
Abstract
Nuclear epigenetics has been a major area of research through decades, involved in the pathogenesis of several human diseases. Current studies brought to light the role of mitochondrial epigenetics in disease initiation and progression. Mitochondrial DNA methylation mark, considered as one of the major epigenetic modifications, has been lately studied in various health outcomes. Herein, this review aims to explore the effects of several environmental factors (including air particulate matter, chemical compounds and heavy metal exposures) and to find their role in influencing diseases through epigenetic alteration, especially focusing on mitochondrial DNA methylation. However, little is known about mitochondrial gene regulation with disease association, which opens up a novel area of research. In addition to this, potential epi-therapeutic approaches have also been discussed. Further, in-depth mito-epigenetic studies will be helpful for clinical research.
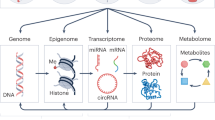
Graphical Abstract
Schematic representation of mitochondrial DNA methylation regulation in response to environmental toxicants and diseases.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epigenetics can be explained as the study related to alteration in gene expression with unchanged DNA sequences. Presently, the field of epigenetics is rapidly expanding. Understanding its role in various diseases and therapeutic interventions is an emerging field of research worldwide [22, 62]. Alteration in expression of target genes depends on the epigenetic status of the chromatin structure, involving DNA promoter methylation or hydroxymethylation, histone modifications, promoter–enhancer interactions, and noncoding RNA–mediated regulation. De-regulation of epigenetic machinery leads to inappropriate onset of genes causing either activation or inhibition of downstream protein complexes [22, 62].
Present researches on epigenetics can be divided into two key areas: epigenetic-regulation of nuclear DNA and mitochondrial DNA [89]. Epigenetic-regulation of nuclear DNA involves DNA methylation, post-translational histone modifications and non-coding RNAs; whereas epigenetic-regulation of mitochondrial DNA seems to be alike in the aspect of DNA methylation and noncoding RNAs but lacks histone moieties. Previously it has been established that mitochondrial epigenetic-mechanisms in coordination with nuclear epigenetic regulation influence transcription activation or repression of crucial genes and protein synthesis [20].
Mitochondria possess their own genome, along with its transcription-translation machinery; communicates intricately with the nuclear genome for maintaining normal cellular homeostasis [20]. Human mtDNA consists of ∼16,569 base pairs (bp) double stranded circular DNA that encompasses 37 genes. The Heavy strand (or H-strand) consisting of 28 genes is guanine rich; whereas the Light strand (or L-strand) consisting of 9 genes is cytosine rich. Out of these, 13 genes encode for polypeptides that comprises the electron transport chain (ETC). These include 7 mitochondrial genes encoding for complex I subunit (MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND4L, MT-ND5, MT-ND6), one for complex III subunit (MT-CYB), three for complex IV subunits (MT-CO1, MT-CO2, MT-CO3) and two for complex V subunits (MT-ATP6, MT-ATP8), along with two ribosomal RNAs (MT-RNR1 and MT-RNR2) and 22 transfer RNAs [20, 26, 90]. Nuclear DNA possess single promoter for the transcription of a target gene, whereas total mtDNA has only three promoter regions (LSP, HSP1 and HSP2) responsible for transcribing all the genes from both L-strand and H-strand. Mitochondrial displacement loop (D-loop) is a unique region which contains all three promoters along with the replication start site (or RSS) controlling mitochondrial gene expression. Replication and transcription of mtDNA is regulated by multiple nuclear encoded proteins, like TFAM, POLRMT, TFB1M, TFB2M and mTERF etc. [90].
Based on previously established facts, mitochondrial epigenetics comprises of four areas: mtDNA methylation/hydroxymethylation, mitochondrial nucleoid modifications, mitochondrial RNA modifications and mtDNA-derived or nuclear DNA-derived non-coding RNA modulations during mtDNA-encoded gene transcription and protein synthesis [26, 90]. In this review, we highlighted the role of mtDNA methylation in various diseases including cancer and in exposures to environmental pollutants in order to provide an in-depth mechanistic insight.
Mitochondrial epigenetics
Despite decades of research work, mitochondrial epigenetics remained a controversial area of study. Major modifications in mitochondrial epigenome include DNA methylation or hydroxymethylation, demethylation and alteration in mitochondrial non-coding RNAs [16, 43, 68, 88]. However, as in nuclear genome, mitochondrial genome lacks histone packaging; therefore, post translational histone modifications are not a part of mitochondrial epigenetic regulation as in nuclear epigenetics. It is already established that, there exists a cross-talk between mitochondria and nucleus which plays an important role in maintaining overall cellular homeostasis. Key regulatory genes for biogenesis, fission-fusion, mitophagy and also mtDNA methyltransferase enzymes (responsible for mito-epigenetic modification) are encoded by nuclear genome. In response to specific cellular micro-environmental condition, synchronized anterograde (nuclear to mitochondrial) and retrograde (mitochondrial to nuclear) signalling pathways are found to be activated. Epigenetic changes in mitochondrial DNA may induce alterations in the nuclear epigenome leading to an array of downstream activities including differential gene expression, genetic instability and tumorigenesis [34, 61, 71]. Furthermore, in response to extracellular signals like environmental exposure towards toxic substances, mitochondria may play an essential role in modification of nuclear epigenetic regulation, which in turn affect mitochondrial functionality [68]. Thus, the reciprocity between nuclear and mitochondrial epigenome thought to be a crucial factor in understanding the aspects of mitochondrial functionality under normal and pathological conditions [51].
Mitochondrial DNA methylation and associated methyltransferases
Earlier, DNA methylation occurring in the mitochondrial epigenome has been a topic of debate since 1970s. In 1973, Nass MM had first reported the evidences of mtDNA methylation in in-vivo and in-vitro model systems [73, 74]. Gradually, technical advancements along with new methodologies have evolved for quantifying methylation on CpG- or non-CpG-specific sites that substantiated the existence of mtDNA methylation mark [90]. The conclusive evidence suggesting the presence of methylated CpG islands came from the study of Infantino and co-workers [44] who used a mass spectrometry-based system which overcame the technical limitations of former studies. On the contrary, few studies showed the absence of mtDNA methylation mark [27, 42, 59, 69]. Several factors such as circular nature of mitochondrial genome, different experimental model systems, disease occurrences, exposures to various external and internal stimuli, stressors etc. play a vital role in determining the presence or absence of mitochondrial cytosine methylation [27, 42, 59, 69].
DNA methylation occurs on cytosine residues that are followed by guanine residues near the promoter regions or transcriptional start sites (CpG islands or CpG clusters or CpG sequences) catalyzed by DNA methyltransferases (DNMTs) in presence of methyl donor, S-adenosylmethionine or SAM [86, 87]. Presence of mtDNA methyltransferase activity was first recorded by Nass MM [73]. Nuclear DNA methyltransferase 1 (DNMT1), the most abundant methyltransferase, is translocated to the mitochondria via signalling by a mitochondrial targeting sequence (MTS) located immediately upstream of the translational start site (TSS). MTS has been found to be conserved across mammals, encodes a peptide required for translocation of nuclear DNMT1 to mitochondrial matrix. Mitochondrial DNMT1 (mtDNMT1) binds to mtDNA, proving the presence of mtDNMT1 in the mitochondrial matrix. However, MTSs are not present in the primary sequences of de novo methyltransferases DNMT3A and DNMT3B that are specifically found in mitochondria [87].
Mitochondrial DNA methyltransferase 1 or mtDNMT1 plays a significant role in the epigenetic mechanism of mtDNA methylation and in the regulation of transcription of mitochondrial genes. MtDNMT1 regulates the mitochondrial gene expression through mtDNMT1 binding to the mitochondrial D-loop control region and promoting transcription of target genes [27, 42, 59, 69, 87]. Interestingly, recent studies have highlighted the role of DNMT1 mutations and its effect on mitochondrial epigenome [36, 67]. Dominant mutations in Replication foci targeting sequence (RFTS), present in the N-terminal domain of DNMT1 has been previously reported, deletion of which has been shown to alter DNMT1-dependent DNA methylation in cancerous cells [96]. Currently, a study by Maresca et al., 2020 showed that DNMT1 mutations lead to mitochondrial hyper-function and increased oxidative stress, resulting in neurodegeneration [67]. Also, DNMT1 mutation was reported to cause epigenetic de-repression of the γ-globin gene in β-thalassemia elevating the fetal hemoglobin levels [36]. In addition to this, mutations in DNMT3A and DNMT3B have also been reported to cause significant alterations in mtDNA methylation thereby affecting mitochondrial functionality [24, 31, 48, 52, 78, 80].
Apart from mtDNA methylation, hydroxymethylation has also been detected in mitochondrial genome. MtDNA hydroxymethylation occurs through direct addition of 5-hydroxymethyl group to cytosine residues catalyzed by mtDNMT1. Also, mitochondrial DNMT3A and DNMT3B contribute to mtDNA hydroxymethylation. Current reports also suggested the existence of N6-Deoxyadenosine methylation in mammalian mtDNA [40]. METTL4, a mammalian methyltransferase mediated mtDNA N6-Deoxyadenosine (6mA) methylation promotes reduced mtDNA transcription along with a decreased mitochondrial biogenesis and mtDNA copy number [40].
Technologies involved in the detection of mtDNA methylation
MtDNA methylation is associated with various physiological aspects such as cardiovascular diseases, neurodegenerative diseases, cancer and many more. Various technical approaches are involved for the assessment of methylation of mtDNA. The most widely used technique for the study of mtDNA methylation is bisulphite sequencing. Prior to this, bisulphite conversion of mtDNA has been done that converts the unmethylated cytosine into uracil, whereas the methylated cytosines remain unchanged. The bisulphite converted DNA undergoes PCR amplification prior to bisulphite sequencing procedure. The methylation level is assessed using the percentage of methylated cytosine over the total number of methylated and non-methylated cytosine residues. MtDNA methylation analysis has been commonly done through bisulphite PCR pyrosequencing [46]. Global DNA m5C content can be assessed using ELISA assay by capture and detection antibodies [21, 37]. Such technique allows the detection of methylated mtDNA fluorimetrically by measuring the intensity of fluorescence. MtDNA methylation can also be evaluated using targeted next-generation bisulphite sequencing (TNGBS) [87, 98]. MassARRAY platform also can be used to detect the methylation of mtDNA which is more efficient as compared to methylation specific PCR and bisulphite sequencing. hMeDIP Assay can be performed using hMeDIP kit which provide three spikes or internal immunoprecipitation controls such as, an unmethylated DNA, a methylated DNA, and a hydroxymethylated DNA [6]. Post bisulphite conversion, the DNA fragments can be hybridized to the Infinium Human Methylation 450 BeadChip array [95]. In addition to this, there are targeted bisulphite deep sequencing and shotgun bisulphite deep sequencing methods to assess the methylation pattern of mtDNA [75]. Another option for evaluation mtDNAmethylation pattern is to use immunoprecipitation reaction using specific antibodies for 5mC followed by microarray hybridization technique [90]. Sequence specific restriction enzymes may be helpful in detecting the methylation sites residing in mtDNA as these enzymes are inhibited by 5mC, they can indicate the methylation pattern more effectively [90]. In combination with high-throughput sequencing, methylation specific restriction enzyme digestion has also been reported for the detection of specific methylation sites [38]. An alternative approach to hybridization and sequencing is to use mass spectrometry; however, it requires gene specific amplification. Also, nanopore sequencing is one of the most effective methods which allow direct sequencing of 5mC without the need for the bisulphite conversion of mtDNA [89, 90]. New approaches such as ELISA along with restriction digestion procedure results in better yield of mitochondrial methylation pattern [90]. Recent advancements include Bisulphite-PCR–single stranded DNA conformation polymorphism (SSCP) and methylation sensitive-high resolution melting (MSHRM) that uses a CFX96 Real-Time PCR detection system [70, 92]. Recently identified easiest methods for mtDNA methylation analysis is Illumina Infinium Bead Chip array and Methylation Epic Bead Chip [79]. Interestingly, it has been found that the closed circular topology of mtDNA inhibits bisulphite conversion resulting in false-positive detection of mtDNA methylation [75]. At times, incomplete bisulphite conversion produces false-positive measurement which becomes a major drawback [75]. The adequacy of mtDNA methylation can be affected by the purity of the bisulphite converted pyrosequencing templates, presence of incompletely converted DNA and the topology of the mtDNA. To avoid these discrepancies and false-positive results it is suggested to use selective primers and the evaluation of conversion efficiency must be calculated accurately for identification and analysis of mtDNAmethylation [75].
Regulation of mtDNA methylation in response to environmental pollutants
Humans are being continuously exposed to environmental toxins in daily life which is a growing concern affecting cellular and genomic integrity. Nuclear and mitochondrial DNA damage is the most widely studied impact of environmental or occupational exposures to xenobiotics [12, 23, 32, 65, 66]. Mitochondria play a crucial role in the cellular response to environmental pollutants or stressors, including both intrinsic and extrinsic pathways. Earlier, few reports have revealed how mitochondrial epigenome gets frequently altered primarily through DNA methylation in response to various environmental toxins [2, 53, 72, 81, 83]. Differential tissue specific mitochondrial DNA methylation has been previously reported in placenta, brain, liver and blood cells as a result of environmental exposures involving heavy metal exposures, air particulate matter, various chemical compounds [2, 53, 72, 81, 83]. In the following sections, a detailed account of association of altered mtDNA methylation and its downstream cellular effects have been discussed.
MtDNA methylation in response to air particulate matter exposure
Epigenetic mechanism tune gene expression in response to various environmental and occupational pollutants such as different air particulate matters [3, 10, 13, 19, 84]. Such exposuresare associated with several human diseases including neurodegeneration, diabetes, obesity, cardiovascular diseases and different types of cancers. Environmental exposures to air particulate matter (PM) or smoke has been previously reported to induce ROS generation that is shown to cause oxidative stress and promote mitochondrial damage. Evidences support alteration in methylation pattern of mitochondrial DNA affects mitochondrial biogenesis [15, 17, 47, 94].
Interestingly, some studies (Table 1) on PM exposure have previously reported a higher level of mtDNA methylationalong with increase in mtDNA copy number [15, 47, 94]. Earlier, a study was performed on steel workers exposed to metal-rich particulate matter demonstrated higher mtDNA methylation(in MT-TF and MT-RNR1 genes) detected through bisulphite pyrosequencing and higher mtDNA copy number [16]. Consistent with the earlier results, exposures to PM (2.5) induced higher oxidative stress and mtDNA methylation that affects mitochondrial biogenesis with increased risk for cardiovascular diseases [15]. However, mother-newborn pairs exposed to PM (2.5) during the gestational period revealed hypermethylation of placental mtDNA tested through bisulphite pyrosequencing which resulted in lower mtDNA content [46]. An in-vivo and in-vitro study performed by Breton et al., 2019 reported that exposure to PM (2.5) caused the hypermethylation of mitochondrial D loop regions that affect mitochondrial respiratory function and mtDNA in SH-SY5Y cells and in liver tissues from C57B1/6 males [10].
MtDNA methylation in response to chemical exposures
Exposure to different chemical compounds has impactful effects on mtDNA methylation and thus in turn can influence various disease progressions (Table 2). Earlier, an in vivo study by Byun et al., 2015 [13] experimentally demonstrated the exposures to PBDEs (Polybrominated diphenyl ethers) and mtDNA methylation in rats PBDE induced ROS generation and hypomethylation of mitochondrial Mt-CO2 (Cytochrome c oxidase gene) led to mitochondrial dysfunction in the frontal brain of rats [13]. Maternal smoking during pregnancy has been reported to affect placental trophoblast tissues, placental activity and foetal development by altering the methylation pattern of mtDNA identified by bisulphite pyrosequencing [3]. Doxorubicin, a well-known chemotherapeutic drug, was found to be associated with decreased SAM levels and consequently the hypomethylation of PGC1α and TFAM genes [29]. Next generation bisulphite sequencing (TNGBS) and ELISA of 5mc DNA content has helped the identification of methylation alteration of mtDNA by the psycho stimulant drug cocaine [25]. Cocaine has been reported to alter mtDNA methylation pattern, lowering the protein levels of DNMT3A and DNMT3B affecting mitochondrial biogenesis [25]. Also, tobacco smoking and air pollution during pregnancy has been reported to cause epigenetic modifications of mtDNA. Hypermethylation of mtDNA in two regions of displacement loop control region (D-loop and LDLR2) has been reported to lower the mtDNA content in placental tissues [94]. Exposures to PAH (Polycyclic aromatic hydrocarbons), also caused significant alterations in the expression of epigenetic modifiers and thus is responsible for the altered level of methylation of mtDNA. Methyl Flash Global DNA methylation (5-mC) ELISA is used for the determination of this epigenetic modification of mtDNA [7].
However, the exact mechanisms responsible for changes in mitochondrial methylation due to different exposures need to be investigated and further studies are needed to substantiate the role of DNA methylation as potential mechanism for different adverse health effects and toxicities.
MtDNA methylation in response to heavy metal exposure
Environmental or occupational heavy metal exposures are associated with diverse mitochondrial dysfunctions. Extensive research has been performed in the field of heavy metal exposure and nuclear DNA epigenetics. However, information regarding how heavy metal contaminants affect mitochondrial epigenome through DNA methylation remains very limited (Table 3). Previous research work demonstrated the role of mtDNA hypomethylation through Mass ARRAY Platform in response to chromium in chrome-plate workers. The methylation levels of MT-TF and MT-RNR1 genes were negatively associated with blood Cr ion concentrations; however, no change in mitochondrial copy number was observed [58]. A recent study by a group reported a significant association between arsenic contamination and hypomethylation (of D-loop region and ND6) in exposed individuals, as detected through Methylation specific PCR. Hypomethylation of D loop and ND6 led to the transcriptional activation of mitochondrial genes along with higher mitochondrial copy number in arsenic exposed individuals [2, 81]. Consistent with the above results, significant hypomethylation of both TFAM and PGC1α in arsenic exposed individuals along with the higher level of gene expression and increased mtDNA copy number was also observed [83]. Transcriptional activation of these crucial genes are important mediating factors for mitochondrial biogenesis and are associated with tumour cell proliferation and development of arsenical skin cancer [81, 83]. Another study have reported Cd2+ and Cr6+ induced methylation in the mitochondrial DNA in an invertebrate, Exopalaemon carinicauda. Heavy metal stress in E. Carinicauda has resulted in been reduced gene expression through mtDNA methylation [63].
Mitochondrial epigenetics and its relation with different health disorders
Nuclear epigenetics and its relation with various diseases have been studied thoroughly; however, the association between mitochondrial epigenetics and different diseases has not been given much importance. Here in, we have tried to shed light over this area to look into various means of association between altered mtDNA methylation and different human diseases (Table 4). Mitochondrial epigenetic alterations have been linked to various disease types including Type 2 Diabetes mellitus, neurological disorders (Alzheimer’s disease, Parkinson disease), Down Syndrome, NASH (Nonalcoholic Steatohepatitis), cardiovascular diseases (CVD), cancer etc. [5, 8, 45, 55, 77]. Increased oxidative stress has been shown to damage to mitochondrial genome and epigenome leading to mitochondrial dysfunction as observed in neurodegenerative disorders [8, 47, 97].
Mitochondrial DNA methylation and its association in disease outcomes have evolved as a new area of research in the last few years. Higher mtDNA methylation was observed in ND6 and D loop region of mitochondria during the early stage of diabetes through Methylation specific-PCR. Altered mtDNA methylation was found to be responsible for the development of insulin resistance in individuals with diabetes [99, 100]. The abnormalities in methyl group donors such as S-adenosylmethionine (SAM) and mtDNA methylation status probably led tomitochondrial dysfunction as widely documented through mass spectrometry in patients with Down syndrome [44]. Higher mtDNA methylation at ND6 region due to high level of DNMT1 mRNA in the liver of NASH (Nonalcoholic steatohepatitis) patients has been reported by Pirola et al., 2013 [77] along with increased oxidative stress. Hypermethylation of ND6 gene in the mitochondrial genome caused its transcriptional inactivation that promoted mitochondrial dysfunction. Higher mitochondrial methylation in platelets has been reported to promote the dysfunction of mitochondria and in turn has been shown tobe responsible for platelet dysfunction and cardiovascular risks [5]. These mitochondrial epigenetic de-regulations are associated with the prognosis of the various diseases. A greater understanding of how DNA methylation in these regions of mitochondria affects mtDNA genes, evolution that needs to be investigated and pharmacologic manipulations is needed which could provide better insights to the patho-physiology of the diseases.
Mitochondrial DNA methylation and cancer
Nuclear epigenetics and its association with diseases especially cancer has been studied widely and given much importance till date; however, the field of mitochondrial epigenetics and its relation with cancer has not been studied and reported thoroughly. Herein, we have made an attempt to elaborate the relationship between mtDNA methylation and cancer progressions (Table 5) elucidated in recent studies [28, 33, 45, 91, 93]. Previously, studies have reported that colorectal cancer tissues with hypomethylated mitochondrial D-loop might be associated with higher expression of mtDNA along with higher mtDNA copy number [28, 33]. MtDNA methylation study was done using Methylation specific PCR and mitochondrial copy number was estimated using quantitative PCR (qPCR). A study by Feng et al., 2012 [28] showed increased expression of ND2 (a subunit of NADH, encoded by mtDNA) is associated with hypomethylation of mtDNA D-loop region, a crucial factor for mtDNA replication and transcription. Hypomethylation of D loop region is associated with the binding of TFAM on mtDNAand thus the expression of ND2 is upregulated leading to oxidative phosphorylation which is required for the enhanced growth of cancer tissues [28]. Consistent with the earlier results, similar finding was observed in colorectal cancer cases and Caco-2 cell line (colorectal cancer cell line) in a study by Gao et al., 2015 [33]. In this study, the demethylation of the D-loop region has facilitated the binding of the TFAM thus increasing the mtDNA expression i.e., the upregulation of ND2. Increased ND2 expression is responsible for higher oxidative phosphorylation thus enhancing the growth of the colorectal cancer cells [33]. Further validation was done by a DNA hypomethylating agent, 5-aza-2’-deoxycytidine (5-AZA). It was performed to examine the effect of DNA methylation on the mtDNA copy number and the biological behaviours of cells by Sequenom MassARRAY platform using various colorectal cell lines [93]. 5-AZA treatment induced hypomethylation of mitochondrial D loop region at specific sites on CpG islands were found to be associated with higher mtDNA copy number that in turn is probably a triggering factor for increased cell proliferation in colorectal cancer cell lines [93]. Similarly, in breast cancer, it has been revealed that DNA methylation in D loop region is maternally inherited and eight aberrant methylation sites are associated with cancer progression detected through whole genome DNA methylation array and bisulphite sequencing[39]. According to a study by Lee et al., 2015 [54], cancer stem-like cells were found to be hypermethylated at exon 2 of the human mtDNA-specific polymerase (DNA polymerase gamma A (POLGA)) that accounted for low mtDNA copy number. Also, a study by Sun et al., 2018 [91], revealed a negative correlation between mtDNA methylation and mtDNA copy number during the progression of tumorigenesis in osteosarcoma and glioblastoma. Mitochondria, being a prognostic marker in carcinogenesis, further studies on the relationship between mtDNAepigenetic regulation and cancer should be clearly elucidated to better understand the clinical significance, function and mode of actions of mitochondrial epigenomein response to disease progression.
Mitoepigenetics, especially mtDNA methylation directly regulate gene expression, which was previously studied in different cancers [28, 33, 39, 54, 91, 93]. MtDNA possess only 13 protein coding genes and all of them encode the subunits of respiratory complexes associated with ETC. As a result, any small change in these gene expressions affects the cellular respiration. For malignant transformation and cancer progression, alterations in glucose metabolism pathway are one of the major phenomena that have been reported earlier [14, 30]. Up-regulation in the glycolytic pathway under hypoxic conditions is an important measure for tumour cell survival. Interestingly, even in the presence of sufficient oxygen, tumour cells preferentially use glycolysis rather than oxidative phosphorylation for energy production known as Warburg effect, leading to a shift in metabolism from aerobic respiration toward glycolysis [11, 30, 35]. This occurs via the induction of HIF-1 (hypoxia-inducible factor-1) which was shown to suppress mitochondrial respiratory activity, as a consequence, to suppress Krebs cycle and mitochondrial respiration [50, 76]. Also, mitoepigenetics may play essential role in mitochondria mediated biological processes. Alteration of mitoepigenetics alters the mtDNA encoded proteins which play essential roles in ETC/OXPHOS including glucose, lipid and amino acid metabolism [50, 76]. MtDNA of cancer stem-like cells has shown hypermethylation and low mtDNA copy number which provokes them to use glycolysis for cell proliferation [55]. It is reported that methylation of mtDNA may differentially suppress mtDNA encoded genes that are necessary for oxidative phosphorylation [85]. Oxidative stress such as ROS can trigger cellular proliferation and carcinogenesis by inducing mutation in mtDNA which further reduce the efficiency of ETC and OXPHOS [18, 56, 60]. Taken together, changes in mitoepigenetics affect downstream gene expression, which in turn may cause altered cellular respiration associated with malignant transformation under certain circumstances.
Therapeutic interventions targeting mtDNA methylation
Epigenetic regulation has a reversible mode of action and can be regulated through direct epigenetic modifications. Several on-going drug therapies under research might provide a novel way to regulate mitochondrial epigenetic regulation particularly focussing on mitochondrial DNA methylation mark. Though several therapeutic drugs are validated that Potential ‘epitherapeutic’ drugs that are currently available include 5-Azacytosine, Decitabine, Guadecitabine, Belinostat, Panobinostat, Vorinostat, and Romidepsin have shown promise in clinical and preclinical trials [49]. Azacitidine and Decitabine have been approved by FDA (Food and Drug Administration) and European Medicines Agency (EMA), are known to function as potent DNA demethylating agents [1]. Certain dietary components play a significant role and have a promising field of research in modulating epigenetic phenomenon in lessening the disease burden. Specific dietary factors and supplements can activate or inhibit DNA methyltransferase activity directly or indirectly to influence mitochondrial DNA methylation [9, 41].Previous studies have suggested the role of folate and Vitamin B12 supplementation that can regulate mitochondrial DNA methylation [64]. Few DNA methyltransferase inhibitors (DNMT inhibitors) include dietary polyphenols, (-)-epigallocatechin-3-gallate (EGCG, from green tea), genistein (from soybean) and isothiocyanates (from plant foods), may be used for cancer prevention and therapy [57]. Implementation of these dietary factors in clinical trials against mitochondrial epigenetic de-regulations might prove to be helpful for therapeutic requirements. An important study done by Lee et al., 2015 [54] have demonstrated that cancerous cells with hypermethylated human mitochondrial POLGA region at exon 2 has been demethylated by applying DNA demethylating agent, 5-azacytidine. Various therapeutic drugs that are in medical practice have been reported to trigger mitochondrial toxicity leading to a wide range of clinical symptoms; hence development of non-toxic mitochondrial epitherapeutic drugs must be a primary concern. Further in-depth high-throughput studies are warranted in this field along with several preclinical and clinical trials and accurate validation is required to obtain maximum health benefits.
Discussion
DNA methylation is a vital epigenetic mark affecting our physiological and biochemical process. Methylation-demethylation phenomenon influence gene expression of crucial signalling pathways required for normal functioning of cells. This review primarily aims to bring a holistic picture of the crosstalk between environmental factors, lifestyle disorders and adverse health outcomes including cancer with altered mtDNA methylation. MtDNA methylation is largely influenced by various environmental toxicants such as airborne particulate matter, exposures to heavy metals or chemical compounds or diseases [87]. These factors perturb the normal cellular homeostasis by genetic and epigenetic de-regulation of mitochondrial genome affecting the transcriptional-translational machinery and protein synthesis [4]. Mitochondrial dysfunction indirectly elevates ROS generation leading to an increased oxidative stress and in-turn damage to the mitochondrial genome [17]. However, in-depth mechanistic insight about how these exposures alter the methylation pattern of mitochondrial DNA needs to be explored more in future studies. A global (country-wise) representation of methylation changes in mitochondrial genome in human studies has been shown (Fig. 1).

A global (Country-wise) representation of methylation changes in mitochondrial DNA in human studies
Environmental or occupational exposure to different chemical compounds or drugs, heavy metal pollutants possesses potential risk factors for alterations in mitochondrial epigenome through differential mtDNA methylation pattern that affect the expression of significant mitochondrial genes [12, 53, 66]. Extensive studies are needed to comprehend the exact mechanisms responsible for methylation of mitochondrial genes to understand the role of ‘methylation mark’ as a potential risk factor. Previous studies have reported the association between mtDNA methylation and altered mtDNA copy number in response to various diseases, environmental exposures and cancer. Both hyper and hypo methylation of specific regions in mtDNA was found to be associated with higher mtDNA copy number [47, 70]. Those findings clearly indicate towards a complex mechanism involving not only the mitochondrial epigenetic modifications but also involvement of other confounding factors like nucleus-mitochondrial retrograde and anterograde signalling, accumulation of ROS, rate of mitochondrial biogenesis, fission-fusion, mitophagy status and individual genetic make-up, environmental exposure, cancer microenvironment (in case of malignant transformations) etc. in mtDNA copy number regulation. However, the exact intracellular molecular signalling pathway and identification of regulatory factors still remain obscure as majority of the studies regarding this area were associative in nature. Evident from previous studies, that differential mtDNA methylation pattern can be used as a prognostic marker for cancer detection [28, 33, 93]. MtDNA methylation also play crucial roles in several other diseases like cardiovascular diseases, neurodegenerative diseases etc. which revealed that how disease progression is often associated with the altered mtDNA methylation pattern leading to an alteration in mitochondrial structural and functionality [5, 44, 77]. As epigenetic changes are known to be reversible, accurate implication of epitherapeutic drugs might reverse the alterations associated with neurodegenerative diseases, cancers etc.
Conclusions
MtDNA methylation plays an important role in disease development and progression. Further in-depth studies are needed to comprehend the alterations of mtDNA methylation pattern in response to various environmental stressors influencing various disease outcomes. Detailed research in the evolving field of mtDNA methylation may pave its way towards the development of potential epitherapeutic approaches in the immediate future.
Abbreviations
- MtDNA:
-
Mitochondrial DNA.
- D-Loop:
-
Displacement Loop
- TFAM:
-
Transcription and mtDNA Maintenance Factor
- TERF:
-
Mitochondrial Transcription Termination Factor
- POLRMT:
-
Mitochondrial RNA polymerase
- TFB1M:
-
Mitochondrial Transcription Factor B1
- TFB2M:
-
Mitochondrial Transcription Factor B2
- L-strand:
-
Light Strand
- H-strand:
-
Heavy strand
- ETC:
-
Electron Transport Chain
- OXPHOS:
-
Oxidative phosphorylation
- NADH:
-
Nicotinamide Adenine Dinucleotide Hydrogen
- SAM:
-
S-adenosylmethionine
- DNMT:
-
DNA methyltransferases
- ROS:
-
Reactive Oxygen Species
References
Ahuja N, Easwaran H, Baylin SB. Harnessing the potential of epigenetic therapy to target solid tumors. J Clin Investig. 2014;124(1):56–63.
Ameer SS, Xu Y, Engström K, Li H, Tallving P, Nermell B, Boemo A, Parada LA, Peñaloza LG, Concha G, Harari F. Exposure to inorganic arsenic is associated with increased mitochondrial DNA copy number and longer telomere length in peripheral blood. Front Cell Dev Biol. 2016;4:87.
Armstrong DA, Green BB, Blair BA, Guerin DJ, Litzky JF, Chavan NR, Pearson KJ, Marsit CJ. Maternal smoking during pregnancy is associated with mitochondrial DNA methylation. Environmental epigenetics. 2016;2(3).
Aseervatham G, Sivasudha T, Jeyadevi R, Arul Ananth D. Environmental factors and unhealthy lifestyle influence oxidative stress in humans—an overview. Environ Sci Pollut Res. 2013;20(7):4356–69.
Baccarelli AA, Byun HM. Platelet mitochondrial DNA methylation: a potential new marker of cardiovascular disease. Clin Epigenetics. 2015;7(1):1–9.
Bellizzi D, D’Aquila P, Scafone T, Giordano M, Riso V, Riccio A, Passarino G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013;20(6):537–47.
Bhargava A, Kumari R, Khare S, Shandilya R, Gupta PK, Tiwari R, Rahman A, Chaudhury K, Goryacheva IY, Mishra PK. Mapping the mitochondrial regulation of epigenetic modifications in association with carcinogenic and noncarcinogenic polycyclic aromatic hydrocarbon exposure. Int J Toxicol. 2020;39(5):465–76.
Blanch M, Mosquera JL, Ansoleaga B, Ferrer I, Barrachina M. Altered mitochondrial DNA methylation pattern in Alzheimer disease–related pathology and in Parkinson disease. The American journal of pathology. 2016 Feb 1;186(2):385 – 97.
Bollati V, Favero C, Albetti B, Tarantini L, Moroni A, Byun HM, Motta V, Conti DM, Tirelli AS, Vigna L, Bertazzi PA. Nutrients intake is associated with DNA methylation of candidate inflammatory genes in a population of obese subjects. Nutrients. 2014;6(10):4625-39. 639.
Breton CV, Song AY, Xiao J, Kim SJ, Mehta HH, Wan J, Yen K, Sioutas C, Lurmann F, Xue S, Morgan TE. Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion. 2019;46:22–9.
Brinker AE, Vivian CJ, Beadnell TC, Koestler DC, Teoh ST, Lunt SY, Welch DR. Mitochondrial haplotype of the host stromal microenvironment alters metastasis in a non-cell autonomous manner. Cancer Res. 2020;80(5):1118–29.
Byun HM, Baccarelli AA. Environmental exposure and mitochondrial epigenetics: study design and analytical challenges. Hum Genet. 2014;133(3):247–57.
Byun HM, Benachour N, Zalko D, Frisardi MC, Colicino E, Takser L, Baccarelli AA. Epigenetic effects of low perinatal doses of flame retardant BDE-47 on mitochondrial and nuclear genes in rat offspring. Toxicology. 2015;328:152–9.
Byun JY, Kim MJ, Eum DY, Yoon CH, Seo WD, Park KH, Hyun JW, Lee YS, Lee JS, Yoon MY, Lee SJ. Reactive oxygen species-dependent activation of Bax and poly (ADP-ribose) polymerase-1 is required for mitochondrial cell death induced by triterpenoid pristimerin in human cervical cancer cells. Mol Pharmacol. 2009;76(4):734–44.
Byun HM, Colicino E, Trevisi L, Fan T, Christiani DC, Baccarelli AA. Effects of air pollution and blood mitochondrial DNA methylation on markers of heart rate variability. J Am Heart Assoc. 2016;5(4):e003218.
Byun HM, Panni T, Motta V, Hou L, Nordio F, Apostoli P, Bertazzi PA, Baccarelli AA. Effects of airborne pollutants on mitochondrial DNA methylation. Part. Fibre Toxicol. 2013;10(1):1–8.
Byun HO, Jung HJ, Seo YH, Lee YK, Hwang SC, Hwang ES, Yoon G. GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) β1-induced senescence. Exp Cell Res. 2012;318(15):1808–19. https://doi.org/10.1186/1743-8977-10-18.
Carew JS, Huang P. Mitochondrial defects in cancer. Mol Cancer. 2002;1(1):1–2.
Castegna A, Iacobazzi V, Infantino V. The mitochondrial side of epigenetics. Physiol Genomics. 2015;47(8):299–307.
Cavalcante GC, Magalhães L, Ribeiro-dos-Santos Â, Vidal AF. Mitochondrial epigenetics: Non-coding RNAs as a novel layer of complexity. Int J Mol Sci. 2020;21(5):1838.
Uzuner SC. Mitochondrial DNA methylation misleads global DNA methylation detected by antibody-based methods. Anal Biochem. 2020;601:113789.
Cheng Z, Almeida F. Mitochondrial alteration in type 2 diabetes and obesity: an epigenetic link. Cell Cycle. 2014;13(6):890–7.
Chestnut BA, Chang Q, Price A, Lesuisse C, Wong M, Martin LJ. Epigenetic regulation of motor neuron cell death through DNA methylation. J Neurosci. 2011;31(46):16619–36.
DiNardo CD, Patel KP, Garcia-Manero G, Luthra R, Pierce S, Borthakur G, Jabbour E, Kadia T, Pemmaraju N, Konopleva M, Faderl S. Lack of association of IDH1, IDH2 and DNMT3A mutations with outcome in older patients with acute myeloid leukemia treated with hypomethylating agents. Leuk Lymphoma. 2014;55(8):1925–9.
Doke M, Jeganathan V, McLaughlin JP, Samikkannu T. HIV-1 Tat and cocaine impact mitochondrial epigenetics: effects on DNA methylation. Epigenetics. 2020 Oct 25:1–20.
Dong Z, Pu L, Cui H. Mitoepigenetics and its emerging roles in cancer. Front. Cell Dev. Biol. 2020 Jan 23;8:4.
Fan LH, Wang ZB, Li QN, Meng TG, Dong MZ, Hou Y, Ouyang YC, Schatten H, Sun QY. Absence of mitochondrial DNA methylation in mouse oocyte maturation, aging and early embryo development. Biochem Biophys Res Commun. 2019;513(4):912–8.
Feng S, Xiong L, Ji Z, Cheng W, Yang H. Correlation between increased ND2 expression and demethylated displacement loop of mtDNA in colorectal cancer. Mol. Med. Rep. 2012 Jul 1;6(1):125 – 30.
Ferreira A, Cunha-Oliveira T, Simões RF, Carvalho FS, Burgeiro A, Nordgren K, Wallace KB, Oliveira PJ. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology. 2017 Sep;1:390:63–73.
Fogg VC, Lanning NJ, MacKeigan JP. Mitochondria in cancer: at the crossroads of life and death. Chin J Cancer. 2011;30(8):526.
Gagliardi M, Strazzullo M, Matarazzo MR. DNMT3B functions: novel insights from human disease. Front Cell Dev Biol. 2018;6:140.
Gao D, Zhu B, Sun H, Wang X. Mitochondrial DNA methylation and related disease. Mitochondrial DNA and Diseases. 2017:117–32.
Gao J, Wen S, Zhou H, Feng S. De-methylation of displacement loop of mitochondrial DNA is associated with increased mitochondrial copy number and nicotinamide adenine dinucleotide subunit 2 expression in colorectal cancer. Mol Med Rep. 2015;12(5):7033–8.
Ghosh S, Singh KK, Sengupta S, Scaria V. Mitoepigenetics: the different shades of grey. Mitochondrion. 2015;25:60–6.
Gogvadze V, Zhivotovsky B, Orrenius S. The Warburg effect and mitochondrial stability in cancer cells. Mol Aspects Med. 2010;31(1):60–74.
Gong Y, Zhang X, Zhang Q, Zhang Y, Ye Y, Yu W, Shao C, Yan T, Huang J, Zhong J, Wang L. A natural DNMT1 mutation elevates the fetal hemoglobin level via epigenetic derepression of the γ-globin gene in β-thalassemia. Blood Am J Hematol. 2021;137(12):1652–7.
Grigoriu A, Ferreira JC, Choufani S, Baczyk D, Kingdom J, Weksberg R. Cell specific patterns of methylation in the human placenta. Epigenetics. 2011;6(3):368–79.
Gupta R, Nagarajan A, Wajapeyee N. Advances in genome-wide DNA methylation analysis. Biotechniques. 2010;49(4):iii–xi.
Han X, Zhao Z, Zhang M, Li G, Yang C, Du F, Wang J, Zhang Y, Wang Y, Jia Y, Li B. Maternal trans-general analysis of the human mitochondrial DNA pattern. Biochem Biophys Res Commun. 2017;493(1):643–9.
Hao Z, Wu T, Cui X, Zhu P, Tan C, Dou X, Hsu KW, Lin YT, Peng PH, Zhang LS, Gao Y. N6-deoxyadenosine methylation in mammalian mitochondrial DNA. Mol Cell. 2020;78(3):382–95.
Hardy TM, Tollefsbol TO. Epigenetic diet: impact on the epigenome and cancer. Epigenomics. 2011;3(4):503–18.
Hong EE, Okitsu CY, Smith AD, Hsieh CL. Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol Cell Biol. 2013;33(14):2683–90.
Iacobazzi V, Castegna A, Infantino V, Andria G. Mitochondrial DNA methylation as a next-generation biomarker and diagnostic tool. Mol Genet Metab. 2013;110(1–2):25–34.
Infantino V, Castegna A, Iacobazzi F, Spera I, Scala I, Andria G, Iacobazzi V. Impairment of methyl cycle affects mitochondrial methyl availability and glutathione level in Down’s syndrome. Mol Genet Metab. 2011;102(3):378–82.
Jackson T, Chougule MB, Ichite N, Patlolla RR, Singh M. Antitumor activity of noscapine in human non-small cell lung cancer xenograft model. Cancer Chemother Pharmacol. 2008;63(1):117–26.
Janssen BG, Byun HM, Gyselaers W, Lefebvre W, Baccarelli AA, Nawrot TS. Placental mitochondrial methylation and exposure to airborne particulate matter in the early life environment: An ENVIRON AGE birth cohort study. Epigenetics. 2015;10(6):536–44.
Jean SR, Tulumello DV, Riganti C, Liyanage SU, Schimmer AD, Kelley SO. Mitochondrial targeting of doxorubicin eliminates nuclear effects associated with cardiotoxicity. ACS Chem Biol. 2015;10(9):2007–15.
Jiang YL, Rigolet M, Bourc’his D, Nigon F, Bokesoy I, Fryns JP, Hulten M, Jonveaux P, Maraschio P, Megarbane A, Moncla A. DNMT3B mutations and DNA methylation defect define two types of ICF syndrome. Hum Mutat. 2005;25(1):56–63.
Jones PA, Issa JP, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet. 2016;17(10):630–41.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell metab. 2006;3(3):177–85.
Kopinski PK, Singh LN, Zhang S, Lott MT, Wallace DC. Mitochondrial DNA variation and cancer. Nat Rev Cancer. 2021;21(7):431–45.
Kubota T, Furuumi H, Kamoda T, Iwasaki N, Tobita N, Fujiwara N, Goto YI, Matsui A, Sasaki H, Kajii T. ICF syndrome in a girl with DNA hypomethylation but without detectable DNMT3B mutation. Am J Med Genet Part A. 2004;129(3):290–3.
Lambertini L, Byun HM. Mitochondrial epigenetics and environmental exposure. Curr Environ Health Rep. 2016;3(3):214–24.
Lee W, Johnson J, Gough DJ, Donoghue J, Cagnone GL, Vaghjiani V, Brown KA, Johns TG, John JS. Mitochondrial DNA copy number is regulated by DNA methylation and demethylation of POLGA in stem and cancer cells and their differentiated progeny. Cell Death Dis. 2015;6(2):e1664.
Lee HC, Wei YH. Mitochondria and aging. Advances in mitochondrial medicine. 2012:311 – 27.
Lenaz G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Advances in mitochondrial medicine. 2012:93–136.
Li Y, Tollefsbol TO. Impact on DNA methylation in cancer prevention and therapy by bioactive dietary components. Curr Med Chem. 2010;17(20):2141–51.
Linqing Y, Bo X, Xueqin Y, Hong D, Desheng W, Huimin Z, Gaofeng J, Jianjun L, Zhixiong Z. Mitochondrial DNA hypomethylation in chrome plating workers. Toxicol Lett. 2016;243:1–6.
Liu B, Du Q, Chen L, Fu G, Li S, Fu L, Zhang X, Ma C, Bin C. CpG methylation patterns of human mitochondrial DNA. Sci Rep. 2016;6(1):1–0.
Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem. 2002;80(5):780–7.
Lopes AF. Mitochondrial metabolism and DNA methylation: a review of the interaction between two genomes. Clin Epigenetics. 2020;12(1):1–3.
Lu Y, Chan YT, Tan HY, Li S, Wang N, Feng Y. Epigenetic regulation in human cancer: the potential role of epi-drug in cancer therapy. Mol Cancer. 2020;19(1):1–6.
Ma H, Sun J, Xu W, Dai Q, Hu G, Lai X, Yan B, Gao H. Profiles of mitochondrial DNA methylation in the ridgetail white prawn, Exopalaemon carinicauda (Holthuis, 1950) (Decapoda, Palaemonidae) under heavy metal stress from chromium and cadmium. Crustaceana. 2019;92(8):997–1005.
Mahajan A, Sapehia D, Thakur S, Mohanraj PS, Bagga R, Kaur J. Effect of imbalance in folate and vitamin B12 in maternal/parental diet on global methylation and regulatory miRNAs. Sci Rep. 2019;9(1):1–21.
Manev H, Dzitoyeva S, Chen H. Mitochondrial DNA: a blind spot in neuroepigenetics. Biomol. 2012;3(2):107–15.
Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007;18(5):190–8.
Maresca A, Del Dotto V, Capristo M, Scimonelli E, Tagliavini F, Morandi L, Tropeano CV, Caporali L, Mohamed S, Roberti M, Scandiffio L. DNMT1 mutations leading to neurodegeneration paradoxically reflect on mitochondrial metabolism. Hum Mol Genet. 2020;29(11):1864–81.
Matilainen O, Quirós PM, Auwerx J. Mitochondria and epigenetics–crosstalk in homeostasis and stress. Trends Cell Biol. 2017;27(6):453–63.
Mechta M, Ingerslev LR, Fabre O, Picard M, Barrès R. Evidence suggesting absence of mitochondrial DNA methylation. Front Genet. 2017;8:166.
Migheli F, Stoccoro A, Coppede F, Wan Omar WA, Failli A, Consolini R, Seccia M, Spisni R, Miccoli P, Mathers JC, Migliore L. Comparison study of MS-HRM and pyrosequencing techniques for quantification of APC and CDKN2A gene methylation. PLoS ONE. 2013;8(1):e52501.
Minocherhomji S, Tollefsbol TO, Singh KK. Mitochondrial regulation of epigenetics and its role in human diseases. Epigenetics. 2012;7(4):326–34.
Mohammed SA, Ambrosini S, Lüscher T, Paneni F, Costantino S. Epigenetic control of mitochondrial function in the vasculature. Front Cardiovasc Med. 2020;7:28.
Nass MM. Differential methylation of mitochondrial and nuclear DNA in cultured mouse, hamster and virus-transformed hamster cells in vivo and in vitro methylation. J Mol Biol. 1973;80(1):155–75.
Nass MM. The circularity of mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 1966;56(4):1215.
Owa C, Poulin M, Yan L, Shioda T. Technical adequacy of bisulfite sequencing and pyrosequencing for detection of mitochondrial DNA methylation: Sources and avoidance of false-positive detection. PLoS ONE. 2018;13(2):e0192722.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–97.
Pirola CJ, Gianotti TF, Burgueño AL, Rey-Funes M, Loidl CF, Mallardi P, San Martino J, Castaño GO, Sookoian S. Epigenetic modification of liver mitochondrial DNA is associated with histological severity of nonalcoholic fatty liver disease. Gut. 2013;62(9):1356–63.
Qu Y, Lennartsson A, Gaidzik VI, Deneberg S, Karimi M, Bengtzén S, Höglund M, Bullinger L, Döhner K, Lehmann S. Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics. 2014;9(8):1108–19.
Rana AK. Crime investigation through DNA methylation analysis: methods and applications in forensics. Egypt J Forensic Sci. 2018;8(1):1–7.
Russler-Germain DA, Spencer DH, Young MA, Lamprecht TL, Miller CA, Fulton R, Meyer MR, Erdmann-Gilmore P, Townsend RR, Wilson RK, Ley TJ. The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer cell. 2014 Apr 14;25(4):442 – 54.
Sanyal T, Bhattacharjee P, Bhattacharjee S. Hypomethylation of mitochondrial D-loop and ND6 with increased mitochondrial DNA copy number in the arsenic-exposed population. Toxicology. 2018 Sep 1;408:54–61.
Sanyal AJ, Campbell–Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, Luketic VA, Shiffman ML, Clore JN. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology. 2001;120(5):1183–92.
Sanyal T, Paul M, Bhattacharjee S, Bhattacharjee P. Epigenetic alteration of mitochondrial biogenesis regulatory genes in arsenic exposed individuals (with and without skin lesions) and in skin cancer tissues: A case control study. Chemosphere. 2020;258:127305.
Sharma J, Parsai K, Raghuwanshi P, Ali SA, Tiwari V, Bhargava A, Mishra PK. Emerging role of mitochondria in airborne particulate matter-induced immunotoxicity. Environ Pollut. 2021;270:116242.
Sharma N, Pasala MS, Prakash A. Mitochondrial DNA. Epigenetics and environment. Environ Mol Mutagen. 2019;60(8):668–82.
Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci. 2011;108(9):3630–5.
Stathopoulos S, Gaujoux R, Lindeque Z, Mahony C, Van Der Colff R, Van Der Westhuizen F, O’Ryan C. DNA methylation associated with mitochondrial dysfunction in a south African autism Spectrum disorder cohort. Autism Res. 2020;13(7):1079–93.
Stimpfel M, Jancar N, Virant-Klun I. New challenge: mitochondrial epigenetics? Stem Cell Rev Rep. 2018;14(1):13–26.
Stoccoro A, Coppedè F. Mitochondrial DNA Methylation and Human Diseases. Int J Mol Sci. 2021;22(9):4594.
Stoccoro A, Mosca L, Carnicelli V, Cavallari U, Lunetta C, Marocchi A, Migliore L, Coppedè F. Mitochondrial DNA copy number and D-loop region methylation in carriers of amyotrophic lateral sclerosis gene mutations. Epigenomics. 2018;10(11):1431–43.
Sun X, Vaghjiani V, Jayasekara WS, Cain JE, John JC. The degree of mitochondrial DNA methylation in tumor models of glioblastoma and osteosarcoma. Clin Epigenetics. 2018;10(1):1–7.
Tannorella P, Stoccoro A, Tognoni G, Petrozzi L, Salluzzo MG, Ragalmuto A, Siciliano G, Haslberger A, Bosco P, Bonuccelli U, Migliore L. Methylation analysis of multiple genes in blood DNA of Alzheimer’s disease and healthy individuals. Neurosci Lett. 2015;600:143–7.
Tong H, Zhang L, Gao J, Wen S, Zhou H, Feng S. Methylation of mitochondrial DNA displacement loop region regulates mitochondrial copy number in colorectal cancer. Mol Med Rep. 2017;16(4):5347–53.
Vos S, Nawrot TS, Martens DS, Byun HM, Janssen BG. Mitochondrial DNA methylation in placental tissue: A proof of concept study by means of prenatal environmental stressors. Epigenetics. 2021;16(2):121–31.
Wang P, Castellani CA, Yao J, Huan T, Bielak LF, Zhao W, Haessler J, Joehanes R, Sun X, Guo X, Longchamps RJ. Epigenome-wide association study of mitochondrial genome copy number. Hum Mol Genet. 2022;31(2):309–19.
Wu BK, Mei SC, Brenner C. RFTS-deleted DNMT1 enhances tumorigenicity with focal hypermethylation and global hypomethylation. Cell Cycle. 2014;13(20):3222–31.
Xu Y, Xu L, Han M, Liu X, Li F, Zhou X, Wang Y, Bi J. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an APP/PS1 transgenic mouse model of Alzheimer disease. Biochem Biophys Res Commun. 2019;520(1):41–6.
Yang Y, Xie B, Yan J. Application of next-generation sequencing technology in forensic science. Genom Proteom Bioinform. 2014;12(5):190–7.
Zheng LD, Linarelli LE, Brooke J, Smith C, Wall SS, Greenawald MH, Seidel RW, Estabrooks PA, Almeida FA, Cheng Z. Mitochondrial epigenetic changes link to increased diabetes risk and early-stage prediabetes indicator. Oxidative medicine and cellular longevity. 2016. 5290638.
Zheng LD, Linarelli LE, Liu L, Wall SS, Greenawald MH, Seidel RW, Estabrooks PA, Almeida FA, Cheng Z. Insulin resistance is associated with epigenetic and genetic regulation of mitochondrial DNA in obese humans. Clin Epigenetics. 2015;7(1):1–9.
Acknowledgements
The authors are grateful to University of Calcutta, Council for Industrial and Scientific Research (for providing Junior Research Fellowship to AD) and Department of Biotechnology (DBT-Twinning to PB).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Corresponding Editor : Sandipan Brahma; Reviewers : Srinivas Animireddy, Somnath Paul
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sanyal, T., Das, A., Bhowmick, P. et al. Interplay between environmental exposure and mitochondrial DNA methylation in disease susceptibility and cancer: a comprehensive review. Nucleus 66, 53–68 (2023). https://doi.org/10.1007/s13237-022-00392-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13237-022-00392-5