
Abstract
Although genome-wide association (GWA) studies have provided valuable insights into the genetic architecture of human disease, they have elucidated relatively little of the heritability of complex traits. A significant part of the missing heritability might be explained by rare combinations of common SNPs. We hypothesized that epistasis among 15 genes (148 SNPs) involved in lipoprotein metabolism would influence HDL-cholesterol (HDL-C) and LDL-cholesterol (LDL-C) levels. Using SNPwinter software with the various epistatic models, we identified 58 association signals with HDL-C levels for SNPs in eleven genes and 118 associations with LDL-C for SNPs in fourteen genes. These associations were discovered in the urban Ansan cohort (n = 4,102) and replicated in a rural cohort (n = 3,434), the Ansung. We found replicated associations with new genes (SOAT1, APOB, HMGCR, and FDFT1 for HDL-C, and SOAT1, FDFT1, LPL, SQLE, ABCA1, LRP1, SCARB1, and PLTP for LDL-C), in addition to those (CETP, LIPC, LPL, ABCA1, PLTP, SCARB1, and LRP1 for HDL-C, and CETP, LIPC, LDLR, APOB, CYP7A1, and HMGCR for LDL-C) identified by GWA studies, through investigating pairwise interactions between candidate genes of biological and clinical importance. Interestingly, we found that some genes were more likely to be involved in epistatic interactions (ABCA1 and LIPC for HDL-C, and ABCA1, SCARB1, and LIPC for LDL-C).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
High LDL-cholesterol (LDL-C) levels and low HDL-cholesterol (HDL-C) levels are risk factors for cardiovascular disease (CVD) (Aguilera et al. 2008; Jacobs et al. 1990), and the likelihood of coronary heart disease development can be determined from their levels (Assmann et al. 2002). However, although statins can be used to aggressively reduce LDL-C levels, significant cardiovascular risk remains and, therefore, HDL-C levels have also been targeted to prevent and treat CVD (Auer et al. 2004; Singh et al. 2007). Extensive research has provided evidence that increases in HDL-C reduce the risk of CVD (Barter et al. 2007; Belalcazar et al. 2003; Gordon et al. 1989; Morehouse et al. 2007). In addition, the use of genetic screening could be useful to guide selection of lipid-lowering therapy, as it has been shown that individuals carrying heterozygous genotypes at two HMGCR SNPs may have significantly smaller reductions in LDL-C levels than those with homozygous genotypes when treated with pravastatin (Chasman et al. 2004).
Epistasis is defined as a departure from a linear model that describes the relationship between a phenotypic result and two or more predictors (Cordell 2009). It has been recognized that epistasis in complex trait studies is a common phenomenon (Carlborg and Haley 2004; Cordell 2009), and evidence for epistatic interaction between QTL (quantitative trait loci) for body mass index (BMI) and HDL-C levels, respectively, was reported (Ma et al. 2012; Wei et al. 2012).
Cordell (2009) examined the methodology and related software tools used to detect epistasis between genetic loci contributing to human disease, which can also provide information about underlying biological and biochemical pathways. PLINK (Purcell et al. 2007) was selected as the most computationally feasible tool as, of the methods investigated, it provided the clearest statistical significance through a semi-exhaustive search of two-locus interactions (Cordell 2009). Unfortunately, however, the PLINK tool can currently only test allelic interactions.
Epistasis of associations with human BMI was investigated through exhaustive two-locus genome scans and a number of epistatic interactions could be replicated (Wei et al. 2012). An interaction effect on HDL-C levels between HMGCR and a locus near LIPC was identified through knowledge-driven analysis and was replicated in a number of independent multiple-ethnic populations (Ma et al. 2012).
Although genome-wide association (GWA) studies have provided valuable insights into the genetic architecture of human disease, they have elucidated relatively little of the heritability of complex traits; for example, only 5.2 and 5.7 % of the heritability of HDL- and LDL-C, respectively, has been explained in this way (Kathiresan et al. 2008; Manolio et al. 2009). One strategy to identify factors underlying the missing heritability is to investigate epistasis among genes (Eichler et al. 2010; Manolio et al. 2009). A substantial proportion of the missing heritability may be due to rare combinations of common variants, rather than individual common or rare variants (Eichler et al. 2010).
We used statistical replication to detect the most reliable results in single locus and epistasis tests of associations with HDL- and LDL-C levels, and based on the results of this analysis, we focused on two key questions: (1) would epistasis among 15 genes (148 SNPs) involved in lipoprotein metabolism influence HDL-C and LDL-C levels; and (2) could the patterns of their epistatic interactions differentiate between HDL-C and LDL-C according to different pairwise interaction models?
We used a novel method employing our in-house developed software, ‘SNPwinter’, to detect epistasis between two loci. In our previous work, we used the classical stratified analysis technique to detect epistasis between two genetic variants associated with premature ovarian failure (Kim et al. 2011a, b; Pyun et al. 2012). This technique has also been used to detect gene-environment and gene–gene interaction (Morgan et al. 2009; Perdigones et al. 2010; Zhou et al. 2012). The SNPwinter method extends the analysis of epistasis used by PLINK, which uses only an allele-(additive)-by-allele model without the adjusted terms, facilitating nine epistatic models with the adjusted terms where many more pairwise interactions are considered. Besides the three typical epistatic models (additive-by-additive, dominant-by-dominant, and recessive-by-recessive), we added six further models (additive-by-dominant, additive-by-recessive, and dominant-by-recessive, and their reversed counterparts), which can be considered relevant models in biological or functional epistasis.
We used SNPwinter to investigate the impact of epistasis among 15 candidate genes (148 SNPs) involved in lipoprotein metabolism on HDL-C and LDL-C levels in a Korean population consisting of 7,536 individuals. The urban Ansan sample (n = 4,102) was used as the discovery cohort for detection of pairwise interactions, and replication was performed in the rural Ansung cohort (n = 3,434).
Materials and methods
Subjects
The population data used in this study was provided by the Korea Association Resource (KARE) project, from the Korean Genome Epidemiology Study (KoGES), which was conducted by the Korean National Institute of Health (KNIH). The study protocol was approved by the Institutional Review Board of KNIH and written informed consent was obtained from all participants. A cross-sectional analysis of samples from urban and rural communities in Korea was conducted. The populations examined (Cho et al. 2009) were recruited from rural (Ansung) and urban (Ansan) communities in South Korea that were part of the KoGES established in 2001. There were 5,018 subjects from the Ansung and 5,020 from the Ansan community included in this study. The age of the participants ranged from 40 to 69 years. A total of 8,842 subjects remained after selection of samples for quality control purposes (Cho et al. 2009). An additional 1,306 subjects who were undergoing treatment for hypertension, diabetes, myocardial infarction, or hyperlipidemia were excluded from the study, since therapy for these conditions could influence LDL-C or HDL-C levels. A detailed list of the characteristics of the study participants is shown in Table 1.
Genotyping
Genotype data from all 148 SNPs was available to the research community through the KARE project from KoGES and was included in this study. Genotyping of samples from the Ansung and Ansan cohorts was performed using the Affymetrix Genome-Wide Human SNP Array 5.0 (Affymetrix Inc., Santa Clara, CA, USA) with genotype calling using the Bayesian Robust Linear Model with the Mahalanobis distance genotype calling algorithm. SNPs were filtered if any of the following criteria were met: (i) a call rate lower than 95 %; (ii) a minor allele frequency (MAF) lower than 0.01; or (iii) a significant deviation from the Hardy–Weinberg equilibrium (HWE; P < 1 × 10−6).
Biochemical measures
Biochemical data from the KoGES were obtained through the KARE project. Blood samples were collected from the participants after at least 8 h of overnight fasting. For the Ansung and Ansan cohorts, serum HDL-C concentration was measured with the Advia 1650 analyzer (Siemens, Tarrytown, NY, USA). LDL-C values were calculated using the equation formulated by Friedewald et al. (1972) for individuals with triglycerides levels lower than 400 mg/dl.
Statistical analyses
To examine possible associations between genotypes (148 SNPs and their pairwise interactions) and phenotypes (HDL-C and LDL-C levels), linear regression analyses were conducted with adjustments for age, sex, and BMI in the Ansan and Ansung cohorts (with the addition of geographical area in the combined cohort). Log transformation was applied to HDL-C (but not LDL-C) level values to normalize their distribution. Results obtained from the Ansan cohort were tested for replication in the Ansung. Replications were confirmed if P < 0.05, given that only a single replication test was required. Replicated results were reconfirmed in the combined cohort.
Statistical analyses were performed using ‘SNPwinter’ for epistasis testing and ‘SNPassociator’ for single locus testing. Both programs were implemented with Python version 2.7.1 and R version 2.15.1. Considering a pair of SNPs denoted as SNP1 and SNP2, the following genetic model was considered:
where y is a quantitative trait in linear regression or a binary trait in logistic regression, and β 0 is the intercept coefficient, β 1 and β 2 are the main effect coefficients at loci 1 and 2, respectively, β 3 is the interaction coefficient, α 1 … α k are the covariate coefficients, and e is the error term. Our tool, SNPwinter, uses nine pairwise interaction models. First, using an additive-(allele)-by-additive interaction model, each genotype was coded as values (0, 1, or 2) according to the number of minor alleles at loci 1 and 2, respectively. Second, using a dominant-by-dominant interaction model, genotypes at both loci were coded as ‘1’ if they had at least one minor allele and as ‘0’ otherwise. Next, using a recessive-by-recessive model the genotypes at both loci were coded as ‘1’ if they were minor allele homozygotes and as ‘0’ otherwise. Finally, each genotype at both loci was coded as described above, using additive-by-dominant, dominant-by-additive, additive-by-recessive, recessive-by-additive, dominant-by-recessive, and recessive-by-dominant models.
Our approach has two main procedures. The first procedure is to code genotypes according to a selected one of nine interaction models. The second procedure is to calculate two-way interactions using linear or logistic regression a user select with adjusted terms. SNPwinter implemented generalized linear models (GLMs) using the ‘glm’ module in the R package to perform genotypic association analysis in the nine different pairwise interaction models. The GLMs make it possible to use various regression models by extending the familar regression models like linear models. It is freely available to download from the website http://code.google.com/p/snpsinteractor, and is supported on Linux and Windows.
The IMPUTE program was used for SNP imputation (Marchini et al. 2007). A reference panel from International HapMap (phase II/release 22/NCBI build 36 and dbSNP build 126) data consisting of 2.2 million SNPs from Japanese and Han Chinese individuals was utilized as a template for inferring untyped genotypes. Imputed SNPs with low quality were eliminated according to the following criteria: (i) low genotype information content (information lower than 0.5), (ii) a posterior probability score lower than 0.90, (iii) a call rate lower than 0.95, (iv) MAF lower than 0.01, and (v) a HWE P value lower than 1 × 10−6.
Results
All 148 SNPs across 15 candidate genes, available to the research community through the KARE project, from the KoGES, were investigated. The selected genes had roles in mediating the impact of statins on hepatic cholesterol metabolism, and consequential impacts on plasma lipoprotein transport (Klein et al. 2001). The features of the included genes are summarized in Supplementary Table S1 and have been described previously (http://www.genecards.org/). The characteristics of the subjects included in the study are presented in Table 1. Supplementary Tables S2–S3 show the single locus association results of 148 SNPs in Ansan (n = 4,102), Ansung (n = 3,434), and combined cohorts (n = 7,536) with HDL-C and LDL-C levels, respectively, in additive, dominant, and recessive models. Supplementary Tables S4–S9 contain the results of replication analyses of the single locus associations with HDL-C and LDL-C levels in the three models. All SNPs replicated in both dominant and recessive models also showed association in the additive model in the single locus tests. The genes with replicated associations with HDL-C levels were LPL, ABCA1, LIPC, and CETP, and for LDL-C they were HMGCR and LDLR.
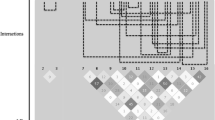
All possible pairwise epistatic interactions were scanned in the Ansan cohort and investigated for replication in the Ansung. The number of tests of association for epistatic effects in the Ansan was 97,902 for HDL-C and LDL-C levels, respectively. No SNP pairs withstood the Bonferroni-corrected threshold (P < 5.1 × 10−7). Nevertheless, using the consensus threshold (P < 0.05), 58 and 118 results for HDL-C and LDL-C, respectively, were replicated in the Ansung cohort using the nine epistatic models (Supplementary Tables S10–S11 and Figs. 1, 2, 3, 4). The figures illustrate epistatic interactions between pairs of genes using data extracted from SNP–SNP interactions determined using all nine epistatic models. Figures 1 and 3 illustrate pairwise interactions separated by the nine different models for HDL-C and LDL-C, respectively. Although only four and two of the 15 candidate genes were found to be associated with HDL-C and LDL-C, respectively, in single locus tests, pairwise interaction tests identified 11 and 14 of them, respectively. Interestingly, the number of epistatic interactions (14 genes and 19 links between them) associated with LDL-C was greater than those with HDL-C (11 genes and 15 links between them), while the number of genes associated with LDL-C in single locus tests was just half of that with HDL-C. Figure 1 illustrates the seven genes and six links (six gene–gene interactions) associated with HDL-C detected under the additive-by-additive model and also exactly the same results under the dominant-by-dominant model. Three genes (HMGCR, APOB, and FDFT1) and two links (HMGCR vs. APOB and FDFT1 vs. LPL) were newly detected as associated with HDL-C levels in the recessive-by-recessive model, with one more link (ABCA1 vs. FDFT1) in the additive-by-dominant model, a new gene (CETP) and three new links (LIPC vs. CETP, LIPC vs. PLTP, and SCARB1 vs. ABCA1) in the additive-by-recessive model, and new links in the dominant-by-dominant (CETP vs. ABCA1), dominant-by-additive (APOB vs. CETP), and recessive-by-additive (PLTP vs. SOAT1) models. Figure 3 shows the seven genes and four links associated with LDL-C detected under the additive-by-additive model, three more genes (SACRB1, CETP, and FDFT1) and three more links (SCARB1 vs. LIPC, CETP vs. ABCA1, and FDFT1 vs. ABCA1) were detected in the dominant-by-dominant model, another gene (LRP1) and two more links (LRP1 vs. LIPC and LRP1 vs. LDLR) in the recessive-by-recessive model, a new gene (SOAT1) and four new links (SOAT1 vs. SCARB1, LIPC vs. ABCA1, LIPC vs. LDLR, and SCARB1 vs. HMGCR) in the additive-by-dominant model, a new gene (CYP7A1) and two new links (SCARB1 vs. CYP7A1 and APOB vs. FDFT1) in the additive-by-recessive model, three new links (FDFT1 vs. SCARB1, LIPC vs. HMGCR, and SCARB1 vs. ABCA1) in the recessive-by-additive model, and a new gene (LPL) and a new link (APOB vs. LPL) in the recessive-by-dominant model.

Pairwise genetic interactions associated with HDL-C levels in the various models. Pairwise genetic interactions, produced separately by the nine different epistatic interaction models, between the SNP pairs of 11 genes involved in lipoprotein metabolism, which are associated with HDL-C levels in the urban Ansan cohort and replicated in the rural Ansung cohort, are illustrated. The nine models are additive-by-additive, dominant-by-dominant, recessive-by-recessive, additive-by-dominant, additive-by-recessive, dominant-by-recessive, dominant-by-additive, recessive-by-additive, and recessive-by-dominant

Pairwise genetic interactions associated with HDL-C levels. Pairwise genetic interactions between the SNP pairs of 11 genes involved in lipoprotein metabolism, which are associated with HDL-C levels in the urban Ansan cohort and replicated in the rural Ansung cohort, are illustrated

Pairwise genetic interactions associated with LDL-C levels in the various models. Pairwise genetic interactions, produced by the nine different epistatic interaction models, between the SNP pairs of 14 genes involved in lipoprotein metabolism, which are associated with LDL-C levels in the urban Ansan cohort and replicated in the rural Ansung cohort, are illustrated. The nine models are additive-by-additive, dominant-by-dominant, recessive-by-recessive, additive-by-dominant, additive-by-recessive, dominant-by-recessive, dominant-by-additive, recessive-by-additive, and recessive-by-dominant

Pairwise genetic interactions associated with LDL-C levels. Pairwise genetic interactions between the SNP pairs of 14 genes involved in lipoprotein metabolism, which are associated with LDL-C levels in the urban Ansan cohort and replicated in the rural Ansung cohort, are illustrated
The best replicated pair were rs4652363 (SOAT1) - rs10773111 (SCARB1) in the dominant-by-dominant model for HDL-C levels. The epistatic interaction in the combined cohort was shown with a 0.082 mg/dl decrease per the unit value of S 1·S 2 in the multiple regression model (P = 3.54 × 10−5). For LDL-C, the best replicated pair were rs7025776 (ABCA1) - rs11631342 (LIPC) in the recessive-by-additive model, and showed the epistatic interaction with a 12.9 mg/dl increase per the unit value of S 1·S 2 (P = 1.21 × 10−4) in the combined cohort.
Discussion
Using SNPwinter software developed in-house, we identified 58 replicated pairwise epistatic association signals with HDL-C levels in eleven genes: SOAT1, APOB, HMGCR, FDFT1, LPL, ABCA1, LRP1, SCARB1, LIPC, CETP, and PLTP (Supplementary Table S10). For LDL-C levels, 118 replicated pairwise associations in fourteen genes (SOAT1, APOB, HMGCR, FDFT1, LPL, CYP7A1, SQLE, ABCA1, LRP1, SCARB1, LIPC, CETP, LDLR, and PLTP) were identified (Supplementary Table S11). Notably, the two genes, ABCA1 and LIPC, were identified as contributing to many pairwise interaction effects (six and four links, respectively) influencing HDL-C levels (Fig. 2). For LDL-C, the ABCA1, SCARB1, and LIPC genes provided multiple epistatic effects (six links each) (Fig. 4). Interestingly, considering the fact that in single locus tests the number of the genes with the replicated associations with LDL-C was half of that with HDL-C, the genes commonly involved in epistatic interactions were more frequently identified as associated with LDL-C than HDL-C levels.
It was reported that the bulk (~93 %) of disease- and trait- associated variants emerging from hundreds of GWAS lay within noncoding sequence, and 76.6 % of all noncoding GWAS SNPs either were within regulatory regions (57.1 %) or were in complete linkage disequilibrium (LD) with SNPs in the regulatory site (19.5 %) (Maurano et al. 2012). In the present study, the many epistatic SNPs associated with HDL-C or LDL-C were also clearly within regulatory regions of SOAT1, HMGCR, CYP7A1, SQLE, CETP, and PLTP genes as investigated in the LD blocks containing epistatic SNPs. After investigating whether the SNPs identified in this study were in regulatory regions or were in strong LDs with the regulatory region, using the UCSC Genome Browser, we found that many blocks containing epistatic SNPs overlapped with promoter and/or enhancer regions as shown in Supplementary Figs. 1–14. In addition, the epistatic SNPs in LIPC, SCARB1, LRP1, ABCA1, and FDFT1 genes might be within regularoty or coding regions. On the other hand, the remainder of epistatic SNPs were within coding regions of LDLR, LPL, and APOB genes, some coding SNPs of which could lead to an amino acid change.
The lipid metabolism genes apolipoprotein B (APOB), cholesteryl ester transfer protein (CETP), and LDL receptor (LDLR) have been evaluated in clinical studies addressing pharmacogenetic interactions with statins (Chasman et al. 2004). In this study, all three of these genes (APOB, CETP, and LDLR) had replicated signals in the epistatic interaction tests for HDL-C and LDL-C, respectively, while only each gene (CETP for HDL-C, and LDLR for LDL-C) was replicated in the single locus associations. The target of statin therapy is 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR), and squalene synthase, encoded by the FDFT1 gene, is an alternative target for lipid-lowering therapy (Chasman et al. 2004). While in single locus tests only the HMGCR gene association with LDL-C levels was replicated, both HMGCR and FDFT1 demonstrated replicated interaction tests for HDL-C and LDL-C, respectively. The four genes, CETP, LIPC, LPL, and ABCA1, play a key role in both reverse cholesterol transport and HDL metabolism (Singh et al. 2007). While all of them were replicated for only HDL-C levels in the single locus associations, they, in the epistatic interaction tests, had replicated associations for HDL-C and LDL-C, respectively, demonstrating many interactions of the two genes in particular (LIPC and ABCA1). GWA findings (Kathiresan et al. 2008; Willer et al. 2008; Hegele 2009; Aulchenko et al. 2009; Kathiresan et al. 2009; Teslovich et al. 2010) have increased confidence in the significance of the associations with genes identified in previous studies, including, for HDL-C, those of CETP, LIPC, lipoprotein lipase (LPL), ATP-binding cassette (ABCA1), phospholipid transfer protein (PLTP), Scavenger receptor class B member 1 (SCARB1), and LDL receptor-related protein 1 (LRP1), and, for LDL-C, CETP, hepatic lipase (LIPC), LDLR, APOB, CYP7A1 (encoding cholesterol 7-alpha-hydroxylase), and HMGCR. All GWA findings were replicated in our pairwise interaction tests for HDL-C and LDL-C, while only four (CETP, LIPC, LPL, and ABCA1) and two (LDLR and HMGCR) genes were replicated in single locus tests of association with HDL-C and LDL-C levels, respectively. Of the 15 genes selected for this study (Supplementary Table S1), the remaining four (SOAT1, APOB, HMGCR, and FDFT1) for HDL-C and eight (SOAT1, FDFT1, LPL, SQLE, ABCA1, LRP1, SCARB1, and PLTP) for LDL-C have previously not reached genome-wide levels of significance for association with levels of lipoproteins, despite their involvement in lipoprotein metabolism. This is of particular note for FDFT1 as its product, squalene synthase, is involved in cholesterol biosynthesis and has been nominated as a strong candidate for lipid-lowering therapy. In this study, all of the remaining genes showed replicated interaction associations with HDL-C and LDL-C levels, respectively.
Given sufficient computing power, the SNPwinter could be used for GWA studies using distributed computing to tackle the huge amount of SNP data. PLINK is able to perform calculations using large datasets very rapidly; however, it only provides analysis of two-way interactions based on allele-(additive)-by-allele model, which limits the diversity of association analyses compared with SNPwinter, which uses nine epistatic models. In addition to the typical three models (additive-by-additive, dominant-by-dominant, and recessive-by-recessive) explained in detail (Cordell 2009), we added six more models (additive-by-dominant, additive-by-recessive, dominant-by-recessive, and the three reverse counterparts of these), which may provide an indication of biological relevance in addition to statistical significance. Our empirical results indicate that the epistatic interactions identified using the new six models are indeed better than those using the three standard models for HDL-C and LDL-C levels, based on the number of replications observed.
Although the 15 candidate genes investigated here are involved in lipoprotein metabolism and the majority are of biological and clinical importance, only four (LPL, ABCA1, LIPC, and CETP) for HDL-C and two (HMGCR and LDLR) for LDL-C were found to be replicated in single locus tests, suggesting substantial missing heritability for HDL- and LDL-C levels. However, we found replicated associations in new genes, in addition to those identified by GWA studies, through investigating pairwise interactions between candidate genes using SNPwinter with the various epistatic models.
References
Aguilera CM, Gil-Campos M, Canete R, Gil A (2008) Alterations in plasma and tissue lipids associated with obesity and metabolic syndrome. Clin Sci (Lond) 114:183–193
Assmann G, Cullen P, Schulte H (2002) Simple scoring scheme for calculating the risk of acute coronary events based on the 10-year follow-up of the prospective cardiovascular Munster (PROCAM) study. Circulation 105:310–315
Auer J, Weber T, Eber B (2004) Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med 351:714–717
Aulchenko YS et al (2009) Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet 41:47–55
Barter P et al (2007) HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med 357:1301–1310
Belalcazar LM et al (2003) Long-term stable expression of human apolipoprotein A-I mediated by helper-dependent adenovirus gene transfer inhibits atherosclerosis progression and remodels atherosclerotic plaques in a mouse model of familial hypercholesterolemia. Circulation 107:2726–2732
Carlborg O, Haley CS (2004) Epistasis: too often neglected in complex trait studies? Nat Rev Genet 5:618–625
Chasman DI, Posada D, Subrahmanyan L, Cook NR, Stanton VP Jr, Ridker PM (2004) Pharmacogenetic study of statin therapy and cholesterol reduction. JAMA 291:2821–2827
Cho YS et al (2009) A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet 41:527–534
Cordell HJ (2009) Detecting gene–gene interactions that underlie human diseases. Nat Rev Genet 10:392–404
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11:446–450
Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18:499–502
Gordon DJ et al (1989) High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation 79:8–15
Hegele RA (2009) Plasma lipoproteins: genetic influences and clinical implications. Nat Rev Genet 10:109–121
Jacobs DR Jr, Mebane IL, Bangdiwala SI, Criqui MH, Tyroler HA (1990) High density lipoprotein cholesterol as a predictor of cardiovascular disease mortality in men and women: the follow-up study of the Lipid Research Clinics Prevalence Study. Am J Epidemiol 131:32–47
Kathiresan S et al (2008) Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 40:189–197
Kathiresan S et al (2009) Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 41:56–65
Kim S, Pyun JA, Cha DH, Ko JJ, Kwack K (2011a) Epistasis between FSHR and CYP19A1 polymorphisms is associated with premature ovarian failure. Fertil Steril 95:2585–2588
Kim S, Pyun JA, Kang H, Kim J, Cha DH, Kwack K (2011b) Epistasis between CYP19A1 and ESR1 polymorphisms is associated with premature ovarian failure. Fertil Steril 95:353–356
Klein TE et al (2001) Integrating genotype and phenotype information: an overview of the PharmGKB project. Pharmacogenetics Research Network and Knowledge Base. Pharmacogenomics J 1:167–170
Ma L, Brautbar A, Boerwinkle E, Sing CF, Clark AG, Keinan A (2012) Knowledge-driven analysis identifies a gene–gene interaction affecting high-density lipoprotein cholesterol levels in multi-ethnic populations. PLoS Genet 8:e1002714
Manolio TA et al (2009) Finding the missing heritability of complex diseases. Nature 461:747–753
Marchini J, Howie B, Myers S, McVean G, Donnelly P (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39:906–913
Maurano MT et al (2012) Systematic localization of common disease-associated variation in regulatory DNA. Science 337:1190–1195
Morehouse LA et al (2007) Inhibition of CETP activity by torcetrapib reduces susceptibility to diet-induced atherosclerosis in New Zealand White rabbits. J Lipid Res 48:1263–1272
Morgan AW et al (2009) Reevaluation of the interaction between HLA-DRB1 shared epitope alleles, PTPN22, and smoking in determining susceptibility to autoantibody-positive and autoantibody-negative rheumatoid arthritis in a large UK Caucasian population. Arthritis Rheum 60:2565–2576
Perdigones N et al (2010) Evidence of epistasis between TNFRSF14 and TNFRSF6B polymorphisms in patients with rheumatoid arthritis. Arthritis Rheum 62:705–710
Purcell S et al (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Pyun JA, Kim S, Cha DH, Ko JJ, Kwack K (2012) Epistasis between the HSD17B4 and TG polymorphisms is associated with premature ovarian failure. Fertil Steril 97:968–973
Singh IM, Shishehbor MH, Ansell BJ (2007) High-density lipoprotein as a therapeutic target: a systematic review. JAMA 298:786–798
Teslovich TM et al (2010) Biological, clinical and population relevance of 95 loci for blood lipids. Nature 466:707–713
Wei WH et al (2012) Genome-wide analysis of epistasis in body mass index using multiple human populations. Eur J Hum Genet 20:857–862
Willer CJ et al (2008) Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 40:161–169
Zhou XJ et al (2012) Gene-gene interaction of BLK, TNFSF4, TRAF1, TNFAIP3, and REL in systemic lupus erythematosus. Arthritis Rheum 64:222–231
Acknowledgments
This study was supported by a grant from the Korea Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (A110749), grants from Korea Centers for Disease Control and Prevention (4845-301, 4851-302, 4851-307), and Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (No. 2009-0093821).
Conflict of interest
The authors have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kim, S., Shin, C., Cho, N.H. et al. Epistasis between SNPs in genes involved in lipoprotein metabolism influences high- and low-density lipoprotein cholesterol levels. Genes Genom 36, 809–817 (2014). https://doi.org/10.1007/s13258-014-0216-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13258-014-0216-7