
ABSTRACT
Chimeric antigen receptor (CAR) is a recombinant immunoreceptor combining an antibody-derived targeting fragment with signaling domains capable of activating cells, which endows T cells with the ability to recognize tumor-associated surface antigens independent of the expression of major histocompatibility complex (MHC) molecules. Recent early-phase clinical trials of CAR-modified T (CAR-T) cells for relapsed or refractory B cell malignancies have demonstrated promising results (that is, anti-CD19 CAR-T in B cell acute lymphoblastic leukemia (B-ALL)). Given this success, broadening the clinical experience of CAR-T cell therapy beyond hematological malignancies has been actively investigated. Here we discuss the basic design of CAR and review the clinical results from the studies of CAR-T cells in B cell leukemia and lymphoma, and several solid tumors. We additionally discuss the major challenges in the further development and strategies for increasing anti-tumor activity and safety, as well as for successful commercial translation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
“Natural forces within us are the true healers of disease.”—Hippocrates (de Coana et al., 2015).
Undoubtedly, the immune system is the right cancer healer, especially in the context of currently available therapies such as chemotherapy, radiotherapy, and targeted therapy, which have been less successful than anticipated. Harnessing the immune system to kill cancer is a durable concept that has more than 100 years of history; it was first demonstrated in 1891 by William Coley’s use of Coley’s toxin, a mixture of heat-killed bacteria to elicit regression of inoperable sarcomas (Elert, 2013). Despite this early beginning, efforts to reliably manipulate the immune system to promote tumor regression have been universally disappointing. In recent decades, with the significant progress in understanding the inherent immune biology related to cancer, effective immunotherapy treatments for cancer have gradually emerged (Fyfe et al., 1995; Atkins et al., 1999; Kantoff et al., 2010) and reached an important turnover in the history of cancer treatment as named by Science magazine the “breakthrough of 2013” due to the striking proof-of-concept data of immune checkpoint anti-CTLA-4 and PD-1 antibodies as well as CAR therapy (Couzin-Frankel, 2013). Subsequently, a spectrum of encouraging outcomes of those modalities in other tumors have attracted more big players during the past 2 years, denoting that cancer immunotherapy is coming of age.
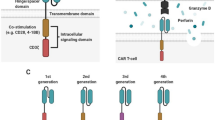
The presented concept of CAR is based on two seminal research studies as the increasing understanding of the construct and function of T cell receptor (TCR) complex (Fig. 1). First, in 1989 Gross et al. constructed a chimeric TCR (cTCR) gene made by replacing the Vα and Vβ extracellular domains of the TCR chains with their VH and VL immunoglobulin homologs (CαVH + CβVL or CαVL + CβVH). The resulting cTCR was expressed on the surface of cytotoxic T lymphocytes, recognized antigen in a non-MHC-restricted manner, and effectively transmitted the transmembrane signal for T cell activation (Gross et al., 1989). These results proved that replacing the variable region of TCR with those of antibody for endowing the T cells with antibody-type specificity is viable (Eshhar, 2014), and was subsequently followed by Goverman et al. with a consistent outcome (Goverman et al., 1990). Another pioneering study mainly focused on the chimeric proteins constructed between either CD8, CD4, or CD25 (also called α chain of the human interleukin-2 receptor) and cytoplasmic tails of ζ (Irving and Weiss, 1991; Romeo and Seed, 1991; Letourneur and Klausner, 1991). Those chimeric proteins have resulted in biochemical events of early T cell activation such as interleukin-2 (IL-2) production and Ca2+ influx, which validated that cytoplasmic tails of ζ could replicate much of the TCR signaling (van der Stegen et al., 2015). Taking advantage of these advances, in 1993 Eshhar et al. pioneered to design a gene composed of a single chain variable fragment (scFv) of an antibody linked with ζ chains, which is aimed to overcome the difficulty in activating anti-tumor T cells through the TCR (Eshhar et al., 1993). The transfected cytolytic T cell hybridoma triggered IL-2 secretion upon encountering antigen and mediated non-MHC-restricted hapten-specific target cell lysis. This new artificial receptor called T-body is known as the first-generation CAR. Subsequent experiments after this initial report further demonstrated the anti-tumor potential of the T cells transfected with these fusion receptors (Brocker et al., 1993; Hwu et al., 1993; Stancovski et al., 1993; Gross et al., 1995; Hwu et al., 1995). However, these fusion receptors are devoid of costimulatory elements that are required for full T cell activation and only induce limited cytokine production and cannot activate resting or naïve lymphocytes (Brocker and Karjalainen, 1995). Furthermore, in the absence of costimulatory signaling by CD28, resting T lymphocytes typically undergo anergy or apoptosis (Boussiotis et al., 1996). To address these issues, the introduction of costimulatory element CD28 (the best characterized costimulatory molecule) to the first-generation CAR was first described by Finney et al. in 1998. This second-generation CAR is capable of mediating up to 20 times more IL-2 production on stimulation with solid-phase Ag when compared to first-generation CAR. Moreover, constructs with the CD28 signaling domain proximal and the ζ -chain distal to the membrane were found to express more efficiently in Jurkat than constructs with the opposite orientation (Finney et al., 1998), thus determining the signaling element arranging pattern adopted by other researchers in the years since. Other than CD28, other costimulatory molecules such as CD134/CD137 also have been incorporated into the first-generation CAR by Finney et al. (2003). Second-generation CAR is superior for inducing cytokine production and proliferation of CAR-T cells compared to the first-generation CAR, which was proved in several preclinical studies (Haynes et al., 2002a, b; Imai et al., 2004; Kowolik et al., 2006) and was further verified in one clinical trial to directly compare such two generation CARs (Savoldo et al., 2011). The initial pilot clinical studies of CAR were opened in solid tumors (Lamers et al., 2006; Kershaw et al., 2006). However, substantial clinical efficacy has been shown in hematological malignancies treated with second-generation CARs (Kochenderfer et al., 2010; Porter et al., 2011; Kalos et al., 2011; Wang et al., 2014; Maude et al., 2014a; Davila et al., 2014; Dai et al., 2015; Lee et al., 2015a; Kochenderfer et al., 2015; Porter et al., 2015; Turtle et al., 2016a; Wang et al., 2016; Zhang et al., 2016a). Third-generation CAR contains two costimulatory domains, which result in more potent persistence and other T cell functions in preclinical studies (Wang et al., 2007; Zhong et al., 2010; Carpenito et al., 2009; Choi et al., 2014). However, the clinical benefit is not as good as expected (Till et al., 2012) and more studies are needed. To further improve the anti-tumor effect of CAR-T cells, engineering CAR-T cells to additionally express cytokines or co-stimulatory ligands (fourth-generation CAR, also called armored CAR) has been employed (Di Stasi et al., 2009) and actively researched (Pegram et al., 2014), yet no results of clinical trial have been published so far. In this article, we briefly review the common structure of CAR and emerging clinical activity, toxicities, and challenges of this novel technology.

Evolution of CAR
COMMON CAR STRUCTURES
CAR is artificial type I transmembrane protein assembled from a series of modular compositions including an amino terminal ectodomain and carboxy-terminal endodomain, as well as a transmembrane domain (TM) (Fig. 2) (Eshhar, 2008; Curran et al., 2012; Dai et al., 2016). Ectodomain usually consists of a target-binding domain most commonly derived from the scFv of a monoclonal antibody (mAb) specific for a surface molecule on the tumor cell (Eshhar et al., 1993; Kershaw et al., 2005), and a spacer (also known as a hinge) domain typically comprises immunoglobulin-like CH2-CH3 (Fc) domains from the constant region of immunoglobulin G (IgG) (Finney et al., 1998; Till et al., 2012), CD8 (Porter et al., 2011; Wang et al., 2014; Zhang et al., 2016a) or CD28 (Kochenderfer et al., 2010; Kochenderfer et al., 2015), which extends the antigen-binding domain out from the T cell membrane. Endodomain acts to transmit T cell signals and typically comprises 0 or 1 or 2 costimulatory domains such as CD28, CD134 (OX40) or CD137 (4-1BB) (van der Stegen et al., 2015; Finney et al., 1998, 2003) and activation domain representing the CD3 ζ (Ghorashian et al., 2015; Sadelain et al., 2013). Such synthetic tumor-targeting receptors provide a choice of specificity and controlled T cell activation that is mainly attributed to extracellular antigen-binding component and intracellular-signaling components that have received the most attention and have been well described (van der Stegen et al., 2015; Ghorashian et al., 2015; Sadelain et al., 2013; Gill and June 2015; Jackson et al., 2016). However, the spacer domain should not be overlooked; it is equally crucial for effective initiation of T cell signaling as it provides flexibility and optimizes T cell and target cell engagement by overcoming the structural constraints in T cells: target cell interactions (Guest et al., 2005; Hudecek et al., 2013, 2015; Srivastava and Riddell 2015). The optimal length of the spacer domain for each CAR may differ depending on the dimensions of the cell surface antigen that is targeted by the scFv (Harris and Kranz, 2016). The transmembrane domain is considered to be a purely structural requirement for anchoring the CAR to the cell membrane and has little to no effect on the function of CAR (van der Stegen et al., 2015).

Anatomy of a second-generation CAR
By arming the T cells with CAR, the engineered T cells can directly recognize cancer cell surface antigens in an MHC-independent fashion and undergo activation, providing an alternative to conventional TCR and enabling them to circumvent the major hurdles suffered by cancer patients, including tumor escapes resulting from downregulation or loss of HLA expression as well as T cell anergy due to decreasing or loss of the expression of costimulatory molecules required for triggering the full potency of T cells (Gross and Eshhar, 2016). Compared to native TCR, scFv-based antigen recognition has both benefits and limitations. CAR only recognizes target antigens expressed on the cell surface rather than internal antigens that are processed and presented by the cells’ MHC, but various cell-surface molecules such as proteins, carbohydrate (Lewis-Y, TAG-72) (Peinert et al., 2010; Ritchie et al., 2013; Hombach et al., 1997; McGuinness et al., 1999), and glycolipid (GD2, GD3) structures (Pule et al., 2008; Louis et al., 2011; Rossig et al., 2001; Yun et al., 2000) can be recognized by CAR, which is a compensation for the limited target selection.
The CAR format provides an opportunity to recognize practically any desired target antigen by changing only the corresponding binding moiety while retaining the backbone structure. Moreover, owing to the modular design, sophisticated engineering of diverse domain components becomes possible. These unique features promote the versatility of CAR structures (Fig. 3); for instance, several second-generation CD19-specific CARs are being tested in clinical trials. The major structural difference between currently applying second-generation CARs is costimulatory domain (van der Stegen et al., 2015). CD28 costimulatory component has been used by the National Cancer Institute (NCI; USA) (Lee et al., 2015a), Memorial Sloan Kettering Cancer Center (MSKCC; USA) (Davila et al., 2014), and Baylor College of Medicine (BCM; USA) (Savoldo et al., 2011), while the 4-1BB costimulatory component has been incorporated by the University of Pennsylvania (Upenn; USA) (Maude et al., 2014a), Chinese PLA General Hospital (PLAGH; CHINA) (Wang et al., 2014; Dai et al., 2015; Zhang et al., 2016a), and the Fred Hutchinson Cancer Research Center (FHCRC; USA) (Turtle et al., 2016a). Accordingly, the second-generation CARs could be classified as two categories of receptors based on the costimulatory domain, referred to as 28ζ and BBζ CAR. Both 28ζ and BBζ CAR have been used to successfully treat multiple blood cancers (Zhang et al., 2015). BBζ CAR appears to favor persistence and memory T cell formation, while 28ζ CAR presents more potent cytotoxic activity and early tumor eradication. So, combining the benefits of 4-1BB and CD28 costimulation could be a good option to best optimize CAR (Holohan et al., 2015). Rather than the strategy of combining CD28 and 4-1BB costimulation, Zhao et al. demonstrated that 28ζ CAR-T cells that constitutively express 4-1BB ligand (4-1BBL) promote T cell expansion and tumor eradication while reducing exhaustion (Zhao et al., 2015), providing valuable implications for evolving CAR-T cell therapies. More studies are required to better understand the kinetics of each of the costimulatory domains and their relative clinical effects.

Common second-generation CARs. Abbreviations: B-ALL, B cell acute lymphoblastic leukemia; BCM, Baylor College of Medicine; B-NHL, B cell non -Hodgkin’s lymphoma; CCA, cholangiocarcinoma; CLL, chronic lymphocytic leukemia; CMPC, castrate metastatic prostate cancer; EGFR, epidermal growth factor receptor; EGFRvIII, variant III of the epidermal growth factor receptor; FHCRC, Fred Hutchinson Cancer Research Center; GBM, glioblastoma multiforme; HER2, human epidermal growth factor receptor-2; HL, Hodgkin’s lymphoma; MM, multiple myeloma; MPD, malignant pleural disease; MPM, malignant pleural mesothelioma; MSKCC, Memorial Sloan Kettering Cancer Center; NCI, National Cancer Institute; NSCLC, non-small cell lung cancer; PC, pancreatic cancer; PDAC, pancreatic ductal adenocarcinoma; PLAGH, Chinese PLA General Hospital; PSMA, prostate-specific membrane antigen; scFv, single chain variable fragment; Upenn, University of Pennsylvania
Besides the costimulatory component, variability is present in scFv fragments, hinge, and transmembrane domains, in addition to differences in CAR transduction approaches, so it can be difficult to compare results from different studies.
CLINICAL OUTCOMES OF HEMATOLOGICAL MALIGNANCIES
Following a decade of preclinical optimization, CAR-T cell therapy has produced impressive clinical results in treating patients with relapsed or refractory B cell leukemia and lymphoma whose treatment options are limited and prognosis is poor. To date, there are more than 30 publications and a large number of congress abstracts reporting clinical trials of CAR-T cells in hematologic malignancies. Although the initial clinical evaluation of CAR-T cells focused on B cell non-Hodgkin’s lymphoma (B-NHL) (Kochenderfer et al., 2010; Till et al., 2008), the most striking outcomes have been obtained in B-ALL by targeting CD19, a B cell-lineage antigen expressed on the surface of normal B cells and many malignant B cells (Scheuermann and Racila, 1995; Depoil et al., 2008). Another pan-B cell marker, CD20, is also an attractive target for CAR-T cell therapy in B cell malignancies (Raufi et al., 2013). Preliminary clinical trials evaluating anti-CD20 CAR-T cells for patients with B-NHL revealed minimal toxicities with modest efficacy (Wang et al., 2014; Till et al., 2008, 2012), until recently a phase IIa clinical trial performed at PLAGH demonstrated an objective remission rate (ORR) of 82% (complete remission (CR) 6/11, partial remission (PR) 3/11) with well-tolerated toxicity (Zhang et al., 2016a). This promising finding provides an effective alternative to address the challenge of antigen escape in anti-CD19 CAR-T cell therapy (Maude et al., 2014a; Davila et al., 2014; Lee et al., 2015a; Turtle et al., 2016a) by using CD19/CD20 bi-specific CAR, which has been proven by Eugenia et al. in a preclinical study (Zah et al., 2016). Other promising B cell lineage of antigens for CAR-T cell therapy in B cell malignancies such as CD22, inactive tyrosine-protein kinase transmembrane receptor (ROR1), and the immunoglobulin kappa chain (Igκ) are still undergoing clinical testing without results reported yet (Jackson et al., 2016). Here we review clinical results from trials investigating CAR-T cells in B-ALL, B-NHL, chronic lymphocytic leukemia (CLL), and Hodgkin’s lymphoma (HL) (Table 1).
B cell acute lymphoblastic leukemia
Up to now, CAR-T cell therapy has been most effective in patients with B-ALL as significant CR rates of 70%–94% were observed even in a post-allogeneic-hematopoietic stem cell transplantation (allo-HSCT) setting (Maude et al., 2014a; Davila et al., 2014; Lee et al., 2015a; Turtle et al., 2016a). Based on the promising results of the initial phase I trials, several pivotal phase II clinical trials evaluating CAR-T cell therapy for B-ALL are underway (US National Library of Science, 2016a, b, c, d, e). However, it is difficult to draw a conclusion about which is better according to the clinical response rate as these trials varied significantly in the key factors that determine the final efficacy, including CAR structure, preconditioning regimen, infused T cell product, T cell dose, etc. Even relapsed or refractory B-ALL (R/R B-ALL) was adopted by almost all institutions for patient selection, patient age, risk features, prior treatment history, and degree of tumor burden at the time of CAR-T cell infusion has been widely discrepant, which led to the trials being more heterogeneous.
MSKCC was the first to publish results of CD19-targeted CAR for adults with R/R B-ALL (NCT01044069) (Brentjens et al., 2011). Updated results of this study were reported by Brentjens et al. (2013), where all five adult patients treated with anti-CD19 28ζ CAR-T (19-28ζ CAR-T) cells at a dose of 1.5–3 × 106 19-28ζ CAR-T cells/kg achieved minimal residual disease(MRD)-negative CR(MRD-CR). Clinical efficacy of this approach has been confirmed and reached a climax in the follow-up study of an additional 11 adult patients treated with R/R B-ALL (Davila et al., 2014). A total of 14 of 16 (88%) patients achieved morphologic CR or CR with incomplete blood count recovery (CRi), and 12 of the 16 (75%) were classified as MRD-negative following CAR-T cell infusion. However, two patients already had been rendered MRD-negative by salvage therapy prior to CAR-T cell infusion, potentially confounding the role of 19-28ζ CAR-T cells. 19-28ζ CAR-T cell expansion in vivo peaked within 12 weeks and persisted for 2–3 months post-infusion in most patients, supplying a window of time following transplant; hence researchers defined the 19-28ζ CAR-T cell therapy as a “bridge” to transplant. This study also first defined the diagnostic criteria for severe cytokine release syndrome (sCRS) secondary to CAR-T cell infusion, and identified C-reactive protein (CRP) as a potential laboratory indicator for CRS severity that could be used as a surrogate for cytokines. The long-term outcome containing survival data for this study was demonstrated in a larger cohort of 22 evaluable patients; the median overall survival (OS) is 9 months and 5 patients have relapsed, including 1 with CD19-negative disease (Park et al., 2014). At the 2015 annual meeting of the American Society of Hematology (ASH), they updated their experience in 44 adults with 43 evaluable patients who received lymphodepleting chemotherapy followed 2 days later by 1–3 × 106 19-28ζ CAR-T cells/kg (Park et al., 2015). The potent anti-tumor efficacy of 19-28ζ CAR-T cells in adults with R/R B-ALL has been confirmed as a similar CR rate of 84% (36/43) and MRD-CR rate of 67% (29/43) was observed in this larger cohort study. Median overall survival (OS) of all patients and those who achieved MRD-CR was 8.5 months and 10.8 months, respectively. Therefore, the researchers concluded that MRD negativity following the 19-28ζ CAR-T cell treatment was highly predictive of survival. It is worth noting that allo-HSCT post-CAR-T cell infusion had no significant impact on the survival of the patients who achieved CR as the OS at 6 months is similar (70% vs. 64%) between the patients who underwent post-CAR allo-HSCT and those who did not. This finding is amazing in the context of the persistence of 19-28ζ CAR cells is 2–3 months post-infusion as reported previously (Davila et al., 2014), which warrants further investigation.
On the basis of an initial successful experience of using CD19-specific BBζ CAR transduced T cells (termed CTL019) to treat three patients with CLL (NCT01029366) (Porter et al., 2011; Kalos et al., 2011), researchers at the Children’s Hospital of Philadelphia and the University of Pennsylvania (CHOP/UPenn) conducted a phase I trial to investigate CTL019 cells for children with R/R B-ALL (NCT01626495) and presented a case report on the first two patients in 2013 (Grupp et al., 2013). CR was observed in both patients and was ongoing in one patient at 11 months after treatment (Ongoing CR at 3 years post-CTL019 cell infusion has been described (Tasian and Gardner 2015)), while the other patient experienced a CD19-negative relapse 2 months post-CAR-T cell infusion. Although only two patients were reported, it made sense to CAR-T cell therapy development as it not only provided an effective approach to the management of sCRS by incorporating tocilizumab (a recombinant humanized monoclonal antibody against interleukin-6 receptor (IL-6R)) without compromising efficacy, but also highlighted the significant threat to a successful CAR regimen, namely, tumor antigen loss escape. The updated outcomes of an expanded cohort of 25 children and 5 adults with R/R B-ALL who received CTL019 cells at a dose of 0.76–20.6 × 106 CTL019 cells/kg following the investigator’s choice lymphodepleting regimen were reported by Maude et al. (2014a). Morphologic CR was achieved in 27 patients (90%), including 2 with blinatumomab-refractory disease and 15 who had undergone stem-cell transplantation; 22 of 27 (81%) patients achieved MRD-CR. Seven patients achieving CR subsequently experienced relapse (3 with CD19-negative disease) between 6 weeks and 8.5 months after the infusion of CTL019 cells. However, it was noted that prolonged persistence of CTL019 cells and B cell aplasia for as long as 2 years was seen in this study, implying CTL019 cells could be proposed as a potential treatment alternative for patients who are ineligible for stem-cell transplantation. The investigators also reported the outcomes and longer follow-up of the first 53 children or young adults with R/R B-ALL treated with a median of 4.3 × 106 CTL019 cells/kg (1–17.4 × 106 cells /kg) (Grupp et al., 2015). At day 28 post CAR-T cell infusion, 50 patients (94%) achieved morphologic CR, including 45 patients who achieved MRD-CR measured by clinical flow cytometry. Intriguingly, two additional patients in morphologic CR at day 28 achieved MRD-CR by 3 months without further therapy. However, 12 of 53 evaluable patients had already been MRD-negative attributed to lymphodepleting chemotherapy at the time of CTL019 cell infusion, which is similar to the aforementioned MSKCC report (Davila et al., 2014). Twenty of fifty patients with CR at day 28 had subsequently relapsed (relapse-free survival is 44% at 12 months), thirteen of whom experienced CD19-negative disease relapse, which should be the most frequent CD19-negative relapse in the available data to date (Maude et al., 2014a; Davila et al., 2014; Lee et al., 2015a, b; Turtle et al., 2016a; Park et al., 2015). They also believed rapid loss of CTL019 cells (prior to 3 months) was associated with a high risk of CD19+ relapse.
The 2 aforementioned institutions all infused CAR-T cells at a relatively broad dose range, while a fixed dose of either 1 × 106 or 3 × 106 CAR-T cells/kg was employed in a phase I dose escalation study (NCT01593696) performed at the NCI to investigate anti-CD19 28ζ CAR-T cells for children and young adults with R/R B-ALL (Lee et al., 2015a). Twenty patients with R/R B-ALL and one patient with diffuse large B cell lymphoma (DLBCL) were treated; the maximum tolerated dose (MTD) for the entire cohort was defined as 1 × 106 CAR-T cells/kg by using a 3 + 3 dose-escalation schema. Fourteen of twenty (70%) patients with B-ALL achieved morphologic CR, with twelve achieving MRD-CR. No CAR-T cells were detected at day 68 post-CAR-T cell infusion in any patient; the persistence of CAR-T cells is similar to those observed by investigators at MSKCC. Ten of twelve patients who were MRD-negative went on to HSCT and all remained disease-free at a median follow-up of 10 months. Therefore, the researchers concluded that anti-CD19 CAR-T cell therapy was an effective bridge to HSCT in patients with refractory B-ALL, which simultaneously explained why the researchers believed that long-term persistence was not necessary to induce meaningful anti-tumor effects. However, the other two patients who did not receive allo-HSCT experienced CD19-negative disease relapse at 3 and 5 months. This study also provided the first evidence that anti-CD19 CAR-T cell could eradicate leukemia in cerebrospinal fluid without long-term toxicity. Additional 18 children or young adults with R/R B-ALL were treated with a selected dose of 1 × 106 CAR-T cells/kg, and updated experiences with the first 38 patients were reported at ASH 2015 (Lee et al., 2015b). Thirty-eight patients across both cohorts showed a morphological CR and MRD-CR rate of 61% and 53%, 13/16 (81%) of low-burden patients had a morphological CR, while 10/22 (45%) of high-burden patients attained a morphological CR. Of the 20 patients achieving an MRD-CR, the median leukemia-free survival (LFS) was 17.7 months, with a 45.5% probability of LFS beginning at 18 months.
In contrast to the strategy of infusion of a mixture of T cell products without preselecting, which is well accepted and widely used by most institutions (Gill and June, 2015), investigators at FHCRC believed that preselecting specific T cell subsets and using defined formulations would be informative for enhancing the potency and reproducibility of cancer immunotherapy, according to their preclinical data (Wang et al., 2011; Terakura et al., 2012; Sommermeyer et al., 2016). Therefore, Turtle et al. conducted a phase I/II trial (NCT01865617) evaluating CD19-targeted BBζ CAR-T cell therapy for advanced CLL, ALL, and lymphoma, in which the T cell products were formulated in a defined 1:1 ratio of CD8+ and CD4+ T cell subsets (Turtle et al., 2016a). Promising preliminary results were achieved in 29 evaluable adults with R/R B-ALL who received 2 × 105, 2 × 106, or 2 × 107 CAR-T cells/kg in a defined 1:1 ration of CD4:CD8 composition. 27 of 29 patients (93%) achieved bone marrow (BM) remission, as leukemia was undetectable by high-resolution flow cytometry. Investigators observed a marked increase in CAR-T cell expansion and persistence in 17 patients who received cyclophosphamide and fludarabine (Cy/Flu) lymphodepletion compared with 12 patients who received lymphodepletion with Cy alone or with etoposide. As a consequence of these enhancements, an improvement in overall and disease-free survival also was observed in the Cy/Flu cohort, wherein only 2 of 17 (12%) patients relapsed (1 CD19+ relapse, 1 CD19− relapse) post-CAR-T cell infusion. In contrast, 7 of 12 patients (58%) relapsed (6 CD19+ relapses, 1 CD19− relapse) post-CAR-T cell infusion in the cohort without Flu. While these data are encouraging, additional patient accrual and longer follow-up periods are required. Moreover, researchers identified a T cell-mediated anti-CAR immune response specific for murine scFv epitopes in the patients in whom CAR-T cells failed to persist after the second infusion. This result is similar to the finding recorded in detail in the other CAR trials (Lamers et al., 2011; Maus et al., 2013; Jensen et al., 2010), highlighting the immunogenicity of murine CAR, especially when it is administered using an intermittent dosing schedule.
B cell non-Hodgkin’s lymphoma
To date, successful experience in patients with B-NHL still mainly generated from the clinical trial using anti-CD19 CAR-T cells; however, CD20-targeted CAR-T cells have also been employed (Zhang et al., 2016a; Till et al., 2008, 2012; Jensen et al., 2010) and have demonstrated potential therapeutic value (Zhang et al., 2016a). Patients with DLBCL and follicular lymphoma (FL) represent of the majority in those clinical trials (Batlevi et al., 2016). The clinical efficacy of CAR-T cell therapy for patients with B-NHL is not as robust as those with R/R B-ALL, for reasons that are not well-defined, but disease-driven depletion of early lineage cells in lymphoma may be a contributing factor (Singh et al., 2016).
Publication of the early clinical trials to evaluate first-generation CAR-T cell therapy for B-NHL occurred in 2008 (Till et al., 2008) and 2010 (Jensen et al., 2010), but there was no evidence of clinical benefit. The researchers at BCM have attempted to simultaneously infuse first- and second-generation CAR-T cells targeting CD19 into patients with active FL or DLBCL; still no clinical benefit was observed, but CAR including CD28 costimulatory domains led to enhanced in vivo expansion and the persistence of CAR-T cells has been demonstrated (Savoldo et al., 2011).
So far, investigators at the NCI have presented the largest data series from clinical trials investigating CD19-targeted CAR-T cells for B-NHL; a cumulative 36 evaluable patients including 27 patients with various DLBCL show an ORR and CR rate of 78% and 44%. In 2010, researchers presented the first PR lasting 32 weeks in a patient with advanced FL who received lymphodepleting chemotherapy followed by an infusion of anti-CD19 28ζ CAR-T cells (NCT00924326) (Kochenderfer et al., 2010). An updated outcome of this trial in four patients with B-NHL and four patients with CLL was reported by Kochenderfer et al. (2012). All the 3 evaluable patients with advanced, progressive B-NHL (2 FL, 1 splenic marginal zone lymphoma (SMZL)) who received conditioning chemotherapy followed by an infusion of anti-CD19 28ζ CAR-T cells and a course of IL-2 obtained PR. Durations of response ranged from 8 to 18 months, and two remissions were ongoing. Of note, the first patient obtaining PR previously reported (Kochenderfer et al., 2010) developed progressive CD19+ lymphoma 32 weeks after his first infusion of anti-CD19 28ζ CAR-T cells, whereas B cell dysplasia lasted 39 weeks and 36 weeks after the first CAR-T cell infusion in the peripheral blood (PB) and BM, respectively. This patient was retreated on the same protocol and was in an 18-month ongoing PR after the second treatment (Kochenderfer et al., 2012; Kochenderfer and Rosenberg, 2011). More impressive results of this study were observed in a larger cohort of 15 patients with B-NHL (9 DLBCL, containing 4 primary mediastinal B cell lymphoma, 1 SMZL, and 1 low-grade NHL) and 4 patients with CLL who were treated with lymphodepleting chemotherapy followed by infusion of anti-CD19 28ζ CAR-T cells at a dose of 1–5 × 106 CAR-T cells/kg without IL-2 (Kochenderfer et al., 2015). Lymphodepleting chemotherapy was Cy at a total dose of either 120 or 60 mg/kg, followed by five daily doses of Flu 25 mg/m2. Of the seven evaluable patients with DLBCL, four obtained CR, two obtained PR; in three of these four CR are ongoing, with durations ranging from 9 to 22 months. The patients with SMZL were previously treated on their anti-CD19 CAR-T cell protocol and obtained a PR lasting 12 weeks (Kochenderfer et al., 2012), then were retreated with the same regimen and obtained a PR with an ongoing response of 23 months as of the time of writing. The most troublesome toxicities were hypotension and neurologic toxicities that can be resolved within 3 weeks after cell infusion. One patient died 16 days after cell infusion from an undetermined reason. In 2016, at the American Society of Clinical Oncology (ASCO) annual meeting, researchers presented an updated outcome of this study, wherein 22 patients with B-NHL were treated with a low dose of FC lymphodepleting chemotherapy regimen of Cy either 300 or 500 mg/kg daily for 3 days and Flu 30 mg/m2 daily for 3 days on the same days as Cy followed by a single infusion of anti-CD19 28ζ CAR-T cells (Kochenderfer et al., 2016). Of the 19 patients treated with various subtypes of DLBCL, 8 had CR, 5 had PR, 2 achieved stable disease (SD), and the other 3 (1 mantle cell lymphoma (MCL), 2 FL) obtained CR. Durations of response ranged from 1 to 20 months, and 10 remissions were ongoing. However, only 4 of all 22 treated patients had either chemotherapy refractory lymphoma or lymphoma that had relapsed after autologous stem cell transplant, undoubtedly comprising the significant efficacy of this regimen. Neurologic toxicities were still the most prominent toxicities; fever and hypotension were also observed in some patients. Intriguingly, the researchers found that patients obtaining CR or PR had higher peak blood CAR+ cell levels than patients experiencing SD or PD. This group also reported the result of a trial (NCT01087294) to evaluate donor-derived CD19-targeted 28ζ CAR-T cells without prior lymphodepleting chemotherapy for patients with B-NHL or CLL in whom tumor lesions persisted after allo-HSCT and standard donor lymphocyte infusions (DLIs) (Kochenderfer et al., 2013). Of the 10 treated patients (2 DLBCL, 4 MCL, and 4 CLL), only 1 patient with CLL obtained a 9-month ongoing CR; 2 patients with MCL experienced PR. This less encouraging outcome could be attribute to no prior lymphodepleting chemotherapy, resulting in less than 1 month persistence of CAR-T cells. No graft-versus-host disease (GVHD) was observed in any of the patients. Toxicities included transient hypotension and fever. Updated results of the first 20 patients with B cell malignancies (5 CLL, 10 B-NHL, and 5 B-ALL) that progressed after allo-HSCT who received allogeneic T cells transduced with CAR targeting CD19 were reported by Brudno et al. (2016). An ORR of 40% with 30% CR was observed among 20 treated patients. Of the 10 treated patients with B-NHL, 1 CR and 1 PR were achieved. None of the treated patients has experienced new-onset acute GVHD post-CAR-T cell infusion.
Investigators at Upenn also updated their preliminary results of a phase IIa trial (NCT02030834) evaluating CTL019 cells for patients with relapsed or refractory lymphomas (Schuster et al., 2015). A total of 38 patients (21 DLBCL, 14 FL, and 3 MCL) were enrolled, and eventually 24 patients (13 DLBCL, 9 FL, and 2 MCL) were treated with physician’s choice conditioning therapy followed by a single infusion of CTL019 at a median dose of 5.84 × 106 CAR+ T cells/kg (range: 3.08–8.87 × 106 CAR+ T cells/kg). A 68% (15/22) ORR was achieved at 3 months post CTL019 infusion in the 22 evaluable patients (13 DLBCL, 7 FL, and 2 MCL). Progression-free survival (PFS) at the median follow-up of 11.7 months was 62% (DLBCL 43%, FL 100%), at which time the response duration was 83% for DLBCL and 100% for FL. However, more detailed efficacy data such as CR rate and CAR-T cell persistence in vivo were not presented in the abstract. A total of 16 of 24 (67%) treated patients developed grade 2–4 CRS with 1 grade 4 CRS. Three patients developed neurologic toxicity, including transient delirium (1 grade 2, 1 grade 3) and 1 possibly related grade 5 encephalopathy.
Differing from the above-mentioned notion that defines CAR-T cell therapy as a treatment alternative for patients with R/R B-NHL, investigators at MSKCC tried to evaluate whether those patients who have been treated with high-dose therapy and autologous stem cell transplant (HDT-ASCT) can benefit from CAR-T cell consolidation. At the 2015 ASCO annual meeting, researchers reported the safety data of a phase I dose-escalation study (NCT01840566) in 8 patients with poor-risk R/R aggressive B-NHL who received BEAM-conditioned HDT-ASCT followed by infusion of anti-CD19 28ζ CAR-T cells at 1 of 3 dose levels (5 × 106, 1 × 107 or 2 × 107 CAR+ T cells/kg) at days +2 and +3 (Sauter et al., 2015). Besides one patient who received dose level 2 (1 × 107 CAR+ T cells/kg), others were treated at dose level 1 (5 × 106 CAR+ T cells/kg). Half of the patients had CRS, which could be well managed with tocilizumab and/or corticosteroids. One patient died from non-relapse mortality (NRM) of mucormycosis pneumonia at day 38 after HDT-ASCT. Five of eight patients with PET(+) PR prior to HDT-ASCT obtained CR with duration ranging from 10 to 18 months post-HDT-ASCT, but whether CAR-T cell therapy contributed to this higher CR rate and longer duration of response still needs further exploration. Investigators at FHCRC reported the outcome of the aforementioned phase I/II trial (NCT01865617) (Turtle et al., 2016a) in 32 patients with R/R B-NHL (22 DLBCL, 6 FL, and 4 MCL) who were treated with the same protocol for patients with B-ALL (Turtle et al., 2016b). Twelve and twenty patients with B-NHL received Cy-based conditioning regimens without Flu or with Flu, respectively. A 50% ORR with 8% CR rate was obtained among 12 evaluable patients in the Cy-based without Flu group, whereas a 72% ORR with 50% CR rate was observed in the Cy/Flu group (18 evaluable patients). Researchers again observed a CD8-mediated immune response as observed in B-ALL (Turtle et al., 2016a) due to CAR transgene immunogenicity, leading to no significant T cell expansion or clinical responses in five of five patients who received a second reinfusion of CAR-T cells. Investigators believed that this finding provided one potential mechanism for the loss of CAR-T cells observed in other trials and concluded that Cy/Flu could minimize the substantial cellular immune response against CAR. sCRS and grade ≥3 neurotoxicity were observed in 13% and 28% of all patients, respectively. Of note, no patient treated at all three dose levels experienced sCRS in the Cy-based without Flu group, whereas three patients experienced sCRS and four patients developed grade ≥3 neurotoxicity among six patients who received 2 × 107 CAR+ T cells/kg following Cy/Flu, implying the toxicities might be related to the cell dose, especially in the context of that the Cy/Flu conditioning regimen was used. Peak IL-6, interleukin-15 (IL-15), interferon-γ (IFN- γ), and interleukin-10 (IL-10), concentrations on day 1 after CAR-T cell infusion have been determined to have a strong correlation with subsequent sCRS and neurotoxicity; nonetheless, whether those serum biomarkers can be used as accurate predictive biomarkers for sCRS and neurotoxicity remains to be elucidated.
Investigators at PLAGH presented the preliminary result of the study (NCT01735604) to evaluate anti-CD20 BBζ CAR-T cells (referred as CART-20) for R/R B-NHL in 2014 (Wang et al., 2014). Seven heavily pretreated patients with refractory advanced CD20+ DLBCL were treated with CART-20 cells at a dose of 0.36–2.35 × 107 CAR+ T cells/kg alone or following physician’s choice debulking chemotherapy in order to alleviate tumor load as well as conditioning. Four of six evaluable patients had bulk tumor burdens defined as lesion(s) with the longest diameter greater than 5 cm or more than three lesions, and 3 of whom achieved PR with a duration ranging from 3 to 6 months by infusion of CART-20 cells. Among the other two patients with no bulky tumors, a 14-month ongoing CR occurred after CART-20 cells infusion alone. CART-20 cells in PB could persist for at least 4 weeks in most patients with a higher CAR gene copy number (1000/μg DNA), which also could be detected in biopsy tissues derived from three of six evaluable patients even after 10 weeks of cell infusion. Correspondingly, a decrease of the CD20+ B cell count in PB was observed, which could be attributed to the on-target/off-tumor recognition of CART-20 cells. Six of the seven treated patients developed delayed toxicities mainly due to the cytokine elevation related to CART-20 cells 3–8 weeks post-CART-20 cells infusion, except for a grade 4 acute alimentary tract hemorrhage resulting in death. An impressive result with 82% ORR with a 55% CR rate was shown in the phase IIa study of 11 patients with refractory or relapsed CD20+ B-NHL (8 DLBCL, 1 FL, 1 MCL, and 1 primary cutaneous marginal zone lymphoma (PCMZL)) who were treated with the same protocol (Zhang et al., 2016a). Of eight patients with DLBCL, four obtained CR with a duration ranging from 4 to 27 months, three remissions were ongoing, and three achieved PR, including one 13-month ongoing PR. Two CR both lasting 5 months were achieved among the other three patients with indolent B-NHL. The median PFS was 6 months. Five patients who had response relapsed with CD20 positive between 60 days and 6 months after infusion of CART-20 cells when the CAR gene copy number declined to the near lowest value as well as polyclonal B cells recovered from aplasia, illustrating an inverse correlation between CAR molecule levels in PB and the CD20+ target cell. No grade 4 toxicities and CRS developed, which mainly should be attributed to the fact that no patient with defined bulky tumors was enrolled, as the lessons drawn from the phase I study that high tumor burden increased the risks of severe toxicities.
Chronic lymphocytic leukemia
The exploration of CAR-T cells targeting CD19 for patients with CLL is earlier than B-ALL; however, less mature data have been reported. Moreover, although all express CD19, it appears that CLL has a lower response rate than B-ALL, with an ORR of 62% across publications by 2014 (Zhang et al., 2015). In vivo disease-intrinsic mechanisms such as defects in the circulating T cells of CLL patients and/or the inhibitory microenvironment associated with this often bulky disease may contribute to this relative paucity of response (Pegram et al., 2015; Kalos 2016; Khalil et al., 2016).
The largest cohort of CD19-targeted CAR-T cell therapy for CLL has been reported by investigators at Upenn. As of now, CTL019 has treated more than 45 patients with relapsed and refractory CLL (R/R CLL) and has shown an ORR of ~45% (Maude et al., 2015a). Researchers at Upenn in their pilot clinical trial (NCT01029366) first demonstrated that CTL019 could induce dramatic antitumor response for patients with advanced, chemotherapy-resistant CLL, where two ongoing CR and one PR were achieved in the three treated patients (Kalos et al., 2011). Mature results from this pilot clinical trial using CTL019 treatment of 14 patients with R/R CLL at a dose of 0.14–11 × 108 CTL019 cells (median, 1.6 × 108 cells) were presented by Porter et al. (2015). The ORR was 8 of 14 (57%), with 4 CR and 4 PR including the aforementioned 3 outcomes (Kalos et al., 2011). Three of the four patients achieving CR maintained this response for 40 months (range 28–53); the other patient died from infection while in CR 21 months after treatment. However, a relatively shorter duration of response (range 5–13 months) was observed in all four patients who attained PR, which was correlated with the in vivo expansion and persistence of the CAR-T cells. Significantly, CTL019 cells could be detected in the first two patients achieving CR 4 years post CTL019 cells infusion, and CTL019 cells isolated from one who was almost 3 years post-CTL019 cells infusion remained functional, highlighting that CAR-T cells could persist over the long term as memory cells and continually provide immunosurveillance and prevent relapse. This finding increases confidence that CAR-T cell therapy could be defined as a stand-alone therapy, at least for R/R CLL. A phase II dose optimization study (NCT01747486) was opened subsequently, in which 28 patients with R/R CLL were randomized to receive either 5 × 108 or 5 × 107 CTL019 cells following a preconditioning regimen (Porter et al., 2016). This ongoing trial confirmed the initial outcomes of the pilot study, albeit the ORR was slightly lower at 42% (5 CR, 5 PR) among 24 evaluable patients (11 high dose, 13 low dose). Moreover, the researchers identified 5 × 108 CTL019 cells as the optimal dose of CTL019 in patients with R/R CLL on account of a relatively high ORR but with similar toxicity shown in the high-dose cohort compared with the low-dose cohort. Twenty-one patients with R/R CLL have been subsequently treated with the selected dose, and 9 patients had a response with 6 CR among the 17 evaluable patients, including 11 who had been treated at stage 1. Remissions were ongoing in five of six patients achieving CR at a median follow-up of 26 months (range 5–34); the other progressed with CD19 negative disease.
Other institutions, including MSKCC, NCI, and FHCRC, also have conducted initial clinical trials to evaluate autologous CD19-targeted CAR-T cells for R/R CLL. Across all three centers in 30 patients with R/R CLL (Geyer and Brentjens 2016), there was an ORR and CR rate of 53 and 30%, 31 and 13% at MSKCC (Brentjens et al., 2011; Geyer et al., 2016), 88 and 50% at NCI (Kochenderfer et al., 2012, 2015), and 67 and 50% at FHCRC (Turtle et al., 2015).
Hodgkin’s lymphoma
Although Hodgkin’s lymphoma (HL) is a B-cell derived cancer, the tumor cells of HL- Hodgkin and Reed-Sternberg (HRS) cells have lost the B cell phenotypes such as CD19, CD20, or CD22, and are instead characterized by bright, uniform expression of CD30, which is also shared by a small population of activated T cells (Kuppers et al., 2012). Antibody-drug conjugate brentuximab vedotin (BV) directed to CD30 has been approved for treatment of relapsed HL as an objective antitumor response with a well-tolerated toxicity (Younes et al., 2010). Importantly, patients who relapse after prior BV appear to retain CD30 expression on HRS cells (Gill and June 2015). Another concern with targeting CD30 for CAR-T cell therapy in HL is that high concentrations of soluble CD30 have been found in patients with progressed HL, which may compete for CAR binding (Jackson et al., 2016); However, a preclinical study showed that this concern was unwarranted (Hombach et al., 1998). Taken together, it could make sense to develop a CD30-targeted CAR for HL.
Two trials evaluating anti-CD30 CAR-T cells for HL are ongoing at BCM (NCT01192464, NCT01316146). Ramos et al. reported (Ramos et al., 2015) the preliminary results of nine patients with lymphoma (7 HL, 2 anaplastic large cell lymphoma (ALCL)) who received 2 × 107, 1 × 108, or 2 × 108 autologous CD30-specific CAR-T cells/m2 without a conditioning regimen. Eight of these patients had relapsed or progressed post-brentuximab treatment. At 6 weeks after treatment, one CR, one PR, and four SD were achieved among the nine treated patients, while persistence of CD30-specific CAR-T cell was limited as the molecular signal from CAR-T cells declined to near baseline by 4 weeks post-infusion. A dose of 2 × 108 CD30-specific CAR-T cells/m2 was safe and associated with significant in vivo expansion compared to other dose cohorts. No adverse events (AEs) were observed, including CRS correlated with CAR-T cell infusion. The study also showed that the frequency of T cells responding to the virus remained unchanged in the CD30-specific CAR-T cell recipients, which implied fratricide that might have occurred as the transient expression of CD30 in activated T cells had not happened. A preclinical study to explore the risks of targeting CD30 by CAR-T cell therapy in humanized mice also confirmed what Ramos et al. observed as the research illustrated CAR-T cells targeting CD30 could confer a superior therapeutic index in the treatment of CD30+ malignancies, leaving healthy activated lymphocytes and hematopoietic stem and progenitor cells (HSPCs) unaffected (Hombach et al., 2016).
Publication of the first results of CD30-targeted CAR-T cells for patients with HL came from the investigators at PLAGH (NCT02259556) (Wang et al., 2017a). Eighteen heavily pretreated patients with lymphoma (17 HL, 1 primary cutaneous anaplastic large cell lymphoma) were enrolled, 15 of whom had a considerable burden of lymphoma characterized by multiple tumor lesions including extensive abnormal lymph node regions (range: 0–7) and extranodal disease involving the bone, lung, liver, pleura, mammary glands, kidney, and soft tissues. A median of 1.56 × 107 CAR+ T cells/kg (range: 1.1–2.1 × 107 cells/kg) were infused over 3–5 days following physician’s choice conditioning chemotherapy. Among the 18 treated patients, 7 patients achieved PR with durations ranging from 2 to 9 months (Three remissions were ongoing), and 6 had SD. Median PFS was 6 months, and the copy number of CAR transgenes in PB peaked about 1 week after infusion and decreased to the baseline level by 4–8 weeks in most patients, while in which time relatively higher numbers in biopsy tissues were detected, and a corresponding decrease of CD30+ tumor cells was observed in some patients, highlighting that CAR-T cells could traffic to tumor sites and remain functional. Importantly, patients appeared to benefit from second or multiple CAR-T cell infusions as ongoing responses were observed in most of patients who received a second CAR-T cell infusion and the decrease of tumor burden was more significant after the second CAR-T cell infusion compared to the first. It was noted that lymph nodes presented a better response than extranodal lesions; lung lesions were likely to be relatively poor. The infusion was well tolerated and no evidence of CRS occurred in all the treated patients, except two who experienced grade ≥3 toxicities.
CAR-T CELL THERAPY FOR SOLID TUMORS
CAR-T cell therapy has shown enormous promise in B cell malignancies. However, this success has not yet extrapolated to solid tumors as they confer several challenges, especially for selecting appropriate targets. To date, there are only a few publications reporting clinical trials to evaluate second- or third-generation CAR-T cells in solid tumors by targeting human epidermal growth factor receptor-2 (HER2) (Morgan et al., 2010; Ahmed et al., 2015), mesothelin (MSLN) (Maus et al., 2013; Beatty et al., 2014), carcinoembryonic antigen (CEA) (Katz et al., 2015), and epidermal growth factor receptor (EGFR) (Feng et al., 2016). The efficacy is less encouraging, until recently, a significant clinical response with lower toxicities has been elicited in a patient with highly aggressive recurrent multifocal glioblastoma multiforme (GBM) who received both intracavitary and intraventricular administration of the interleukin-13 receptor alpha2 (IL13Rα2)-directed BBζ CAR-T cells (Brown et al., 2016), highlighting that CAR-T cell therapy could be useful for treating solid tumors by continuous optimization. Currently ongoing trials targeting solid tumors are listed in Table 2. In short, using CAR-T cells for solid tumors is still in a “proof-of-concept” stage, and feasibility and efficacy remain to be further established in clinical trials. Herein we review the preliminary outcomes of those early clinical trials for the treatment of solid tumors.
The ErbB family, subclass I of receptor tyrosine kinases (RTK), comprises four members widely expressed in adults at low levels: ErbB1/EGFR/HER1, ErbB2/HER2/Neu, ErbB3/HER3, and ErbB4/HER4 (Hynes and Lane 2005). Of these, EGFR and HER2 have been implicated in the development of a variety of tumors, including breast, lung, prostate, head and neck, pancreas, gastrointestinal tract, and gynecologic tract, so receptors have been intensely pursued as therapeutic targets (Whilding and Maher 2015). Several licensed monoclonal antibodies specific for EGFR (cetuximab, panitumumab, and nimotuzumab) and HER2 (trastuzumab and pertuzumab) are already available and demonstrate important therapeutic benefits. Moreover, these antibodies also present unique toxicities due to the baseline expression of EGFR or HER2 in normal tissues; for instance, most common are skin toxicity from EGFR inhibitors (Pastore et al., 2014) and cardiac toxicity associated with HER2-directed inhibitors, which is related to the physiological roles that EGFR and HER2 signaling is essential in the function of keratinocytes and cardiac myocytes. Taken together, there are grounds to believe that CAR targeting the ErbB family would have greater potential for potent antitumor activity in multiple malignancies; meanwhile, utmost consideration must be given to the safety concern as CAR-T cell-targeting EGFR/HER2 would possess greater avidity than the bivalent soluble antibody (Dotti et al., 2014).
Investigators at PLAGH pioneered an EGFR-directed CAR characterized by a shorted promoter in an effort to minimize the risk of on-target/off-tumor recognition and first tested this receptor in humans (NCT01869166). The preliminary outcome of 11 patients with advanced relapsed/refractory non-small cell lung cancer (NSCLC) who received anti-EGFR CAR-T cells at a dose of 0.45–1.09 × 107 CAR+ cells/kg alone or following investigator’s choice conditioning chemotherapy showed that the treatment was well-tolerated without severe toxicity (Feng et al., 2016). As expected, mild skin toxicities due to on-target/off-tumor recognition were observed. In addition, objective responses including 2 PR lasting 2 to 3.5 months and 5 SD lasting 2 to 8(+) months were observed. Of note, immunohistochemistry (IHC) examination of biopsy tumor tissues from patients achieving either PR or SD illustrated that anti-EGFR CAR-T cells could traffic to tumor sites and infiltrate the tumor tissues and elicit EGFR-specific cytotoxicity even at 3.5 months post-cell infusion, implying that anti-EGFR CAR-T cells could persist and remain functional in an immunosuppression microenvironment. On this basis, this receptor in addition to a conditioning chemotherapy regimen of Cy and Nab-paclitaxel in order to eradicate stroma in other EGFR-positive solid tumors including cholangiocarcinoma (CCA) and pancreatic cancer (PC) are being tested.
Investigators at NCI first conducted a clinical trial to test anti-HER2 third-generation CAR with a CD28.CD137.ζ endodomain in patients with metastatic cancer (NCT00924287). Unfortunately, the first patient with metastatic HER2+ colon cancer who received 1010 T cells (79% CAR+) following conditioning chemotherapy developed rapid respiratory distress within 15 min after cell infusion and ultimately died of multiple organ failure as a result of reactivity against lung epithelial cell expression of low levels of HER2 (Morgan et al., 2010). This unforeseen systemic adverse event has been known as a fatal example of the on-target/off-tumor effect of CAR-T cells targeting non-tumor-specific antigens and lends a cautionary tale to using CAR-T cells in solid tumors. However, encouraging safety data from nine patients with osteosarcoma who received 28ζ CAR targeting HER2 transduced T cells at doses ranging from 104–106 cells/m2 without conditioning (NCT00902044) were reported at the American Association for Cancer Research (AACR) 2012 by Ahmed et al. at BCM (Ahmed et al., 2014). Infusion was well tolerated without systemic side effects and no elevation of pro-inflammatory cytokine. Updated results of this trial in the first 19 patients with HER2-positive sarcoma (including 16 osteosarcomas) further confirmed that anti-HER2 CAR-T cell treatment was safe and feasible as no dose-limiting toxicity (DLT) was observed even in the highest dose level of 1 × 108 CAR+ T cells/m2 (Ahmed et al., 2015). Four of seventeen evaluable patients had SD for 12 weeks to 14 months, and 3 had their residual tumor removed with no further treatment and remained in remission at 6, 12, and 16 months, albeit no post-infusion expansion of anti-HER2 CAR-T cells in the PB was observed in most of the treated patients. Multiple reasons may contribute to the difference in the observed toxicity profile between the BCM and NCI trials, including no prior conditioning chemotherapy, 2-log lower maximum dose of cells, using a 28ζ CAR rather than CD28.4-1BB. ζ CAR. Furthermore, the HER2-specific scFvs of each CAR were derived from different MAbs (FRP5 vs. trastuzumab), which also could account for the substantial differences in safety observed in both trials. Besides sarcoma, Ahmed et al. also conducted a trial of HER2-specific CAR-T cells for GBM (NCT02442297). Furthermore, another two trials determining the safety of virus-specific HER2 re-targeted CAR-T cells (CMV; NCT01109095) (EBV; NCT00889954) are ongoing at BCM.
Variant III of the epidermal growth factor receptor (EGFRvIII), the most common variant of EGFR first identified in human GBM, is also present in many other tumor types, but is not found in healthy tissues (Li and Wong, 2008), which makes EGFRvIII a suitable target for CAR-T cell therapy. Investigators at Upenn reported (NCT02209376) (O’Rourke et al., 2016) the initial outcome of the first 9 patients with EGFRvIII-positive GBM who were treated with anti-EGFRvIII CAR-T cells at a dose of 1–5 × 108 CAR+ cells. The infusion was safe without evidence of off-tumor toxicity or CRS and no cross-reactivity to wild type EGFR, except one patient developed non-convulsive status epilepticus 9 days after infusion, which was resolved with standard treatment and anti-cytokine therapy. Significant expansion of anti-EGFRvIII CAR-T cells between 7 and 10 days post-infusion were observed in all patients, which is a sharp contrast to what Ahmed et al. observed (Ahmed et al., 2015). More importantly, a pathologic evaluation of five patients who had undergone surgical resection of tumors between 6 and 120 days after infusion demonstrated that anti-EGFRvIII CAR-T cells were immunologically active as recruitment of new T cells as well as specific EGFRvIII target antigen loss in GBM cells were observed in some cases. EGFRvIII-specific CARs for patients with GBM are also being tested at several other institutions, including the NCI (NCT01454596), Beijing Sanbo Brain Hospital (NCT02844062), and Duke University (NCT02664363).
MSLN is a tumor-associated antigen named for its low-level expression on mesothelial cells that line the peritoneal, pleural, and pericardial cavities, and yet is overexpressed in malignant pleural mesothelioma, pancreatic, ovarian, and lung cancer (O’Hara et al., 2016). To minimize the potential on-target/off-tumor toxicity of MSLN-specific CAR, investigators at Upenn developed an approach to transiently express the CAR on T cells by using electroporation of CAR mRNA and tested the safety of multiple infusions of MSLN-RNA-CAR-T cells in a first in-human study (NCT01355965) based on the encouraging results of preclinical studies (Zhao et al., 2010; Barrett et al., 2011). Preliminary results in four patients (three malignant pleural mesothelioma, one pancreatic adenocarcinoma) showed that multiple infusions of MSLN-RNA-CAR-T cells was feasible and safe without overt evidence of on-target/off-tumor toxicity against normal tissues, except one patient developed anaphylaxis due to the murine scFv used in the CAR and went into cardiac arrest within minutes of completing the third infusion but rapidly recovered as a consequence of intensive treatment (Maus et al., 2013; Beatty et al., 2014). Researchers also demonstrated the antitumor activity of anti-MSLN CAR-T cells based on the clinical and laboratory evidence such as specific and potent lysis capacity of anti-MSLN CAR-T cells resulting in a decrease in the tumor cells in a patient’s ascites. Updates of another trial to evaluate anti-MSLN CAR-T cells in patients with pancreatic ductal adenocarcinoma (PDAC) (NCT01897415) were presented by Beatty et al. (2015). Well-tolerated toxicity and modest antitumor efficacy with 2 SD among 6 treated patients were demonstrated. In light of the aforementioned safety profile of anti-MSLN CAR-T cells, researchers opened a trial to test anti-MSLN CAR-T cells transduced with lentivirus for PDAC, epithelial ovarian cancer, and malignant epithelial pleural mesothelioma (NCT02159716), and reported the early results of this trial at AACR 2015 (Tanyi et al., 2015). Five patients with advanced stage cancers (two serous ovarian, two epithelial mesothelioma, and one PDAC) were treated with a single dose of 1–3 × 107 CAR+ T cells/m2 without lymphodepletion. Infusions were well tolerated with no acute AE and no evidence of on-target/off-tumor toxicity albeit anti-MSLN CAR-T cells were found to traffic to on-target/off-tumor sites such as the pericardial fluid. The loss of malignant cells in the pleura fluid near 4 weeks post-cell infusion happened in one patient and another experienced stable to decreased burden of disease, suggesting anti-MSLN CAR-T cells possessed direct anti-tumor efficacy. Updated experiences of 6 patients with recurrent serous ovarian cancer were reported by Tanyi et al. at ASCO 2016 (Tanyi et al., 2016). The treatment still was well-tolerated even in two patients who received 3 × 108 CAR+ T cells/m2. Six of six treated patients achieved SD 1 month after anti-MSLN CAR-T cell infusion, and clearance of pleural effusion by anti-MSLN CAR-T cells which trafficked to tumor sites was noted in one patient. Moreover, a variety of MSLN-specific CARs are being tested in other ongoing trials (NCT02414269, NCT01583686, NCT02465983, and NCT02792114).
Prostate-specific membrane antigen (PSMA), a type II transmembrane glycoprotein, is expressed in all forms of prostate tissue, but is upregulated 10-fold in prostate cancer (Ma et al., 2004). PSMA-targeted CAR-T cells for patients with castrate metastatic prostate cancer (CMPC) was tested in a dose escalation study performed at MSKCC (NCT01140373), and early experiences in the first three patients who received dose level 1 (1 × 107 CAR+ T cells/kg) following 300 mg/m2 of Cy one day were presented at ASCO 2012 by Slovin et al. No toxicities occurred, and two of three treated patients had SD for longer than 6 months (Slovin et al., 2012). On this basis, the fourth patient received the same dose with a modified vector with a higher copy number, and an additional three patients in cohort 2 were treated with 3 × 107 CAR+ T cells/kg following the same conditioning regimen. Updated results showed that one of two patients achieving SD in cohort 1 maintained a response for greater than 16 months. All three patients in cohort 2 had elevated levels of cytokine, including interleukin-4 (IL-4), interleukin-8 (IL-8), and IL-6, etc., and up to 2 weeks persistence of CAR-T cells post-CAR-T cell infusion (Slovin et al., 2013). An encouraging early data from trial of PSMA-specific CAR (NCT00664196) was demonstrated by Junghans (Junghans, 2012) at ASCO 2012 as two PR with a decrease in prostate specific antigen (PSA) levels and delayed disease progression were attained among five patients with metastatic prostate cancer who were treated with a non-myeloablative (NMA) conditioning regimen followed by either 109 or 1010 anti-PSMA CAR-T cells and IL-2 given by continuous infusion for 1 month alongside the T cell infusion. Of note, these two PRs were observed at the lowest T cell dose of 109, together with the plasma IL-2 in non-responders was as much as 10-fold lower compared to that in responders, the researchers drew a conclusion that adequate higher IL-2 in vivo plus a higher CAR-T cell dose could be beneficial to anti-tumor activity of anti-PSMA CAR-T cells, which is being tested in a redesign study.
CEA is overexpressed in many epithelial cancers but is also expressed in a variety of normal epithelial cells (Hammarstrom, 1999). Investigators at Roger Williams Medical Center conducted a phase I hepatic immunotherapy for metastases (HITM) trial (NCT01373047) to investigate CAR-T cell targeting CEA for patients with CEA-expressing adenocarcinoma liver metastases and reported the early results in 2015 (Katz et al., 2015). Given that severe transient colitis had been induced by intravenous infusion of CEA-specific TCR-transduced T cells in a previous study (Parkhurst et al., 2011), a regional delivery strategy was adopted aiming to enhance the tolerability and therapeutic efficacy of anti-CEA CAR-T cells. Of the six treated patients, three received anti-CEA CAR-T cells alone in dose-escalation fashion (108, 109, and 1010 cells), whereas an additional three patients received the maximum planned anti-CEA CAR-T cell dose (1010 cells × 3) along with systemic IL-2 support. No grade 3 or 4 AE related to the anti-CEA CAR-T cell infusion developed in all treated patients. One patient had stable disease for 23 months after anti-CEA CAR-T cell infusion and other five patients had progressive disease; however, a median 37% decrease of CEA levels was observed in patients receiving systemic IL-2 support, and four of six treated patients showed necrosis of metastatic liver lesions. Another anti-CEA CAR-T cell for CEA positive cancer is currently being tested at Southwest Hospital in China (NCT02349724); no results of this trial have been published yet.
IL13Rα2, a monomeric high-affinity IL13 receptor, is selectively expressed on GBM while absent in the surrounding normal brain tissue, rendering it can be proposed as an optimal candidate for target selection of CAR-T cell therapy in glioma (Thaci et al., 2014). Building on their previous experience in 3 patients with glioblastoma that multiple intracranial infusions of first generation CAR-T cells targeting IL13Rα2 was well-tolerated (Brown et al., 2015), the investigators at City of Hope National Medical Center (City of Hope; USA) conducted a trial (NCT02208362) to evaluate a IL13Rα2-specific BBζ CAR (IL13 BBζ-CAR) without lymphodepleting chemotherapy in GBM and reported their clinical experience in one patient with recurrent multifocal GBM (Brown et al., 2016). Local control after intracavitary administration of six cycles of IL13BBζ-CAR T cells was observed, whereas other disease foci that were distant from the CAR-T cell injection site continued to progress. Together with new metastatic lesions in the spine, ten additional intraventricular treatment cycles were administered in an effort to effectively control tumor progression at distant sites. After five intraventricular infusions, all tumors including spinal metastases have decreased by 77%–100% and continued to resolve during the five additional intraventricular consolidation infusions. No grade ≥3 toxicities related to intracranial infusions of IL13 BBζ-CAR-T cells occurred during these infusions. The clinical response lasted for 7.5 months, however, the disease eventually recurred at new four lesions. The researchers speculated that this relapse might be attributed to the downregulation of IL13Rα2, which should be an example of tumor editing caused by the selective pressure exerted by CAR-T cell therapy.
TOXICITIES OF CAR-T CELL THERAPY
CAR-T cells offer a promising new therapy for cancers, but the toxicities elicited in the clinical trials are still a great concern. Deaths with CAR-T were reported previously (Morgan et al., 2010; Brentjens et al., 2010) and recently (DeFrancesco 2016), which has been a wake-up call to the potential for toxicity of CAR-T cell therapy (Junghans 2010). The toxicities of CAR-T cell therapy generally fall into several of the following categories.
Cytokine release syndrome
The most significant and life-threatening toxicity following CAR-T cell therapy is CRS, which is attributable to the rapid and extensive activation of infused CAR-T cells upon antigen engagement and results in elevated inflammatory cytokines (Lee et al., 2014). The frequency and severity of CRS vary greatly among different studies, which has been reported in 18%–100% of patients, with sCRS noted in 27%–53% of patients (Batlevi et al., 2016). Since CRS can be successfully ameliorated with the IL-6R inhibitor tocilizumab (Grupp et al., 2013), investigators now have more experience in how to diagnose and manage CRS, and recently several reviews have highlighted and summarized these advances (Lee et al., 2014; Maude et al., 2014b; Xu and Tang 2014; Brudno and Kochenderfer 2016; Bonifant et al., 2016). The strong correlation between the severity of CRS and the tumor burden at the time of infusion has been well-recognized (Maude et al., 2015a), nevertheless, whether to use prophylactic or early tocilizumab remains undetermined (Nellan and Lee 2015). Besides tocilizumab, other cytokine-directed approaches to managing CRS could be considered, and inhibitor of TNF-α infliximab has also been successfully used in our center. Of note, due to the concern that the pre-emptive CRS treatment could impair the anti-tumor efficacy of the infused CAR-T cells, Ruella et al. added kinase inhibitor ibrutinib to anti-CD19 CAR-T cells in an effort to prevent CRS, and proved the feasibility of this strategy in an NOD/SCID/gamma-chain-deficient (NSG) mice model. On this basis, a clinical trial (NCT02640209) was opened to test CTL019 cells in addition to ibrutinib in patients with CLL (Ruella et al., 2017). Significantly, a more complete understanding regarding the biology of the syndrome and to subsequently prevent or abrogate sCRS as well as to determine predictive biomarkers for CRS is of utmost importance. David et al. developed a novel algorithm to predict CRS recently and showed that peak levels of IFN-γ, IL-6, sgp130, and sIL-6R within the first month after infusion could be proposed as a predictive marker for sCRS, which might guide future cytokine-directed therapy (Teachey et al., 2016).
On-target/off-tumor toxicity
This type of toxicity is a direct result of the specific recognition of a target expressed in normal tissues by CAR-T cells, thus its profile is dependent on the antigenic specificity of the engineered T cell and can be predictably seen in a variety of organ systems (Bonifant et al., 2016; Barrett et al., 2015). B cell aplasia is a classical on-target/off-tumor toxicity in patients treated with anti-CD19 or 20 CAR-T cells (Maude et al., 2014a; Zhang et al., 2016a; Kochenderfer et al., 2012), which can easily be managed with intravenously (i.v.) Ig replacement and serve as a surrogate maker for the persistence of CAR-T cells in vivo (Maude et al., 2014a). With respect to solid tumors, the toxicity resulting from on-target/off-tumor recognition may not be so tolerable and acceptable, which was highlighted by an aforementioned anti-HER2 CAR-T death case report (Morgan et al., 2010). However, subsequent trials of ErbB family-specific CAR demonstrated acceptable toxicities (Ahmed et al., 2015; Feng et al., 2016). In order to minimize the risk of on-target/off-tumor toxicity in solid tumors, multiple strategies have been developed and fall in to two major categories: enhancing selectivity of CAR aiming to enhance the tumor recognition and bystander discrimination as well as control CAR-T cell activity in an attempt to provide ways for physician to intervene and either eliminate or modulate the T cell activity when acute severe off-tumor toxicities occurred. The enhancing selectivity of CAR can be achieved via selecting safer antigen (i.e., tumor specific antigen EGFR vIII, aberrantly glycosylated antigens, TCR-like CAR) (O’Rourke et al., 2016; Posey et al., 2016; Zhang et al., 2014; Ma et al., 2016a; Liu et al., 2017a), combinatorial antigen targeting (i.e., complementary signaling, synNotch/CAR circulation, iCAR) (Fedorov et al., 2013; Kloss et al., 2013; Roybal et al., 2016; Wilkie et al., 2012), turning sensitivity of scFv by turning the affinity (Caruso et al., 2015; Liu et al., 2015), and masked CAR (Desnoyers et al., 2013), while the design of limiting CAR expression (i.e., transient mRNA CAR) (Maus et al., 2013; Beatty et al., 2014), switchable CAR-T cell (i.e., dimerizing small molecules, tumor targeting antibody) (Cao et al., 2016; Juillerat et al., 2016; Ma et al., 2016b; Rodgers et al., 2016; Wu et al., 2015) and suicide gene (i.e., inducible Caspase-9, antibody-mediated depletion) (Turtle et al., 2016a, b; Di Stasi et al., 2011) can be introduced to flexibly control the CAR-T cell activity. For a detailed description and analysis of these proof-of concept designs, please refer to the reviews published recently by our group (Wang et al., 2017b) and Upenn group (Lim and June 2017). Overall, most of these strategies are in early stages, the preliminary results from the experimental studies provide the initial evidence of feasibility and pave the road to further optimization. However, the eventual effects of these novel designs still need to be determined in forthcoming clinical trials.
Neurologic toxicities
Neurologic toxicities were described in 13%–52% of patients across institutions, and symptoms ranged from confusion and delirium to aphasia, obtundation, myoclonus, and seizure, which is frequently self-limiting (Maude et al., 2014a; Davila et al., 2014; Lee et al., 2015a; Kochenderfer et al., 2015; Brentjens et al., 2013; Park et al., 2015; Grupp et al., 2015; Schuster et al., 2015). However, the deaths of 3 patients with R/R B-ALL after receiving anti-CD19 CAR-T cells following Cy/Flu conditioning chemotherapy highlights the potential harm of this type of toxicity (DeFrancesco, 2016). The etiology of this syndrome remains unclear; it often accompanies CRS, but also can be present alone (Maude et al., 2014a). The NCI believes that tocilizumab may temporarily worsen neurotoxicity, thus they recommend using high-dose steroids rather than tocilizumab to treat grade ≥3 neurologic toxicity (Brudno and Kochenderfer, 2016). More studies are needed to determine the pathophysiology and subsequently find the best approach to treat or prevent severe neurotoxicity.
Other rare toxicities
Hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) occurred in a subset of patients who received CAR-T cells (Porter et al., 2015; Grupp et al., 2013). HLH/MAS is a rare AE triggered by a cascade of immune activation and characterized by hyperinflammation with prolonged fever, hepatosplenomegaly, and cytopenias. This syndrome has parallels in both clinical and laboratory findings with CRS; however, elevated levels of ferritin and triglycerides can be used to differentiate these two syndromes (Janka, 2012). Genetic predisposition may increase the risk of developing HLH/MAS in some patients (Maude et al., 2014b), but it can be well controlled by tocilizumab.
An IgE-mediated clinical anaphylaxis after the third MSLN-RNA CAR-T cell infusion has been reported, which was suggested by markedly elevated tryptase levels and the presence of human anti-mouse antibodies after cell infusion (Maus et al., 2013). This effect may be attributable to the multiple infusion schedule, which may lead to a substantial humoral immune response against the CAR with murine SS1 scFv. Similarly, a cellular immune response specific for murine scFv epitopes of anti-CD19 CAR was identified in the patients who received the second infusion, resulting in the failure of the second infusion (Turtle et al., 2016a, b). We also observed that anti-EGFR CAR-T cells could not be well proliferated in some patients who received a second infusion. Humanized or fully human scFv will hopefully abrogate or at least reduce the potential for anti-murine immune-mediated rejection, which has been shown to be highly effective in a phase I study of humanized CD19-directed CAR-T cells (CTL119) (Maude et al., 2015b).
Tumor lysis syndrome (TLS) also has been described in some patients (Dai et al., 2015; Grupp et al., 2013; Brudno et al., 2016). A severe TLS occurred after stand-alone low-dose chemotherapy in a patient who relapsed after anti-CD19 CAR-T cell infusion resulting from the loss of CAR-T cells (Zhang et al., 2016b).
CHALLENGES FOR CAR-T CELL THERAPY
CAR-T cell therapy has shown unprecedented initial response rates in advanced B cell malignancies; however, relapse after CAR-T cell infusion is a major hurdle in successful CAR regimens. To date, the main understanding regarding this phenomenon is gained from the trials involving CD19, and two modes of relapse have been seen: CD19 negative and CD19 positive (Maude et al., 2015a). CD19 negative relapses were reported by several groups (Maude et al., 2014a; Lee et al., 2015a; Turtle et al., 2016a; Grupp et al., 2013; Pegram et al., 2015), in which the Upenn group showed the highest incidence of up to 60% (Ruella et al., 2016). This group also demonstrated that splice-based adaptations in tumor cells was an underlying mechanism for tumor antigen loss escape, leading to an outgrowth of tumor escape variant cells (Sotillo et al., 2015). Dual-targeted T cells is a potential strategy to reduce the risk of antigen escape, which has two patterns: (i) T cell expressing a CAR comprising two different scFv in tandem (termed “TanCAR”) (Grada et al., 2013) or expressing two different CARs targeting two different targets (known as “dual-signaling CAR”) (Ruella et al., 2016). Both of these two designs only have one group CAR-T cells; (ii) “pooled” CAR-T, where two groups of CAR-T cells express two different CARs, which can be infused sequentially or simultaneously. Although the preliminary evidence of feasibility of TanCAR and dual-signaling CAR designs were demonstrated in several proof of concept preclinical studies (Zah et al., 2016; Grada et al., 2013; Hegde et al., 2016), which are challenging to implement due to the difficulty of identifying 2 appropriate targets on 1 tumor (Jackson and Brentjens 2015) as well as the constraint of suitable epitopes selection in the setting of TanCAR (Sadelain, 2016). Regarding the “pooled” CAR-T, the development period is longer as is the combination of two groups of CAR-T cells. CD19 positive relapse as a result of loss of CAR-T cell persistence can be prevented by prolonging CAR-T cell persistence, which can be achieved by using preconditioning, optimization of CAR constructs, and increasing the ratio of early lineage T cells (Maude et al., 2015a). Multiple infusions of CAR-T cells is also an effective option for patients who experienced CD19 positive relapse (Maude et al., 2014a; Kochenderfer et al., 2012, 2015). However, failure of the second or third infusion was observed in a subset of patients (Lee et al., 2015a; Turtle et al., 2016a, b), warranting further studies.
It remains a huge challenge for CAR-T cell therapy beyond the hematological malignancies. Besides the aforementioned safety concern due to the risk of on-target/off-tumor recognition, limited therapeutic success is another major hurdle in CAR-T cell treatment of solid tumors. This limitation is mainly attributable to the hostile solid tumor microenvironment characterized by physical/anatomical barriers (i.e., tumor stroma) and immunosuppressive cytokines and immune cells such as regulatory T (TREG) cells and myeloid-derived suppressor cells (MDSCs), which are harmful to the infiltration of infused CAR-T cells into tumor sites and for retaining cytotoxic functionality (Newick et al., 2016). One promising approach to circumvent this obstacle is the use of armored CAR-T cells, which are the fourth-generation CAR-T cells that are further modified to additionally express immune-modulatory proteins, including cytokines (IL-2, IL-12, and IL-15) and ligands (PD-1/CD28 fusion, CD40L, or 4-1BBL) (Fesnak et al., 2016; Khalil et al., 2016). Armored CAR-T cells modified to secrete pro-inflammatory IL-12 have been known as TRUCKs (T cells redirected for universal cytokine killing), which can release IL-12 upon CAR-mediated T cell activation and have yielded encouraging results in several preclinical studies (Chmielewski et al., 2014). The first clinical trial to explore the impact of IL-12 CAR-T cells has been opened by MSKCC (NCT02498912), where IL-12-secreting CAR-T cells transduced with a 28ζ CAR targeting mucin-16 (MUC-16) is being tested in patients with ovarian cancer. Other examples of armored CAR-T cells such as those modified to additionally express ligands are in the proof-of-concept stage and have not yet moved forward to the stage of clinical trials (Khalil et al., 2016).
CAR-T cell therapy is entering advanced phases of clinical trial testing; anti-CD19 CAR-T especially will enter mainstream clinical oncology for patients with B cell malignancies in the near future (Klebanoff et al., 2016). However, these clinical successes thus far have employed autologous cells, which were produced on the campuses of multiple academic facilities for a given recipient on a case-by-case basis. This personalized manufacturing and widely “distributed” approach greatly limit the broad implementation and commercialization of CAR-T cell therapy due to the complicated and time-consuming procedures, great cost to generate one product for one patient, and heterogeneity of T cell products produced for or from individual recipients (Torikai and Cooper 2016). Centralized manufacturing of patient-derived CAR-T cells and distribution to multiple points-of-care have already been adopted by biopharmaceutical companies such as Novartis, Juno, Kite, and CBMG, aiming to reduce the variation of CAR-T cell products and the costs associated with them (Cooper 2015). On this basis, “off-the-shelf” (OTS) CAR-T cell therapy, which is deemed to be the ultimate product formulation, will open of a new chapter in the race to commercialize CAR-T cell therapy (Ratner 2016). OTS CAR-T cell (also known as universal CAR-T cell, or UCAR-T) is defined as a biologic that is pre-prepared in advance from one or more healthy unrelated donors, validated, and cryopreserved (Torikai and Cooper 2016) and then can be shipped in a day or two to patients worldwide. The first clinical application of universal CAR-T cells was reported by Qasim et al. (2015) at ASH 2015; a 1-year-old girl with relapsed leukemia achieved molecular remission without significant toxicity after transcription activator-like effector nucleases (Talen) engineered anti-CD19 UCAR-T cell infusion following lymphodepleting conditioning with Flu 90 mg/m2, Cy 1.5 g/m2, and alemtuzumab 1 mg/kg, providing early proof-of-concept evidence for this strategy. However, more studies are needed to optimize this innovative approach, as many challenges remain (Torikai and Cooper 2016).
CONCLUSIONS AND FUTURE PERSPECTIVES
CAR-T cell therapy, especially CD19-specific CAR-T cell therapy, is poised to shift the treatment paradigm for B cell malignancies as significant response rates and well-tolerated toxicities. For this reason, many researchers are currently developing strategies in an effort to recapitulate this success in solid tumors, albeit the road is unlikely to be straightforward mainly due to the risk of on-target/off-tumor recognition and hostile solid tumor microenvironments resulting in less efficacy. However, strategies are being implemented to address these obstacles, and some encouraging preliminary results have been demonstrated. Despite these advancements, several issues remain to be resolved (Box 1), providing impetus for continuous optimization of CAR as well as appropriately powered, well-designed clinical studies.
Box 1: Unresolved questions in CAR-T cell therapy |
|---|
· What is the suitable dosage range of CAR-T cells, and is it the same in different targets or diseases? |
· What is the optimal ratio of engineered CAR-T cell subsets, including early memory T cells? |
· How great immunogenicity of CAR-modified T cells can be resolved by humanized and or fully human CAR, and what is the optimal multiple infusion regimen? |
· Can smart CAR aiming to reduce on-target/off-tumor recognition provide for adequate safety in clinical testing? |
· What is the optimal management for patients who have received CAR-T cell therapy, and what are the relative roles of CAR-T cells and HSCT in the context of transplant-eligible patients? |
Actually, besides how to enhance efficacy and safety of CAR-T cells, the development of resistance is particularly noteworthy for the optimization of CAR-T cell therapy either in hematological malignancies or solid tumors. As is well known, downregulation of target antigens is one of mechanism that tumor escape from cancer immunotherapy (Marincola et al., 2000; Leen et al., 2007). By tumor editing such as target antigen loss (Evans et al., 2015), mutation (Sotillo et al., 2015) or leukemic lineage switch (Gardner et al., 2016), tumor clone can be invisible to the CAR-T cell therapy, resulting in the tumor cells resistant to the killing mediated by CAR-T cells and disease recurrence. This phenomena occurs not only in hematological malignancies but also in solid tumors (Brown et al., 2016; Hegde et al., 2016), highlighting the shortcoming of single-target CAR-T cell therapy. Generating T cells capable of recognizing multiple antigens may be an effective alternative to address the challenge of resistance and relapse after CAR-T cell therapy; moreover, T cell exhaustion, an acquired state of T cell dysfunction due to the persisting antigenic stimulation during cancer, can also lead to the CAR-T cells failure to eliminate the tumor cells even the target antigens are still present. PD-L1/PD-1 immune inhibitory axis plays a central role in the regulation of T cell exhaustion. Blocking PD-1 can re-invigorate the exhausted T cells and improve control of cancer, which has been seen in a patient with refractory DLBCL whose disease has progressed after anti-CD19 CAR-T cell infusion (Chong et al., 2017). Thus we believe that elucidating the underlying mechanisms of CAR-T cell therapy resistance and development of effective combination therapy that combination of PD-1/PD-L1 blockade with CAR T cell therapy in an effort to reverse T cell exhaustion will be an active research area in CAR-T cell therapy field.
Finally, with the emergence of clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated protein 9 (Cas9) system, a new gene editing tool that can induce targeted genetic alterations and process multiplex genome engineering with a relative ease compared to the Talen system (Maeder and Gersbach 2016; Cong et al., 2013), applying this novel system to disrupt TCRα subunit constant (TRAC) and or beta-2 microglobulin (B2M) of CAR-T cells to avoid GVHD and minimize immunogenicity has been actively investigated (Liu et al., 2017b; Eyquem et al., 2017; Ren et al., 2016). The preliminary data from these experimental studies suggest that CRISPR-Cas9-mediated multiplex gene editing is readily applicable to CAR-T cells even in the setting of triple genes disruption. This would be helpful for the development of OTS donor-derived CAR-T cells. We believe the combination of CAR-T cell therapy and gene editing will revolutionize the industry even if many difficult challenges lie ahead.
References
Ahmed N, Brawley V, Diouf O, Anderson P, Hicks J, Wang L, Dotti G, Wels W, Liu H, Gee A et al (2014) Abstract 3500: T cells redirected against HER2 for the adoptive immunotherapy for HER2-positive osteosarcoma. Cancer Res 72(8 Supplement):3500
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, Liu E, Dakhova O, Ashoori A, Corder A et al (2015) Human epidermal growth factor receptor 2 (HER2)—specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol 33(15):1688–1696
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, Abrams J, Sznol M, Parkinson D, Hawkins M et al (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17(7):2105–2116
Barrett DM, Zhao Y, Liu X, Jiang S, Carpenito C, Kalos M, Carroll RG, June CH, Grupp SA (2011) Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Therapy 22(12):1575–1586
Barrett DM, Grupp SA, June CH (2015) Chimeric antigen receptor- and TCR-modified T cells enter main street and wall street. J Immunol (Baltimore, Md : 1950) 195(3):755–761
Batlevi CL, Matsuki E, Brentjens RJ, Younes A (2016) Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol 13(1):25–40
Beatty GL, Haas AR, Maus MV, Torigian DA, Soulen MC, Plesa G, Chew A, Zhao Y, Levine BL, Albelda SM et al (2014) Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2(2):112–120
Beatty GL, O’Hara MH, Nelson AM, McGarvey M, Torigian DA, Lacey SF, Melenhorst JJ, Levine B, Plesa G, June CH (2015) Safety and antitumor activity of chimeric antigen receptor modified T cells in patients with chemotherapy refractory metastatic pancreatic cancer. ASCO Meet Abstr 33(15_suppl):3007
Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ (2016) Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics 3:16011
Boussiotis VA, Freeman GJ, Gribben JG, Nadler LM (1996) The role of B7-1/B7-2:CD28/CLTA-4 pathways in the prevention of anergy, induction of productive immunity and down-regulation of the immune response. Immunol Rev 153:5–26
Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M (2010) Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther 18(4):666–668
Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, Yeh R, Bartido S, Borquez-Ojeda O et al (2011) Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood 118(18):4817–4828
Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, Bartido S, Stefanski J, Taylor C, Olszewska M et al (2013) CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 5(177):177ra138
Brocker T, Karjalainen K (1995) Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J Exp Med 181(5):1653–1659
Brocker T, Peter A, Traunecker A, Karjalainen K (1993) New simplified molecular design for functional T cell receptor. Eur J Immunol 23(7):1435–1439
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang W-C, Naranjo A, Starr R, Wagner J, Wright C et al (2015) Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res 21(18):4062
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, Ostberg JR, Blanchard MS, Kilpatrick J, Simpson J et al (2016) Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med 375(26):2561–2569
Brudno JN, Kochenderfer JN (2016) Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127(26):3321–3330
Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, Gea-Banacloche JC, Pavletic SZ, Hickstein DD, Lu TL et al (2016) Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J Clin Oncol 34(10):1112–1121
Cao Y, Rodgers DT, Du J, Ahmad I, Hampton EN, Ma JS, Mazagova M, Choi SH, Yun HY, Xiao H et al (2016) Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem Int Ed Engl 55(26):7520–7524
Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, Haines KM, Heitjan DF, Albelda SM et al (2009) Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA 106(9):3360–3365
Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, Mi T, Switzer K, Singh H, Huls H et al (2015) Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res 75(17):3505–3518
Chmielewski M, Hombach AA, Abken H (2014) Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 257(1):83–90
Choi BD, Suryadevara CM, Gedeon PC, Herndon JE 2nd, Sanchez-Perez L, Bigner DD, Sampson JH (2014) Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. J Clin Neurosci 21(1):189–190
Chong EA, Melenhorst JJ, Lacey SF, Ambrose DE, Gonzalez V, Levine BL, June CH, Schuster SJ (2017) PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129(8):1039–1041
Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339(6121):819–823
Cooper LJ (2015) Moving from tinkering in the garage to assembly line production: the manufacture of genetically modified T cells expressing chimeric antigen receptors (CARs) comes on line. Cancer Gene Ther 22(2):64–66
Couzin-Frankel J (2013) Breakthrough of the year 2013. Cancer immunotherapy. Science 342(6165):1432–1433
Curran KJ, Pegram HJ, Brentjens RJ (2012) Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. J Gene Med 14(6):405–415
Dai H, Zhang W, Li X, Han Q, Guo Y, Zhang Y, Wang Y, Wang C, Shi F, Zhang Y et al (2015) Tolerance and efficacy of autologous or donor-derived T cells expressing CD19 chimeric antigen receptors in adult B-ALL with extramedullary leukemia. Oncoimmunology 4(11):e1027469
Dai H, Wang Y, Lu X, Han W (2016) Chimeric antigen receptors modified T-cells for cancer therapy. J Natl Cancer Inst 108(7):439
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M et al (2014) Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 6(224):224ra225
de Coana YP, Choudhury A, Kiessling R (2015) Checkpoint blockade for cancer therapy: revitalizing a suppressed immune system. Trends Mol Med 21(8):482–491
DeFrancesco L (2016) Juno’s wild ride. Nat Biotechnol 34(8):793
Depoil D, Fleire S, Treanor BL, Weber M, Harwood NE, Marchbank KL, Tybulewicz VL, Batista FD (2008) CD19 is essential for B cell activation by promoting B cell receptor-antigen microcluster formation in response to membrane-bound ligand. Nat Immunol 9(1):63–72
Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW, Wong C, Bessette PH, Kamath K, Moore SJ et al (2013) Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med 5(207):207ra144
Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, Heslop HE, Brenner MK, Dotti G, Savoldo B (2009) T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113(25):6392–6402
Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, Liu E, Durett AG, Grilley B et al (2011) Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 365(18):1673–1683
Dotti G, Gottschalk S, Savoldo B, Brenner MK (2014) Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol Rev 257(1):107–126
Elert E (2013) Calling cells to arms. Nature 504(7480):S2–S3
Eshhar Z (2008) The T-body approach: redirecting T cells with antibody specificity. Handb Exp Pharmacol 181:329–342
Eshhar Z (2014) From the mouse cage to human therapy: a personal perspective of the emergence of T-bodies/chimeric antigen receptor T cells. Hum Gene Ther 25(9):773–778
Eshhar Z, Waks T, Gross G, Schindler DG (1993) Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA 90(2):720–724
Evans AG, Rothberg PG, Burack WR, Huntington SF, Porter DL, Friedberg JW, Liesveld JL (2015) Evolution to plasmablastic lymphoma evades CD19-directed chimeric antigen receptor T cells. Br J Haematol 171(2):205–209
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M (2017) Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543(7643):113–117
Fedorov VD, Themeli M, Sadelain M (2013) PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med 5(215):215ra172
Feng K, Guo Y, Dai H, Wang Y, Li X, Jia H, Han W (2016) Chimeric antigen receptor-modified T cells for the immunotherapy of patients with EGFR-expressing advanced relapsed/refractory non-small cell lung cancer. Sci China Life Sci 59(5):468–479
Fesnak AD, June CH, Levine BL (2016) Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer 16(9):566–581
Finney HM, Lawson AD, Bebbington CR, Weir AN (1998) Chimeric receptors providing both primary and costimulatory signaling in T cells from a single gene product. J Immunol (Baltimore, Md : 1950) 161(6):2791–2797
Finney HM, Akbar AN, Lawson ADG (2003) Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR Chain. J Immunol 172(1):104–113
Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC (1995) Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol 13(3):688–696
Gardner R, Wu D, Cherian S, Fang M, Hanafi LA, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG et al (2016) Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127(20):2406–2410
Geyer MB, Brentjens RJ (2016) Review: current clinical applications of chimeric antigen receptor (CAR) modified T cells. Cytotherapy 18(11):1393–1409
Geyer MB, Park JH, Riviere I, Wang X, Purdon T, Sadelain M, Brentjens RJ (2016) Updated results: phase I trial of autologous CD19-targeted CAR T cells in patients with residual CLL following initial purine analog-based therapy. ASCO Meet Abstr 34(15):7526
Ghorashian S, Pule M, Amrolia P (2015) CD19 chimeric antigen receptor T cell therapy for haematological malignancies. Br J Haematol 169(4):463–478
Gill S, June CH (2015) Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev 263(1):68–89
Goverman J, Gomez SM, Segesman KD, Hunkapiller T, Laug WE, Hood L (1990) Chimeric immunoglobulin-T cell receptor proteins form functional receptors: implications for T cell receptor complex formation and activation. Cell 60(6):929–939
Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, Schonfeld K, Koch J, Dotti G et al (2013) TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucl Acid 2:e105
Gross G, Eshhar Z (2016) Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: counteracting off-tumor toxicities for safe CAR T cell therapy. Annu Rev Pharmacol Toxicol 56:59–83
Gross G, Waks T, Eshhar Z (1989) Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA 86(24):10024–10028
Gross G, Levy S, Levy R, Waks T, Eshhar Z (1995) Chimaeric T-cell receptors specific to a B-lymphoma idiotype: a model for tumour immunotherapy. Biochem Soc Trans 23(4):1079–1082
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, Chew A, Hauck B, Wright JF et al (2013) Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 368(16):1509–1518
Grupp SA, Maude SL, Shaw PA, Aplenc R, Barrett DM, Callahan C, Lacey SF, Levine BL, Melenhorst JJ, Motley L et al (2015) Durable remissions in children with relapsed/refractory ALL treated with T cells engineered with a CD19-targeted chimeric antigen receptor (CTL019). Blood 126(23):681
Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, Irlam J, Chester KA, Kemshead JT, Shaw DM et al (2005) The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother (Hagerstown, Md : 1997) 28(3):203–211
Hammarstrom S (1999) The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 9(2):67–81
Harris DT, Kranz DM (2016) Adoptive T cell therapies: a comparison of T cell receptors and chimeric antigen receptors. Trends Pharmacol Sci 37(3):220–230
Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK (2002a) Single-chain antigen recognition receptors that costimulate potent rejection of established experimental tumors. Blood 100(9):3155–3163
Haynes NM, Trapani JA, Teng MW, Jackson JT, Cerruti L, Jane SM, Kershaw MH, Smyth MJ, Darcy PK (2002b) Rejection of syngeneic colon carcinoma by CTLs expressing single-chain antibody receptors codelivering CD28 costimulation. J Immunol (Baltimore, Md : 1950) 169(10):5780–5786
Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, Wakefield A, Fousek K, Bielamowicz K, Chow KK et al (2016) Tandem CAR T cells targeting HER2 and IL13Ralpha2 mitigate tumor antigen escape. J Clin Investig 126(8):3036–3052
Holohan DR, Lee JC, Bluestone JA (2015) Shifting the evolving CAR T cell platform into higher gear. Cancer Cell 28(4):401–402
Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Kruis W, Pohl C, Abken H (1997) T cell targeting of TAG72+ tumor cells by a chimeric receptor with antibody-like specificity for a carbohydrate epitope. Gastroenterology 113(4):1163–1170
Hombach A, Heuser C, Sircar R, Tillmann T, Diehl V, Pohl C, Abken H (1998) An anti-CD30 chimeric receptor that mediates CD3-zeta-independent T-cell activation against Hodgkin’s lymphoma cells in the presence of soluble CD30. Cancer Res 58(6):1116–1119
Hombach AA, Gorgens A, Chmielewski M, Murke F, Kimpel J, Giebel B, Abken H (2016) Superior therapeutic index in lymphoma therapy: CD30(+) CD34(+) hematopoietic stem cells resist a chimeric antigen receptor T-cell attack. Mol Ther 24(8):1423–1434
Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR (2013) Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res 19(12):3153–3164
Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, Jensen MC, Riddell SR (2015) The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res 3(2):125–135
Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, Rosenberg SA, Eshhar Z (1993) Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the Fc receptor gamma chain. J Exp Med 178(1):361–366
Hwu P, Yang JC, Cowherd R, Treisman J, Shafer GE, Eshhar Z, Rosenberg SA (1995) In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res 55(15):3369–3373
Hynes NE, Lane HA (2005) ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer 5(5):341–354
Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, Campana D (2004) Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia 18(4):676–684
Irving BA, Weiss A (1991) The cytoplasmic domain of the T cell receptor zeta chain is sufficient to couple to receptor-associated signal transduction pathways. Cell 64(5):891–901
Jackson HJ, Brentjens RJ (2015) Overcoming antigen escape with CAR T-cell therapy. Cancer Discov 5(12):1238–1240
Jackson HJ, Rafiq S, Brentjens RJ (2016) Driving CAR T-cells forward. Nat Rev Clin Oncol 13(6):370–383
Janka GE (2012) Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med 63:233–246
Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ (2010) Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biology Blood Marrow Transpl 16(9):1245–1256
Juillerat A, Marechal A, Filhol JM, Valton J, Duclert A, Poirot L, Duchateau P (2016) Design of chimeric antigen receptors with integrated controllable transient functions. Sci Rep 6:18950
Junghans RP (2010) Is it safer CARs that we need, or safer rules of the road? Mol Ther 18(10):1742–1743
Junghans RP (2012) Phase IB trial redesign to test role of IL2 with anti-PSMA designer T cells to yield responses in advanced prostate cancer. ASCO Meet Abst 30(5_suppl):70
Kalos M (2016) Chimeric antigen receptor-engineered T cells in CLL: the next chapter unfolds. J Immunother Cancer 4:5
Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH (2011) T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 3(95):95ra73
Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB et al (2010) Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363(5):411–422
Katz SC, Burga RA, McCormack E, Wang LJ, Mooring W, Point GR, Khare PD, Thorn M, Ma Q, Stainken BF et al (2015) Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res 21(14):3149–3159
Kershaw MH, Teng MW, Smyth MJ, Darcy PK (2005) Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol 5(12):928–940
Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, White DE, Wunderlich JR, Canevari S, Rogers-Freezer L et al (2006) A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res 12(20 Pt 1):6106–6115
Khalil DN, Smith EL, Brentjens RJ, Wolchok JD (2016) The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 13(5):273–290
Klebanoff CA, Rosenberg SA, Restifo NP (2016) Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 22(1):26–36
Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M (2013) Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31(1):71–75
Kochenderfer JN, Rosenberg SA (2011) Chimeric antigen receptor-modified T cells in CLL. N Engl J Med 365(20):1937–1938 author reply 1938
Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, Maric I, Raffeld M, Nathan DA, Lanier BJ et al (2010) Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116(20):4099–4102
Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, Phan GQ, Hughes MS, Sherry RM et al (2012) B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 119(12):2709–2720
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, Hakim FT, Halverson DC, Fowler DH, Hardy NM et al (2013) Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122(25):4129–4139
Kochenderfer JN, Dudley ME, Kassim SH, Somerville RP, Carpenter RO, Stetler-Stevenson M, Yang JC, Phan GQ, Hughes MS, Sherry RM et al (2015) Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol 33(6):540–549
Kochenderfer J, Somerville R, Lu T, Shi V, Yang JC, Sherry R, Klebanoff C, Kammula US, Goff SL, Bot A et al (2016) Anti-CD19 chimeric antigen receptor T cells preceded by low-dose chemotherapy to induce remissions of advanced lymphoma. ASCO Meet Abstr 34(15_suppl):LBA3010
Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJ (2006) CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res 66(22):10995–11004
Kuppers R, Engert A, Hansmann ML (2012) Hodgkin lymphoma. J. Clin Investig 122(10):3439–3447
Lamers CH, Sleijfer S, Vulto AG, Kruit WH, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E (2006) Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 24(13):e20–e22
Lamers CH, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, Oosterwijk E, Sleijfer S, Debets R, Gratama JW (2011) Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood 117(1):72–82
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, Grupp SA, Mackall CL (2014) Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124(2):188–195
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, Fry TJ, Orentas R, Sabatino M, Shah NN et al (2015a) T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet (London, England) 385(9967):517–528
Lee DW, Stetler-Stevenson M, Yuan CM, Fry TJ, Shah NN, Delbrook C, Yates B, Zhang H, Zhang L, Kochenderfer JN et al (2015b) Safety and response of incorporating CD19 chimeric antigen receptor T cell therapy in typical salvage regimens for children and young adults with acute lymphoblastic leukemia. Blood 126(23):684
Leen AM, Rooney CM, Foster AE (2007) Improving T cell therapy for cancer. Annu Rev Immunol 25:243–265
Letourneur F, Klausner RD (1991) T-cell and basophil activation through the cytoplasmic tail of T-cell-receptor zeta family proteins. Proc Natl Acad Sci USA 88(20):8905–8909
Li G, Wong AJ (2008) EGF receptor variant III as a target antigen for tumor immunotherapy. Expert Rev Vaccines 7(7):977–985
Lim WA, June CH (2017) The principles of engineering immune cells to treat cancer. Cell 168(4):724–740
Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, Cogdill AP, Li N, Ramones M, Granda B et al (2015) Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 75(17):3596–3607
Liu H, Xu Y, Xiang J, Long L, Green S, Yang Z, Zimdahl B, Lu J, Cheng N, Horan LH et al (2017a) Targeting alpha-fetoprotein (AFP)-MHC complex with CAR T-cell therapy for liver cancer. Clin Cancer Res 23(2):478–488
Liu X, Zhang Y, Cheng C, Cheng AW, Zhang X, Li N, Xia C, Wei X, Liu X, Wang H (2017b) CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res 27(1):154–157
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, Rossig C, Russell HV, Diouf O, Liu E et al (2011) Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118(23):6050–6056
Ma Q, Safar M, Holmes E, Wang Y, Boynton AL, Junghans RP (2004) Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate 61(1):12–25
Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, Sergeeva A, Wood MS, Dotti G, Salvado B et al (2016a) A novel TCR-like CAR with specificity for PR1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy 18(8):985–994
Ma JS, Kim JY, Kazane SA, Choi SH, Yun HY, Kim MS, Rodgers DT, Pugh HM, Singer O, Sun SB et al (2016b) Versatile strategy for controlling the specificity and activity of engineered T cells. Proc Natl Acad Sci USA 113(4):E450–E458
Maeder ML, Gersbach CA (2016) Genome-editing technologies for gene and cell therapy. Mol Ther 24(3):430–446
Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S (2000) Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 74:181–273
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF et al (2014a) Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 371(16):1507–1517
Maude SL, Barrett D, Teachey DT, Grupp SA (2014b) Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J (Sudbury, Mass) 20(2):119–122
Maude SL, Teachey DT, Porter DL, Grupp SA (2015a) CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood 125(26):4017–4023
Maude SL, Barrett DM, Ambrose DE, Rheingold SR, Aplenc R, Teachey DT, Callahan C, Barker CS, Mudambi M, Shaw PA et al (2015b) Efficacy and safety of humanized chimeric antigen receptor (CAR)-modified T cells targeting CD19 in children with relapsed/refractory ALL. Blood 126(23):683
Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, Zhao Y, Kalos M, June CH (2013) T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res 1(1):26–31
McGuinness RP, Ge Y, Patel SD, Kashmiri SV, Lee HS, Hand PH, Schlom J, Finer MH, McArthur JG (1999) Anti-tumor activity of human T cells expressing the CC49-zeta chimeric immune receptor. Hum Gene Ther 10(2):165–173
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA (2010) Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18(4):843–851
Nellan A, Lee DW (2015) Paving the road ahead for CD19 CAR T-cell therapy. Curr Opin Hematol 22(6):516–520
Newick K, Moon E, Albelda SM (2016) Chimeric antigen receptor T-cell therapy for solid tumors. Mol Ther Oncolytics 3:16006
O’Hara M, Stashwick C, Haas AR, Tanyi JL (2016) Mesothelin as a target for chimeric antigen receptor-modified T cells as anticancer therapy. Immunotherapy 8(4):449–460
O’Rourke DM, Nasrallah M, Morrissette JJ, Melenhorst JJ, Lacey SF, Mansfield K, Martinez-Lage M, Desai AS, Brem S, Maloney E et al (2016) Pilot study of T cells redirected to EGFRvIII with a chimeric antigen receptor in patients with EGFRvIII+ glioblastoma. ASCO Meet Abstr 34(15_suppl):2067
Park JH, Riviere I, Wang X, Bernal YJ, Yoo S, Purdon T, Halton E, Quintanilla H, Curran KJ, Sauter CS et al (2014) CD19-targeted 19-28z CAR modified autologous T cells induce high rates of complete remission and durable responses in adult patients with relapsed, refractory B-cell ALL. Blood 124(21):382
Park JH, Riviere I, Wang X, Bernal Y, Purdon T, Halton E, Wang Y, Curran KJ, Sauter CS, Sadelain M et al (2015) Implications of minimal residual disease negative complete remission (MRD-CR) and allogeneic stem cell transplant on safety and clinical outcome of CD19-targeted 19-28z CAR modified T cells in adult patients with relapsed, refractory B-cell ALL. Blood 126(23):682
Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM et al (2011) T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19(3):620–626
Pastore S, Lulli D, Girolomoni G (2014) Epidermal growth factor receptor signalling in keratinocyte biology: implications for skin toxicity of tyrosine kinase inhibitors. Arch Toxicol 88(6):1189–1203
Pegram HJ, Park JH, Brentjens RJ (2014) CD28z CARs and armored CARs. Cancer J 20(2):127–133
Pegram HJ, Smith EL, Rafiq S, Brentjens RJ (2015) CAR therapy for hematological cancers: can success seen in the treatment of B-cell acute lymphoblastic leukemia be applied to other hematological malignancies? Immunotherapy 7(5):545–561
Peinert S, Prince HM, Guru PM, Kershaw MH, Smyth MJ, Trapani JA, Gambell P, Harrison S, Scott AM, Smyth FE et al (2010) Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther 17(5):678–686
Porter DL, Levine BL, Kalos M, Bagg A, June CH (2011) Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365(8):725–733
Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, Bagg A, Marcucci KT, Shen A, Gonzalez V et al (2015) Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 7(303):303ra139
Porter DL, Frey NV, Melenhorst JJ, Hwang W-T, Lacey SF, Shaw PA, Chew A, Marcucci K, Gill S, Loren AW et al (2016) Randomized, phase II dose optimization study of chimeric antigen receptor (CAR) modified T cells directed against CD19 in patients (pts) with relapsed, refractory (R/R) CLL. ASCO Meet Abstr 34(15_suppl):3009
Posey AD Jr, Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, Stone JD, Madsen TD, Schreiber K, Haines KM et al (2016) Engineered CAR T cells targeting the cancer-associated Tn-Glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 44(6):1444–1454
Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, Huls MH, Liu E, Gee AP, Mei Z et al (2008) Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med 14(11):1264–1270
Qasim W, Amrolia PJ, Samarasinghe S, Ghorashian S, Zhan H, Stafford S, Butler K, Ahsan G, Gilmour K, Adams S et al (2015) First clinical application of Talen engineered universal CAR19 T cells in B-ALL. Blood 126(23):2046
Ramos CA, Ballard B, Liu E, Dakhova O, Mei Z, Liu H, Grilley B, Rooney CM, Gee AP, Chang BH et al (2015) Chimeric T cells for therapy of CD30+ Hodgkin and non-Hodgkin lymphomas. Blood 126(23):185
Ratner M (2016) Off-the-shelf CAR-T therapy induces remission in child with ALL. Nat Biotechnol 34(1):12
Raufi A, Ebrahim AS, Al-Katib A (2013) Targeting CD19 in B-cell lymphoma: emerging role of SAR3419. Cancer Manag Res 5:225–233
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y (2016) Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. doi:10.1158/1078-0432.CCR-16-1300
Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K, Chen K, Shin M, Wall DM, Honemann D et al (2013) Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther 21(11):2122–2129
Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, Schulman A, Du J, Wang F, Singer O et al (2016) Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc Natl Acad Sci USA 113(4):E459–E468
Romeo C, Seed B (1991) Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell 64(5):1037–1046
Rossig C, Bollard CM, Nuchtern JG, Merchant DA, Brenner MK (2001) Targeting of G(D2)-positive tumor cells by human T lymphocytes engineered to express chimeric T-cell receptor genes. Int J Cancer 94(2):228–236
Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, Lim WA (2016) Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164(4):770–779
Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, Klichinsky M, Aikawa V, Nazimuddin F, Kozlowski M et al (2016) Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Investig 126(10):3814–3826
Ruella M, Kenderian SS, Shestova O, Klichinsky M, Melenhorst JJ, Wasik MA, Lacey SF, June CH, Gill S (2017) Kinase inhibitor ibrutinib to prevent cytokine-release syndrome after anti-CD19 chimeric antigen receptor T cells for B-cell neoplasms. Leukemia 31(1):246–248
Sadelain M (2016) Tales of antigen evasion from CAR therapy. Cancer Immunol Res 4(6):473
Sadelain M, Brentjens R, Riviere I (2013) The basic principles of chimeric antigen receptor design. Cancer Discov 3(4):388–398
Sauter CS, Riviere I, Bernal Y, Wang X, Purdon T, Yoo S, Moskowitz CH, Giralt S, Matasar MJ, Curran KJ et al (2015) Phase I trial of 19-28z chimeric antigen receptor modified T cells (19-28z CAR-T) post-high dose therapy and autologous stem cell transplant (HDT-ASCT) for relapsed and refractory (rel/ref) aggressive B-cell non-Hodgkin lymphoma (B-NHL). ASCO Meet Abstr 33(15_suppl):8515
Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, Bollard CM, Gee AP, Mei Z et al (2011) CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Investig 121(5):1822–1826
Scheuermann RH, Racila E (1995) CD19 antigen in leukemia and lymphoma diagnosis and immunotherapy. Leuk Lymphoma 18(5–6):385–397
Schuster SJ, Svoboda J, Dwivedy Nasta S, Porter DL, Chong EA, Landsburg DJ, Mato AR, Lacey SF, Melenhorst JJ, Chew A et al (2015) Sustained remissions following chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas. Blood 126(23):183
Singh N, Perazzelli J, Grupp SA, Barrett DM (2016) Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med 8(320):320
Slovin SF, Wang X, Borquez-Ojeda O, Stefanski J, Olszewska M, Taylor C, Bartido S, Scher HI, Sadelain M, Riviere I (2012) Targeting castration resistant prostate cancer (CRPC) with autologous PSMA-directed CAR+ T cells. ASCO Meet Abstr 30(15):TPS4700
Slovin SF, Wang X, Hullings M, Arauz G, Bartido S, Lewis JS, Schoder H, Zanzonico P, Scher HI, Sadelain M et al (2013) Chimeric antigen receptor (CAR+) modified T cells targeting prostate-specific membrane antigen (PSMA) in patients (pts) with castrate metastatic prostate cancer (CMPC). ASCO Meet Abst 31(6_suppl):72
Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, Riddell SR (2016) Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 30(2):492–500
Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR et al (2015) Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 5(12):1282–1295
Srivastava S, Riddell SR (2015) Engineering CAR-T cells: design concepts. Trends Immunol 36(8):494–502
Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z (1993) Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol (Baltimore, Md : 1950) 151(11):6577–6582
Tanyi JL, Haas AR, Beatty GL, Morgan MA, Stashwick CJ, O’Hara MH, Porter DL, Maus MV, Levine BL, Lacey SF et al (2015) Abstract CT105: Safety and feasibility of chimeric antigen receptor modified T cells directed against mesothelin (CART-meso) in patients with mesothelin expressing cancers. Cancer Res 75(15 Supplement):CT105
Tanyi JL, Haas AR, Beatty GL, Stashwick CJ, O’Hara MH, Morgan MA, Porter DL, Melenhorst JJ, Plesa G, Lacey SF et al (2016) Anti-mesothelin chimeric antigen receptor T cells in patients with epithelial ovarian cancer. ASCO Meet Abst 34(15_suppl):5511
Tasian SK, Gardner RA (2015) CD19-redirected chimeric antigen receptor-modified T cells: a promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther Adv Hematol 6(5):228–241
Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, Pequignot E, Gonzalez VE, Chen F, Finklestein J et al (2016) Identification of predictive biomarkers for cytokine release syndrome after chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Cancer Discov 6(6):664–679
Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR (2012) Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood 119(1):72–82
Thaci B, Brown CE, Binello E, Werbaneth K, Sampath P, Sengupta S (2014) Significance of interleukin-13 receptor alpha 2-targeted glioblastoma therapy. Neuro Oncol 16(10):1304–1312
Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, James SE, Raubitschek A, Forman SJ et al (2008) Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood 112(6):2261–2271
Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, Lindgren CG, Lin Y, Pagel JM, Budde LE et al (2012) CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 119(17):3940–3950
Torikai H, Cooper LJ (2016) Translational implications for off-the-shelf immune cells expressing chimeric antigen receptors. Mol Ther 24(7):1178–1186
Turtle CJ, Berger C, Sommermeyer D, Hanafi L-A, Pender B, Robinson EM, Melville K, Budiarto TM, Steevens NN, Chaney C et al (2015) Anti-CD19 chimeric antigen receptor-modified T cell therapy for B cell non-Hodgkin lymphoma and chronic lymphocytic leukemia: fludarabine and cyclophosphamide lymphodepletion improves in vivo expansion and persistence of CAR-T cells and clinical outcomes. Blood 126(23):184
Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, Sommermeyer D, Melville K, Pender B, Budiarto TM et al (2016a) CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Investig 126(6):2123–2138
Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X et al (2016b) Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med 8(355):355ra116
US National Library of Science (2016a) ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02614066.
US National Library of Science (2016b) ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02535364.
US National Library of Science (2016c) ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02228096.
US National Library of Science (2016d) ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02030847
US National Library of Science (2016e) ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT02028455.
van der Stegen SJ, Hamieh M, Sadelain M (2015) The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 14(7):499–509
Wang J, Jensen M, Lin Y, Sui X, Chen E, Lindgren CG, Till B, Raubitschek A, Forman SJ, Qian X et al (2007) Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther 18(8):712–725
Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC (2011) Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood 117(6):1888–1898
Wang Y, Zhang WY, Han QW, Liu Y, Dai HR, Guo YL, Bo J, Fan H, Zhang Y, Zhang YJ et al (2014) Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin Immunol 155(2):160–175
Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, Norris AP, Wong CW, Urak RZ, Chang WC et al (2016) Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood 127(24):2980–2990
Wang CM, Wu ZQ, Wang Y, Guo YL, Dai HR, Wang XH, Li X, Zhang YJ, Zhang WY, Chen MX et al (2017a) Autologous T cells expressing CD30 chimeric antigen receptors for relapsed or refractory hodgkin lymphoma: an open-label phase I trial. Clin Cancer Res 23(5):1156–1166
Wang Z, Wu Z, Liu Y, Han W (2017b) New development in CAR-T cell therapy. J Hematol Oncol 10(1):53
Whilding LM, Maher J (2015) ErbB-targeted CAR T-cell immunotherapy of cancer. Immunotherapy 7(3):229–241
Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira AC, Burbridge SE, Box C, Eccles SA, Maher J (2012) Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 32(5):1059–1070
Wu CY, Roybal KT, Puchner EM, Onuffer J, Lim WA (2015) Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350(6258):aab4077
Xu XJ, Tang YM (2014) Cytokine release syndrome in cancer immunotherapy with chimeric antigen receptor engineered T cells. Cancer Lett 343(2):172–178
Younes A, Bartlett NL, Leonard JP, Kennedy DA, Lynch CM, Sievers EL, Forero-Torres A (2010) Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl J Med 363(19):1812–1821
Yun CO, Nolan KF, Beecham EJ, Reisfeld RA, Junghans RP (2000) Targeting of T lymphocytes to melanoma cells through chimeric anti-GD3 immunoglobulin T-cell receptors. Neoplasia 2(5):449–459
Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY (2016) T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res 4(6):498–508
Zhang G, Wang L, Cui H, Wang X, Zhang G, Ma J, Han H, He W, Wang W, Zhao Y et al (2014) Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci Rep 4:3571
Zhang T, Cao L, Xie J, Shi N, Zhang Z, Luo Z, Yue D, Zhang Z, Wang L, Han W et al (2015) Efficiency of CD19 chimeric antigen receptor-modified T cells for treatment of B cell malignancies in phase I clinical trials: a meta-analysis. Oncotarget 6(32):33961–33971
Zhang W, Wang Y, Guo Y, Dai H, Yang Q, Zhang Y, Zhang Y, Chen M, Wang C, Feng K et al (2016a) Treatment of CD20-directed chimeric antigen receptor-modified T cells in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an early phase IIa trial report. Signal Transduct Target Ther 1:16002
Zhang Y, Zhang W, Dai H, Wang Y, Shi F, Wang C, Guo Y, Liu Y, Chen M, Feng K et al (2016b) An analytical biomarker for treatment of patients with recurrent B-ALL after remission induced by infusion of anti-CD19 chimeric antigen receptor T (CAR-T) cells. Sci China Life Sci 59(4):379–385
Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL, Chew A, Carroll RG, Scholler J, Levine BL et al (2010) Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res 70(22):9053–9061
Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, Plotkin J, Sadelain M (2015) Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer cell 28(4):415–428
Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M (2010) Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther 18(2):413–420
ACKNOWLEDGEMENTS
This research was supported by the grants from the National Natural Science Foundation of China (Grant No. 81230061 to WDH) and the Science and Technology Planning Project of Beijing City (No. Z151100003915076 to WDH) and the National Key Research and Development Program of China (No. 2016YFC1303501 and 2016YFC1303504 to WDH).
COMPLIANCE WITH ETHICS GUIDELINES
Zhenguang Wang, Yelei Guo, and Weidong Han declare that they have no conflict of interest. This article does not contain any studies with human or animal subjects performed by the any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Z., Guo, Y. & Han, W. Current status and perspectives of chimeric antigen receptor modified T cells for cancer treatment. Protein Cell 8, 896–925 (2017). https://doi.org/10.1007/s13238-017-0400-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13238-017-0400-z