obtained from MaizeGDB (http://www.maizegdb.org/).
SSR amplification was conducted in a total volume of 30 µl, and consisted of 20 ng of genomic DNA, 1× PCR buffer, 0.3 µM forward and reverse primers, 0.2 mM dNTPs, and 1 unit of Taq Polymerase (Biotools). The PCR profile consisted of a 5-min initial denaturation period at 94 °C, followed by two 1-min denaturation cycles at 94 °C, a 1-min annealing cycle at 65 °C, and one 2-min extension at 72 °C. After the second cycle, the annealing temperature was decreased in 1 °C increments every second cycle, until a final temperature of 55 °C was reached. The last cycle was then repeated 20 times. A 10-min extension at 72 °C followed the completed cycles.
Five µl of the final reaction product was mixed with 10 µl of electrophoresis loading-buffer (98% formamide, 0.02% BPH, 0.02% Xylene C, and 5 mM NaOH). After denaturation and immediate cooling, 2 μl of the sample was loaded onto a 6% denaturing (7.5 M urea) acrylamide-bisacrylamide gel (19:1) in 1× TBE buffer, and electrophoresed at 1,800 volts and 60 watts for 120 min. The separated fragments were then visualized using a silver-staining kit (Promega, USA).
Data analysis
The number of alleles, allele frequency, major allele frequency (MAF), gene diversity (GD), and polymorphic information content (PIC) for 100 SSR markers were calculated with the PowerMarker 3.25 program (Liu and Muse 2005). GD is defined as the probability that two randomly chosen alleles from the population are different. It can be estimated at the lth locus as:
where f represents the inbreeding coefficient, Plu the frequency of the uth allele, and n the sample size. And PIC (Bostein et al. 1980) was calculated by:
where Plu 2 and Plv 2 are the frequency of the uth and vth alleles of marker l.
The genetic similarities (GS) were calculated for each pair of accessions using the Dice similarity index (Dice 1945). The similarity matrix was used to construct an Unweighted Pair Group Method with Arithmetic Mean Algorithm (UPGMA) dendrogram with the help of SAHN-clustering from NTSYSpc version 2.1 (Rohlf 1998).
The population structure (Q matrix) of 105 maize accessions was analyzed using the model-based program STRUCTURE 2.2 (Pritchard and Wen 2003). The membership coefficient for each individual in each subpopulation was run five times for each cluster (K), ranging from 1 to 10, using the admixture model with a burn-in of 100,000 and a replication of 100,000. Because the estimated log probability of data [LnP(D)] overestimated the number of subgroups, we used the ad hoc criterion (ΔK) described by Evanno et al. (2005) to determine the most probable value of K. The run of the estimated numbers of subgroups showing the maximum likelihood was used to assign maize accessions with membership probabilities ≥0.80 to subgroups. The maize accessions with membership probabilities <0.80 were assigned to an admixed group (Wang et al. 2008).
TASSEL 3.0 (Bradbury et al. 2007) was used to evaluate r 2 and D′ for level of linkage disequilibrium and marker-trait associations using a Q general linear model (GLM) and a Q + K mixed linear model (MLM). The Q GLM method was performed using a Q-matrix derived from the STRUCTURE program. The number of permutation runs was set to 10,000 to obtain a marker significance value of P ≤ 0.01. The Q + K MLM method used a kinship K matrix and the population-structure Q matrix at P ≤ 0.01. To obtain the K matrix, kinship coefficients was calculated with SPAGeDi software (Hardy and Vekemans 2002) using the method by Loiselle et al. (1995).
Results
Phenotypic analysis and correlation analysis
Phenotypic variations for 11 agronomic traits in 105 maize accessions of Korean RDA-Genebank are shown in Table 2. The average GR value was 88.8 ± 14.0%, ranging from 40.0 to 100%. The DT value ranged from 44.0 to 59.0 days, with an average of 52.8 ± 4.4 days. The average DS value was 56.8 ± 6.4 days, ranging from 42.0 to 70.0 days. The SD value ranged from 1.0 to 3.9 cm, with an average of 1.9 ± 0.6 cm. The average PH value was 143.6 ± 23.1 cm, ranging from 76.0 to 215.5 cm. The EH value ranged from 28.0 to 112.0 cm, with an average of 67.3 ± 15.6 cm. The average LW value was 7.4 ± 1.3 cm, ranging from 3.9 to 10.9 cm. The LL value ranged from 46.9 to 89.2 cm, with an average of 63.0 ± 8.0 cm. The average EL value was 14.7 ± 2.6 cm, ranging from 7.5 to 23.5 cm. The ER value ranged from 9.3 to 20.0 row, with an average of 13.9 ± 2.0 row. The average 100 KW value was 21.7 ± 5.0 g, and ranged from 11.6 to 41.9 g. We evaluated correlation coefficients among 11 agronomic traits in 105 maize accessions. Twenty-seven combinations correlated significantly with P < 0.05 or 0.01. Among them, PH and EH (0.799**), DT and DS (0.748**), EH and LL (0.573**), and LW and 100 KW (0.555**) showed comparatively higher correlation coefficients than the other combinations (Table 2).
Genetic diversity among 105 maize accessions of Korean RDA-Genebank
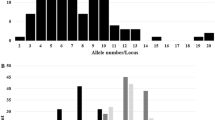
We used a total of 100 SSR loci to evaluate the GD among 105 maize accessions (Fig. 1; Table 3). These loci comprised a total of 1104 alleles in 105 accessions. The number of alleles per locus ranged from 3 to 27, and the average number of alleles per locus was 11.0 (Table 3, Supplementary Table 1). The average GD was 0.73 with a range of 0.18–0.92. In addition, the average PIC value was 0.70 with a range of 0.17–0.91. The average MAF was 0.41 with a range of 0.18–0.90 (Table 3). Of the 1104 alleles, 269 private alleles (24.4%) were each detected in only 1 of the 105 maize accessions. The frequency of rare alleles (frequency <0.05) was 60.6% (669 of 1104 alleles), whereas intermediate (frequency 0.05–0.5) and abundant alleles (frequency >0.5) comprised 37.0% (408 alleles) and 2.4% (27 alleles) of 1104 alleles, respectively (Fig. 2).

Frequency of gene diversity and PIC per locus in 105 maize accessions of Korean RDA-Genebank

Histogram of allele frequencies in 105 maize accessions of Korean RDA-Genebank
Population structure and cluster analysis among 105 maize accessions
For the full set of maize accessions, the highest ΔK value was confirmed for K = 2 in population structure analysis (Fig. 3). Based on a membership threshold of 0.8 (Wang et al. 2008), the maize accessions were divided into group I, group II, or the admixed group. We assigned 35 maize accessions to group I. Group II contained 46 maize accessions. The admixed group, composed of accessions with membership thresholds lower than 0.8, contained 24 maize accessions (Fig. 3). A dendrogram of the 105 maize accessions developed by UPGMA analysis is presented in Fig. 3, which shows three clusters with a GS value of 36%. Group I accounted for 57 accessions, group II contained 47 accessions, and group III contained only one accession (Fig. 3). Among them, 21 accessions were collected in foreign countries: three (IT026994, IT026995, IT026996) from France (FRA), six (IT105365, IT105366, IT105367, IT105368, IT105369, IT105370) from Austria (AUT), nine (IT124200, IT124217, IT124226, IT124236, IT124242, IT124259, IT124273, IT124279, IT124282) from the USA, and three (IT124245, IT124246, IT124247) from Canada (CAN). Our STRUCTURE results placed 12 of the foreign accessions (1 FRA, 4 AUT, 6 USA, 1 CAN) in group II, and assigned the remaining 9 accessions to the admixed group. However, no foreign maize accessions were clustered into group I. The NTSYS results assigned 19 foreign accessions to group I and the remaining two accessions to group II (Fig. 3).

UPGMA dendrogram and population structure in 105 maize accessions of Korean RDA-Genebank based on the SSR markers
Level of linkage disequilibrium and association analysis using Q GLM and Q + K MLM
Level of LD was confirmed based on combinations of the 100 SSR loci in 105 maize accessions. The mean of r 2 value was 0.016 and 0.014 in intra- and inter-chromosome, respectively. The mean of D′ values for intra-chromosome was 0.161, whereas that for inter-chromosome was 0.149 (Table 4). In addition, 2.26% combinations of SSR pairs were showed significant LD (P < 0.01) among total combinations (Table 4).
We performed association analysis between our sets of 100 SSR markers and 11 phenotypic traits in 105 maize accessions by Q GLM and Q + K MLM. We detected 72 marker-trait associations involving 42 SSR markers associated with the 11 agronomic traits using Q GLM (Supplementary Table 2). When we used Q + K MLM, five markers were associated with traits of SD, LW, LL, and ER (Supplementary Table 3). Table 4 presents information on overlapping significant marker trait associations (SMTAs) between Q GLM and Q + K MLM at a significance level of P ≤ 0.01. Among the five SMTAs, umc1062 and nc009 were associated with SD. phi092 was associated with ER, umc1857 was associated with LW, and umc1638 was associated with LL.
Discussion
Erosion of plant GD is a very serious problem caused by modernization and replacement of wild plants or landraces with a few elite varieties (van de Wouw et al. 2010; van Heerwaarden et al. 2009). Therefore, collection and preservation of PGR is increasingly important for crop breeding to support the demands of a growing human population. Effective management and utilization of PGR requires information about strain origins, phenotypic traits, and GD identified by molecular techniques. This study analyzed GD, as well as marker-trait associations, of maize accessions from the RDA-Genebank. To estimate GD and population structure in 105 RDA-Genebank maize accessions, we analyzed 100 SSR loci (10 loci per chromosome) covering the whole maize genome. We detected a total of 1104 alleles, with an average number of 11.0 alleles per locus in 105 maize accessions, and average GD and PIC of 0.73 and 0.70, respectively (Table 3).
So far, many similar studies have been done with us. In the International Maize and Wheat Improvement Center (CIMMYT), GD of 137 maize accessions at 79 SSR loci showed an average 7.2 alleles per locus and an average PIC value of 0.64 (Xia et al. 2005). An analysis of 129 maize accessions from Agriculture and Agri-Food Canada (AAFC) found an average of 3.62 alleles per locus and an average PIC of 0.68 at 105 SSR loci (Reid et al. 2011). In our study of RDA-Genebank accessions, we found that the average allele number and PIC values were higher than in the other two studied groups. Although the number of individuals, markers, and material types were different among the three groups, we concluded that the genetic resources of RDA-Genebank are relatively diverse. A previous report suggested that good populations for association analysis in plants are breeding and genebank collections of cultivars, breeding lines, and germplasm (Malosetti et al. 2007). The materials used in this study were genetic resources from a genebank with relatively higher GD, indicating that our materials are suitable for association analyses.
Determining the GD and population structure in a set of cultivars is very important for marker-assisted selection (MAS) and association analysis (Flint-Garcia et al. 2005; Wang et al. 2008). Our study used two methods to evaluate population structure, a model-based clustering method and a distance-based phylogenetic method. The model-based clustering method using the STRUCTURE program revealed a maximum ΔK value at K = 2, based on the statistical method described by Evanno et al. (2005). In addition, we divided the 105 RDA-Genebank lines into three groups by a distance-based phylogenetic method using the NTSYS program (Fig. 3). As a result, the patterns of population structure calculated by the two methods did not clearly distinguish between Korean and foreign collection regions. In general, population structures and clustering patterns are influenced by many different factors, like the natural history of a population, breeding systems, complexity of breeding practices, and selection by humans (Xie et al. 2008).
Identifying genes that control important agronomic traits is essential for effective maize breeding programs. Many other studies have identified SMTAs using linkage disequilibrium analysis (Borba et al. 2010; Mezmouk et al. 2011; Lorenz et al. 2010; Cui et al. 2015). LD is the non-random association of alleles between different two loci in a population (Flint-Garcia et al. 2005). The success of association analyses depend on the level of LD among the marker alleles related trait (Xie et al. 2008; Yan et al. 2011). The levels of LD in this study confirmed by calculating LD parameters, r 2 and D′, and allowed us to determine significant marker-trait associations. In general, if absolute value of D′ value is one, these two markers or alleles is a complete linkage disequilibrium. Whereas if absolute value of D′ is zero, represents as linkage equilibrium. The range of r 2 is between 0 and 1. If r 2 value is 1, those pair is known as perfect linkage disequilibrium. Based on these theory, we checked value of r 2 and D′ between intra- and inter-chromosome. As a results, both value was higher in intra-than those of inter-chromosome, and percentage of significant LD pair was also higher in intra-chromosome than inter-chromosome (Table 4). These results indicated that linkage is main factor causing higher LD. The level of LD in population was affected from many factors including linkage, experimental materials, marker density, selection, mutation and genetic drift (Wang et al. 2008). When compared with previous studies, level of LD in this study was relatively lower than other studies (Wang et al. 2008; Zhang et al. 2011). These may result from relatively lower marker density or using different experimental materials, i.e. relative higher heterozygosity due to lines in this study is breeding line.
However, false positives (Type I error) caused by spurious associations with population structure (Q matrix) and kinship (K matrix) are critical problems associated with these analyses (Zhang et al. 2010). To avoid false positives and compare two statistical models, we performed Q GLM and Q + K MLM (Supplementary Tables 2 and 3). The Q GLM, using only the Q matrix, identified 72 marker-trait associations, while the Q + K MLM, using both the Q matrix and the K matrix, identified five marker-trait associations. The Q + K MLM model had fewer markers than Q GLM at the same significance level, P ≤ 0.01, but all of the significant associations (P ≤ 0.01) detected by the Q + K MLM were also detected by the Q GLM. These results indicate that the Q + K MLM method is better for reducing the false positive rate in association analyses. Based on overlapping SMTAs between Q GLM and Q + K MLM, we detected two markers significantly (P ≤ 0.01) associated with SD, umc1062 and nc009, located on chromosomes 3 and 6, respectively. A SSR marker, phi092 located on chromosome 4, was associated with the ER trait. Another SSR marker, umc1857 located on chromosome 6, was associated with LW. Finally, umc1638, located on chromosome 8, was significantly associated with LL (P ≤ 0.01) (Table 4).
Among these SSR markers, umc1062, associated with SD in this study, was tightly linked to the phot1 (blue-light receptor phototropin 1) gene in the distal tip of the long arm of chromosome 3 (bin 3.09) (http://www.maizeGDB.org). The phot1 gene of maize plays a role in first positive phototropic curvature of maize coleoptiles through a mechanism involving chloroplast accumulation (Suzuki et al. 2014). In a previous report, umc1062 was a flanking marker for QTL of potassium (K) content in maize stalks (Tang et al. 2015), and K content significantly correlated with lodging as increase of stem strength and with rind thickness in maize (Melis and Farina 1984; Arnold et al. 1974). nc009, also associated with SD in this study, is tightly linked to the pl1 (purple plant 1) gene in the middle part of the long arm of chromosome 6 (bin 6.04) (http://www.maizeGDB.org). Moreover, nc009 is linked to QTL for leaf number (Zhang et al. 2011) and ear width (Peng et al. 2016) (Table 4).
The ER trait was linked with SSR marker phi092, which is also tightly linked to the ssu1 (ribulose bisphosphate carboxylase small subunit 1) or ssu2 (ribulose bisphosphate carboxylase small subunit 2) gene in the middle part of the long arm of chromosome 2 (bin 2.05) or 4 (bin 4.08), respectively (http://www.maizeGDB.org). The phi092 marker maps to regions of both chromosomes 2 and 4 in many separate mapping populations (http://www.maizeGDB.org). Although we did not check its exact chromosomal position in our study, phi092 is associated with QTL for oil/starch ratio (Guo et al. 2013), and QTL for kernel oil concentration on chromosome 2 (Song et al. 2004). This locus is also associated with QTL on chromosome 4 for growing degree units to anthesis, tassel branch number, and tassel length (Mayor 2008) (Table 4). Another SSR, umc1857, related to LW in this study, is linked to QTL for traits related to roots (Cai et al. 2012) and QTL for traits related to anthesis, silking interval (Chen et al. 2012), and maximum root length (Pan et al. 2011) on chromosome 6 (bin 6.04). Finally, umc1638 is linked to QTL for 20-kernel thickness (Liu et al. 2014). Thus, we found several SSR markers associated with different traits compared to other studies, suggesting potential pleiotropy or tight linkage of genes (Table 4).
In conclusion, this study successfully confirmed the GD and established the population structure of 105 maize accessions from the Korean RDA-Genebank PGR. We found relatively high diversity and two- or three-cluster population structures by two different methods, STRUCTURE and NTSYS. In addition, we found marker-trait associations, which can assist in marker-assisted selection (MAS) for breeding programs. We detected a total of five SMATs in our association analysis. These loci may support opportunities for effective preservation and utilization of existing cultivars, and for maize breeders to improve crop quality by MAS.
References
Akagi H, Yokozaki Y, Inagaki A, Fujimura T (1997) Highly polymorphic microsatellites of rice consist of AT repeats, and a classification of closely related cultivars with these microsatellite loci. Theor Appl Genet 94:61–67
Arnold JM, Josephson LM, Parks WL, Kincer HC (1974) Influence of nitrogen, phosphorus, and potassium applications on stalk quality characteristics and yield of corn. Agron J 66:605–608
Borba TCO, Brondani RPV, Breseghello F, Coelho ASG, Mendonça JA, Rangel PHN, Brondani C (2010) Association mapping for yield and grain quality traits in rice (Oryza sativa L.). Genet Mol Biol 33:515–524
Bostein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Genet 32:314–331
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23(19):2633–2635
Cai H, Chen F, Mi G, Zhang F, Maurer HP, Liu W, Reif JC, Yuan L (2012) Mapping QTLs for root system architecture of maize (Zea mays L.) in the field at different developmental stages. Theor Appl Genet 125:1313–1324
Chen Z, Cao Z, Tang B, Li L (2012) Mapping QTL for several drought related traits in maize (Zea mays L.) under field condition. Acta Agric Boreal Sin 27:79–86
Cui D, Xu C, Yang C, Zhang Q, Zhang J, Ma X, Qiao Y, Cao G, Zhang S, Han L (2015) Association mapping of salinity and alkalinity tolerance in improved japonica rice (Oryza sativa L. subsp. Japonica Kato) germplasm. Genet Resour Crop Evol 62:539–550
Dellaporta SL, Wood J, Hicks JB (1983) A simple and rapid method for plant DNA preparation. Version II. Plant Mol Biol Rep 1:19–21
Dice LR (1945) Measures of the amount of ecologic association between species. Ecology 26:297–302
Enoki H, Sato H, Koinuma K (2002) SSR analysis of genetic diversity among maize inbred lines adapted to cold regions of Japan. Theor Appl Genet 104:1270–1277
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Flint-Garcia SA, Thuillet AC, Yu JM, Pressoir G, Romero SM, Mitchell SE, Doebley J, Kresovich S, Goodman MM, Buckler ES (2005) Maize association population: a high-resolution platform for quantitative trait locus dissection. Plant J 44:1054–1064
Govindaraj M, Vetriventhan M, Srinivasan M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genet Res Int 2015:431487. doi:10.1155/2015/431487
Guo Y, Yang X, Chander S, Yan J, Zhang J, Song T, Li J (2013) Identification of unconditional and conditional QTL for oil, protein and starch content in maize. Crop J 1:34–42
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Kashiani P, Saleh G, Panandam JM, Abdullah NAP, Selamat A (2012) Molecular characterization of tropical sweet corn inbred lines using microsatellite markers. Maydica 57:154–163
Khoury MJ, Bertram L, Boffetta P, Butterworth AS, Chanock SJ, Dolan SM et al (2009) Genome-wide association studies, field synopses, and the development of the knowledge base on genetic variation and human diseases. Am J Epidemiol 170:269–279
Kim OG, Hong TK, Ko WR, Woo SY, Ma SJ, Park JW, Sa KJ, Lee JK (2016) Analysis of morphological characteristics for a core set of 194 maize accessions. Korean Breed Sci 48:133–139
Legesse BW, Myburg AA, Pixley KV, Botha AM (2007) Genetic diversity of African maize inbred lines revealed by SSR markers. Hereditas 144:10–17
Li Y, Shi Y, Cao Y, Wang T (2004) Establishment of a core collection for maize germplasm preserved in Chinese National Genebank using geographic distribution and characterization data. Genet Res Crop Evol 51:845–852
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Liu Y, Wang L, Sun C, Zhang Z, Zheng Y, Qiu F (2014) Genetic analysis and major QTL detection for maize kernel size and weight in multi-environments. Theor Appl Genet 127:1019–1037
Loiselle BA, Sork VL, Nason J, Graham C (1995) Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82:1420–1425
Lorenz AJ, Coors JG, Hansey CN, Kaeppler SM, De Leon N (2010) Genetic analysis of cell wall traits relevant to cellulosic ethanol production in Maize (Zea mays L.). Crop Sci 50:842–852
Malosetti M, van der Linden CG, Vosman B, van Eeuwijk FA (2007) A mixed-model approach to association mapping using pedigree information with an illustration of resistance to Phytophthora infestans in potato. Genetics 175:879–889
Mayor ML (2008) Genetic analysis of ear development and tassel architecture in maize (Zea mays L. ssp. mays). Graduate Theses and Dissertations, Paper 10636
Melis M, Farina MPW (1984) Potassium effects on stalk strength, premature death and lodging of maize (Zea mays L.). South Afr J Plant Soil 1:122–124
Mezmouk S, Dubreuil P, Bosio M, De´cousset L, Charcosset A, Praud S, Mangin B (2011) Effect of population structure corrections on the results of association mapping tests in complex maize diversity panels. Theor Appl Genet 122:1149–1160
Pan Q, Liu J, Mi G, Hou J, Chen F (2011) QTL Mapping of root and shoot traits of maize seedlings under different light and nitrogen conditions. J Maize Sci 19:39–42
Peng Q, Xue YD, Zhang XG, Li HM, Sun GY, Li WH, Xie HL, Tang JH (2016) Identification of heterotic loci for yield and ear traits using CSSL test population in maize. Acta Agron Sin 42:482–491
Pritchard JK, Wen W (2003) Documentation for STRUCTURE software: version 2
Rafalski JA, Vogel JM, Morgante M, Powell W, Andre C, Tingey SV (1996) Generating and using DNA markers in plants. In: Birren B, Lai E (eds) Non-mammalian genomic analysis. A practical guide. Academic Press, San Diego, pp 75–134
Reid LM, Xiang K, Zhu X, Baum BR, Molnar SJ (2011) Genetic diversity analysis of 119 Canadian maize inbred lines based on pedigree and simple sequence repeat markers. Can J Plant Sci 91:651–661
Rohlf FJ (1998) NTSYS-pc: Numerical taxonomy and multivariate analysis system. Version: 2.02. Exeter Software, Setauket, New York
Smith JSC, Chin ECL, Shu H, Smith OS, Wall SJ, Senior ML, Mitchell SE, Kresovich S, Ziegler J (1997) An evaluation of the utility of SSR loci as molecular marker in maize (Zea mays L.): comparison with RFLPs and pedigree. Theor Appl Genet 95:163–173
Solomon KF, Zeppa A, Mulugeta SD (2012) Combining ability, genetic diversity and heterosis in relation to F1 performance of tropically adapted shrunken (sh2) sweet corn lines. Plant Breed 131:430–436
Song XF, Song JR, Dai JR, Rocheford T, Li JS (2004) QTL mapping of kernel oil concentration with high-oil maize by SSR markers. Maydica 49:41–48
Steinhoff J, Liu W, Reif JC, Della Porta G, Ranc N, Würschum T (2012) Detection of QTL for flowering time in multiple families of elite maize. Theor Appl Genet 125:1539–1551
Suzuki H, Okamoto A, Kojima A, Nishimura T, Takano M, Kagawa T, Kadota A, Kanegae T, Koshiba T (2014) Blue-light regulation of ZmPHOT1 and ZmPHOT2 gene expression and the possible involvement of Zmphot1 in phototropism in maize coleoptiles. Planta 240:251–261
Tang JH, Wang YP, Hu YM, Wang CL, Tian GW, Liu ZH (2015) Effect of low N-stress to N, P, K contents and quantitative trait locus (QTL) analysis in maize kernels and stalks. Afr J Agric Res 10:2633–2641
van Heerwaarden J, Hellin J, Visser RF, van Eeuwijk FA (2009) Estimating maize genetic erosion in modernized smallholder agriculture. Theor Appl Genet 119:875–888
van de Wouw M, van Hintum T, van Treuren R, Visser B (2010) Genetic erosion in crops: concept, research results and challenges. Plant Genet Resour 8:1–15
Vančetović J, Mladenović-Drinić S, Babić M, Ignjatović-Micić D, Anđjelković V (2010) Maize Genebank collections as potentially valuable breeding material. Genetika 42:9–11
Wang R, Yu Y, Zhao J, Shi Y, Song Y, Wang T, Li Y (2008) Population structure and linkage disequilibrium of a mini core set of maize inbred lines in China. Theor Appl Genet 117:1141–1153
Xia XC, Reif JC, Melchinger AE, Frisch M, Hoisington DA, Beck D, Pixley K, Warburton ML (2005) Genetic diversity among CIMMYT maize inbred lines investigated with SSR markers: II. Subtropical, tropical midaltitude, and highland maize inbred lines and their relationships with elite U.S. and European maize. Crop Sci 45:2573–2582
Xie CX, Warburton M, Li MS, Li XH, Xiao MJ, Hao ZF, Zhao Q, Zhang SH (2008) An analysis of population structure and linkage disequilibrium using multilocus data in 187 maize inbred lines. Mol Breed 21:407–418
Yan J, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L.) genetic improvement. Crop Sci 51:433–449
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Zhang Z, Ersoz E, Lai CQ, Todhunter RJ, Tiwari HK, Gore MA, Bradbury PJ, Yu J, Arnett DK, Ordovas JM, Buckler ES (2010) Mixed linear model approach adapted for genome-wide association studies. Nature Genet 42:355–360
Zhang X, Tang B, Liang W, Zheng Y, Qiu F (2011) Quantitative genetic analysis of flowering time, leaf number and photoperiod sensitivity in maize (Zea mays L.). J Plant Breed Crop Sci 3:168–184
Zhu C, Gore M, Buckler ES, Yu J (2008) Status and prospects of association mapping in plants. Plant Genome J 1(1):5
Acknowledgements
This study was supported by the Golden Seed Project (No. 213009-05-1-WT821, PJ012650012017), the Ministry of Agriculture, Food, and Rural Affairs (MAFRA), the Ministry of Oceans and Fisheries (MOF), the Rural Development Administration (RDA), and the Korea Forest Service (KFS).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
O Gyeom Kim declares that he has no conflict of interest. Kyu Jin Sa declares that he has no conflict of interest. Jung-Ro Lee declares that he has no conflict of interest. Ju Kyong Lee declares that he has no conflict of interest.
Ethical approval
This article does not contain any studies with human subjects or animals performed by any of the above authors.
Additional information
O Gyeom Kim and Kyu Jin Sa have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, O.G., Sa, K.J., Lee, JR. et al. Genetic analysis of maize germplasm in the Korean Genebank and association with agronomic traits and simple sequence repeat markers. Genes Genom 39, 843–853 (2017). https://doi.org/10.1007/s13258-017-0547-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-017-0547-2